




Extracellular Vesicles, Cell-Penetrating Peptides and miRNAs as Future Novel Therapeutic Interventions for Parkinson’s and Alzheimer’s Disease


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
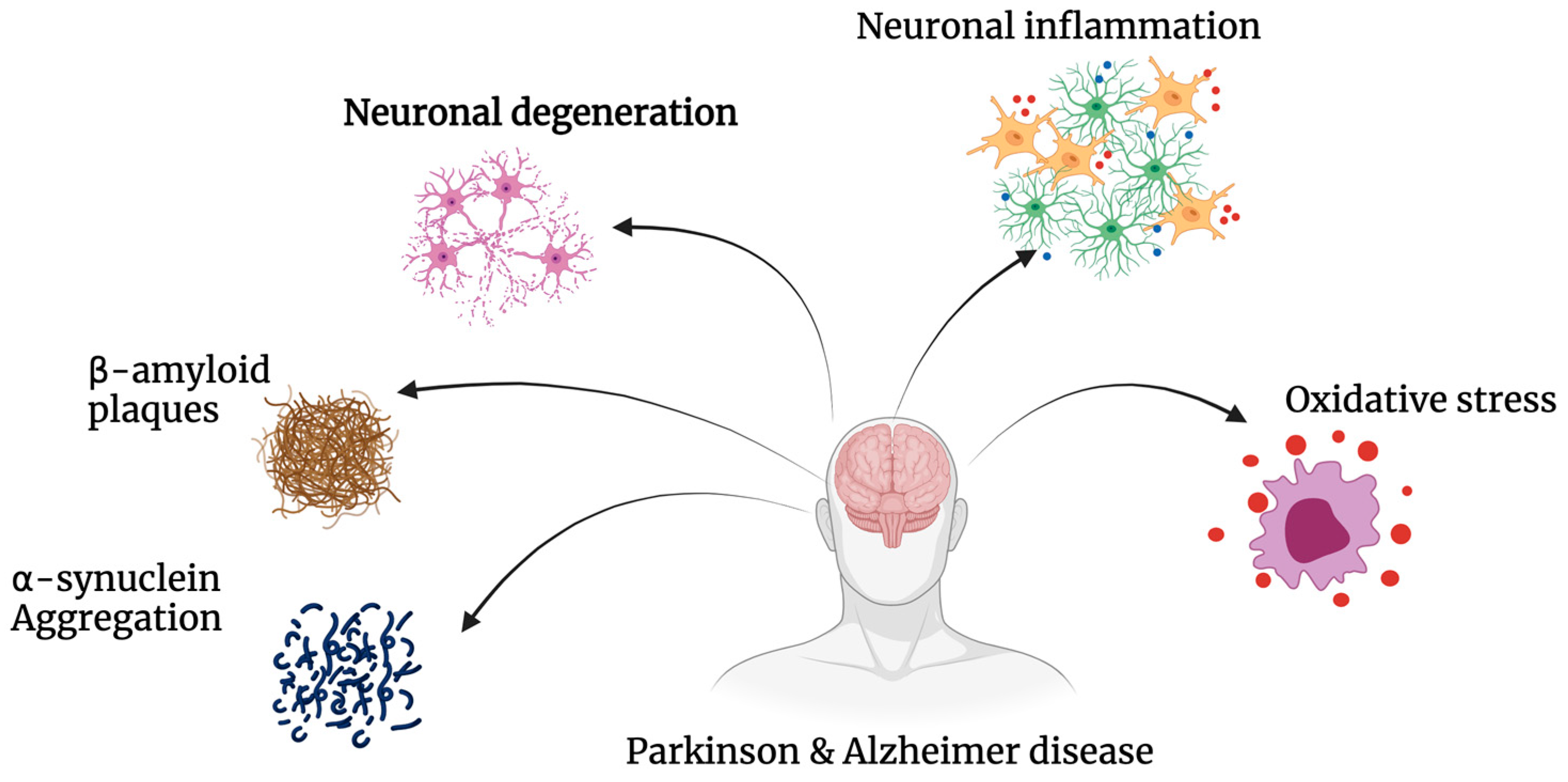
2. Aetiology and Pathophysiology of Disease
3. Current Therapeutic Approaches
3.1. Current Therapeutic Approaches—Alzheimer’s Disease
3.1.1. Pharmacology-Based Therapies
3.1.2. Surgery-Based Therapies
3.2. Current Therapeutic Approaches—Parkinson’s Disease
3.2.1. Pharmacology-Based Therapies
3.2.2. Surgery-Based Therapies
3.2.3. Physical Therapies
4. Potential Future Novel Therapeutic Interventions
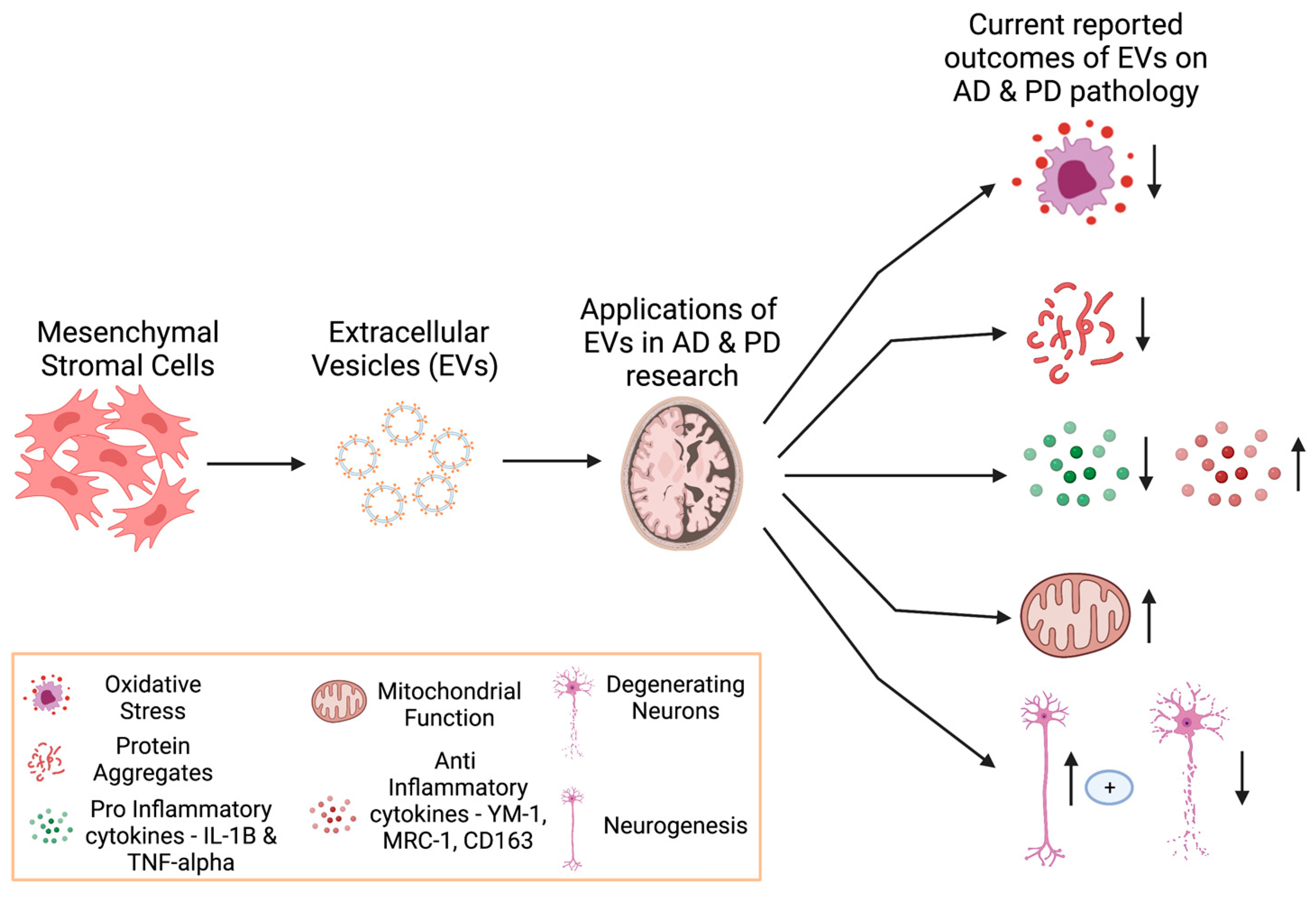
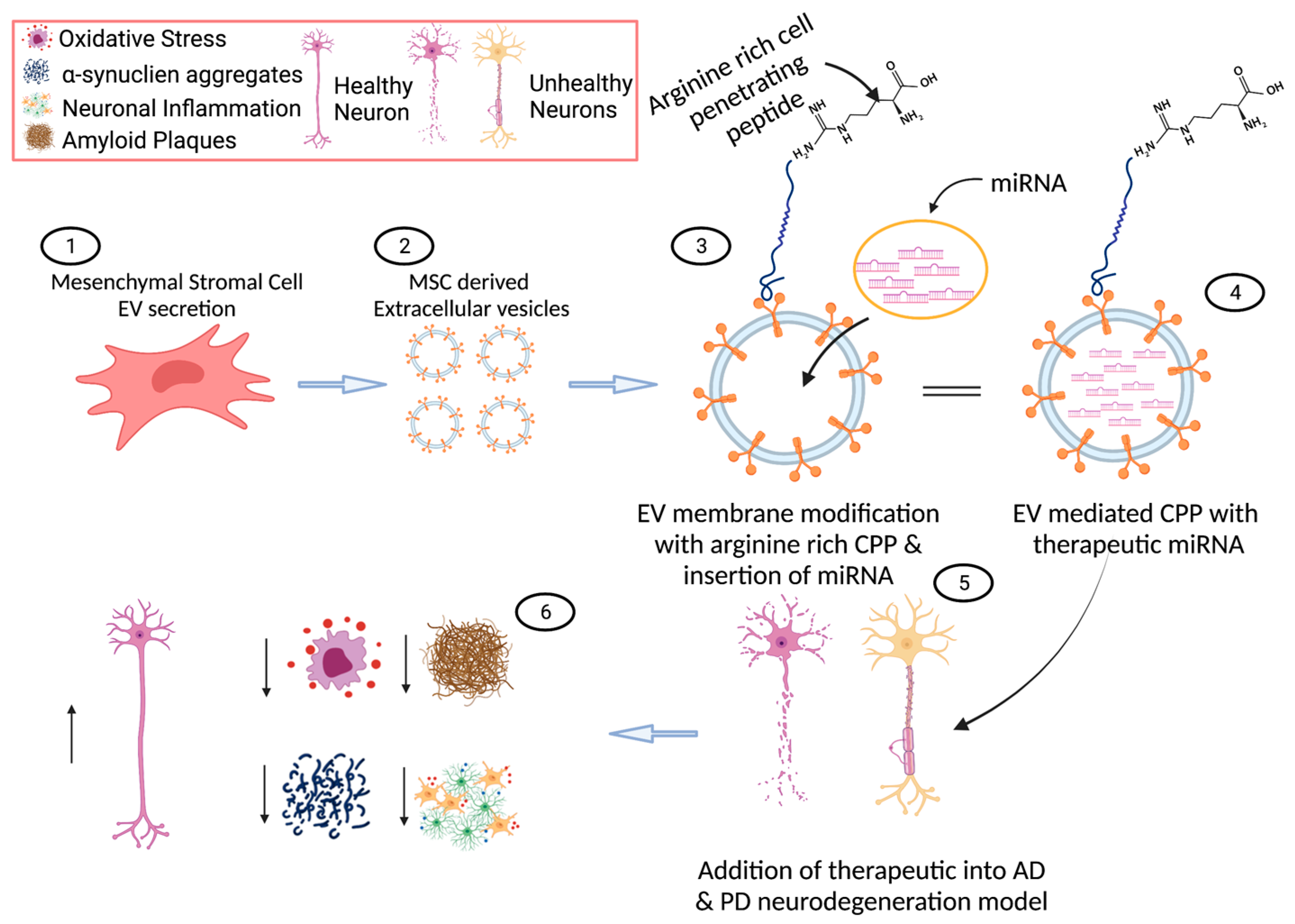
4.1. Extracellular Vesicles in Neurodegeneration
4.2. Extracellular Vesicles as Cargo Delivery Vehicles
- Their ability to migrate across the BBB, in a bi-directional manner, with several studies showing their proficiency in this area, albeit with very little information about their mechanism of action [128,129,130]. A study evaluating 6-hydroxy-dopamine (6-OHDA)-induced ND on dopaminergic neurons in SH-SH5Y cells found that MSC-derived EVs reduced DA neuronal death in this in vitro model. Furthermore, they found these EVs also reduced toxicity and neuronal death in their rat model [131].
- They can carry miRNAs. EVs can mediate the movement and delivery of many miRNAs, including miR-21. This miR is usually associated with the microglial anti-inflammatory (although in other cases it has been reported to be inflammatory) response and indicated as a potential new AD biomarker. This has been demonstrated in in vitro studies of SH-SH5Y cells that were transfected with APP [132,133].
- EVs can be utilized as a cargo vehicle for therapeutic agents that target neurodegeneration. For example, curcumin is a drug that is suggested to reduce neuroinflammation and oxidative stress in AD, yet it lacks the ability to cross the BBB alone. It has been suggested that combining EVs and curcumin may help overcome some of these shortcomings [134,135,136]. In PD, dopamine has been trialled as a potential drug to be carried by EVs to enhance its delivery to cells, which seems to create a less transient expression of dopamine and increased neuronal function in their model [133,137]. Therapeutic proteins are loaded into exosomes ex vivo and can deliver neuroprotective effects on specific cells in the brain, with EVs crossing the BBB. They cross the BBB through targeting endothelial cells via receptor interactions, thus entering the cell through membrane fusion and endocytosis. Catalase loaded into exosomes has shown neuroprotective effects in the microglial cells, with a reduction in microgliosis in their 6OHDA-induced neurodegeneration model, as measured by a reduction in CD111b expression [138]. EVs augmented or transfected with plasmids have also been used in animal models of PD to evaluate their therapeutic effect. EVs with catalase-encoded plasmids have been shown to transverse the BBB, to incorporate with neurons and ameliorate PD symptoms in mouse models [138]. Macrophages that were genetically modified to express glial-derived neurotrophic factor (GDNF) were found to express modified EVs with GDNF and were shown to slow the progression of PD via a reduction in neuro-inflammation [139].
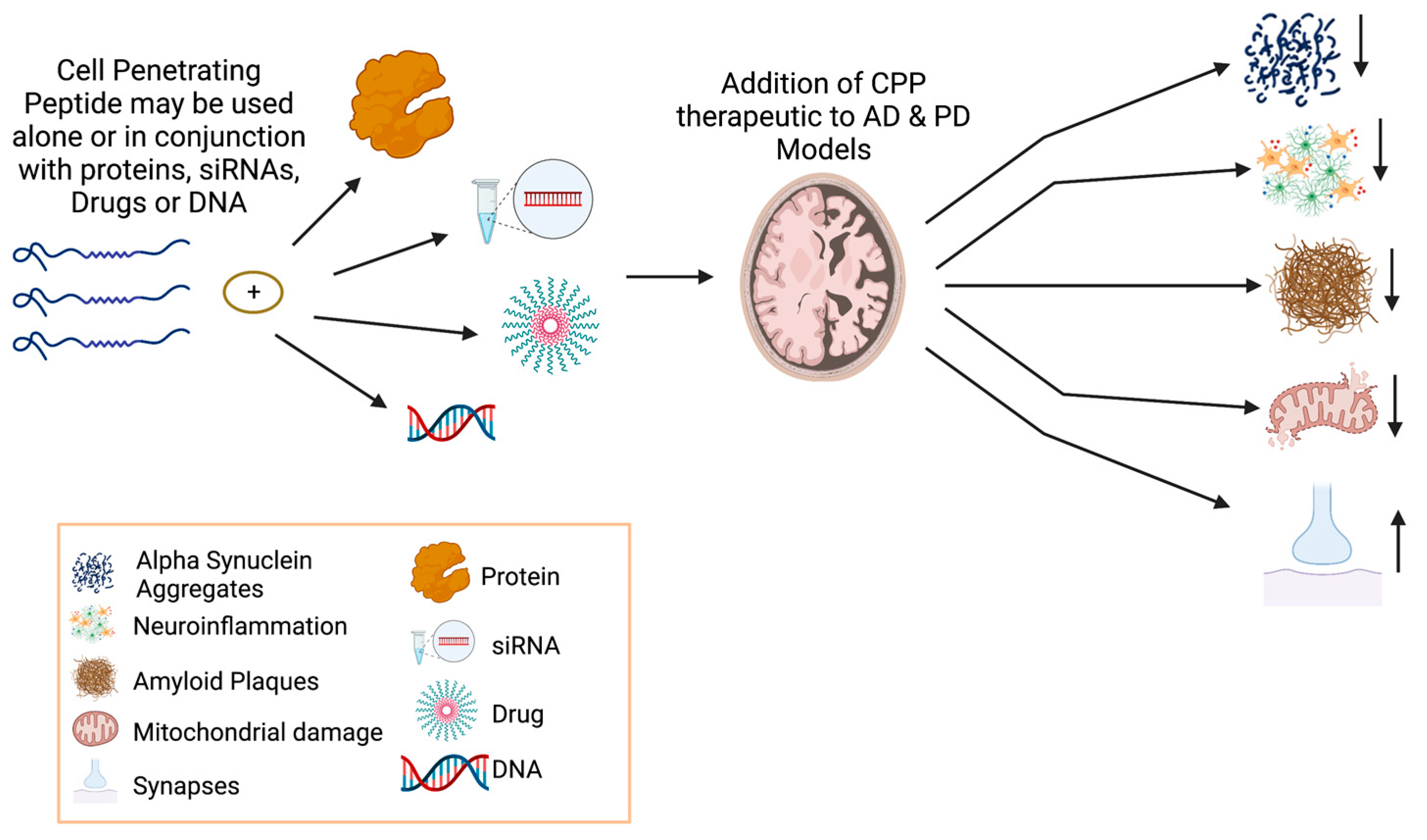
4.3. Cell-Penetrating Peptides (CPPs) in Neurodegeneration
4.4. Cell-Penetrating Peptides (CPPs) as Cargo Delivery Vehicles
4.5. Micro RNAs (miRNAs) in Neurodegeneration
5. Next Generation of Novel Therapeutics in AD and PD
6. Current Barriers to Clinical Translation
7. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Dugger, B.N.; Dickson, D.W. Pathology of neurodegenerative diseases. Cold Spring Harb. Perspect. Biol. 2017, 9, a028035. [Google Scholar] [CrossRef] [PubMed]
- Kurosinski, P.; Guggisberg, M.; Götz, J. Alzheimer’s and Parkinson’s disease—Overlapping or synergistic pathologies? Trends Mol. Med. 2002, 8, 3–5. [Google Scholar] [CrossRef]
- Uttara, B.; Singh, A.V.; Zamboni, P.; Mahajan, R.T. Oxidative Stress and Neurodegenerative Diseases: A Review of Upstream and Downstream Antioxidant Therapeutic Options. Curr. Neuropharmacol. 2009, 7, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.H.; Karri, V.; Tay, N.W.R.; Chang, K.H.; Ah, H.Y.; Ng, P.Q.; Ho, H.S.; Keh, H.W.; Candasamy, M. Emerging pathways to neurodegeneration: Dissecting the critical molecular mechanisms in Alzheimer’s disease, Parkinson’s disease. Biomed. Pharmacother. 2019, 111, 765–777. [Google Scholar] [CrossRef]
- Xie, A.; Gao, J.; Xu, L.; Meng, D. Shared mechanisms of neurodegeneration in Alzheimer’s disease and Parkinson’s disease. Biomed. Res. Int. 2014, 2014, 648740. [Google Scholar] [CrossRef] [PubMed]
- Guadagnolo, D.; Piane, M.; Torrisi, M.R.; Pizzuti, A.; Petrucci, S. Genotype-Phenotype Correlations in Monogenic Parkinson Disease: A Review on Clinical and Molecular Findings. Front. Neurol. 2021, 12, 648588. [Google Scholar] [CrossRef]
- Kiriyama, Y.; Nochi, H. The Function of Autophagy in Neurodegenerative Diseases. Int. J. Mol. Sci. 2015, 16, 26797–26812. [Google Scholar] [CrossRef] [PubMed]
- Scorziello, A.; Borzacchiello, D.; Sisalli, M.J.; Di Martino, R.; Morelli, M.; Feliciello, A. Mitochondrial Homeostasis and Signaling in Parkinson’s Disease. Front. Aging Neurosci. 2020, 12, 100. [Google Scholar] [CrossRef]
- Dias, V.; Junn, E.; Mouradian, M.M. The Role of Oxidative Stress in Parkinson’s Disease. J. Parkinsons Dis. 2013, 3, 461–491. [Google Scholar] [CrossRef] [PubMed]
- Gan, L.; Cookson, M.R.; Petrucelli, L.; La Spada, A.R. Converging pathways in neurodegeneration, from genetics to mechanisms. Nat. Neurosci. 2018, 21, 1300–1309. [Google Scholar] [CrossRef]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.-E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Prim. 2017, 3, 17013. [Google Scholar] [CrossRef] [PubMed]
- Apostolova, L.G. Alzheimer Disease. Continuum 2016, 22, 419–434. [Google Scholar] [CrossRef] [PubMed]
- Wattmo, C.; Minthon, L.; Wallin, K. Mild versus moderate stages of Alzheimer’s disease: Three-year outcomes in a routine clinical setting of cholinesterase inhibitor therapy. Alzheimer Res. Ther. 2016, 8, 7. [Google Scholar] [CrossRef]
- Dubois, B.; Hampel, H.; Feldman, H.H.; Scheltens, P.; Aisen, P.; Andrieu, S.; Bakardjian, H.; Benali, H.; Bertram, L.; Blennow, K.; et al. Preclinical Alzheimer’s disease: Definition, natural history, and diagnostic criteria. Alzheimers Dement. 2016, 12, 292–323. [Google Scholar] [CrossRef]
- McKhann, G.; Drachman, D.; Folstein, M.; Katzman, R.; Price, D.; Stadlan, E.M. Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 1984, 34, 939–944. [Google Scholar] [CrossRef]
- Neugroschl, J.; Wang, S. Alzheimer’s disease: Diagnosis and treatment across the spectrum of disease severity. Mt. Sinai J. Med. 2011, 78, 596–612. [Google Scholar] [CrossRef] [PubMed]
- Breijyeh, Z.; Karaman, R. Comprehensive Review on Alzheimer’s Disease: Causes and Treatment. Molecules 2020, 25, 5789. [Google Scholar] [CrossRef]
- Singh, S.K.; Srivastav, S.; Yadav, A.K.; Srikrishna, S.; Perry, G. Overview of alzheimer’s disease and some therapeutic approaches targeting aβ by using several synthetic and herbal compounds. Oxid. Med. Cell. Longev. 2016, 2016, 7361613. [Google Scholar] [CrossRef]
- Spires-Jones, T.L.; Hyman, B.T. The Intersection of Amyloid Beta and Tau at Synapses in Alzheimer’s Disease. Neuron 2014, 82, 756–771. [Google Scholar] [CrossRef]
- Armstrong, R.A. Risk factors for Alzheimer’s disease. Folia Neuropathol. 2019, 57, 87–105. [Google Scholar] [CrossRef]
- Emamzadeh, F.N.; Surguchov, A. Parkinson’s disease: Biomarkers, treatment, and risk factors. Front. Neurosci. 2018, 12, 612. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, M.J.; Okun, M.S. Diagnosis and treatment of parkinson disease: A review. JAMA 2020, 323, 548–560. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Del Tredici, K.; Rüb, U.; de Vos, R.A.I.; Jansen Steur, E.N.H.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological Alterations in Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef]
- Stoker, T.B.; Torsney, K.M.; Barker, R.A. Emerging Treatment Approaches for Parkinson’s Disease. Front. Neurosci. 2018, 12, 693. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- El-Agnaf, O.M.A.; Salem, S.A.; Paleologou, K.E.; Curran, M.D.; Gibson, M.J.; Court, J.A.; Schlossmacher, M.G.; Allsop, D. Detection of oligomeric forms of α-synuclein protein in human plasma as a potential biomarker for Parkinson’s disease. FASEB J. 2006, 20, 419–425. [Google Scholar] [CrossRef]
- Bekris, L.M.; Mata, I.F.; Zabetian, C.P. The genetics of Parkinson disease. J. Geriatr. Psychiatry Neurol. 2010, 23, 228–242. [Google Scholar] [CrossRef]
- Simón-Sánchez, J.; Schulte, C.; Bras, J.M.; Sharma, M.; Gibbs, J.R.; Berg, D.; Paisan-Ruiz, C.; Lichtner, P.; Scholz, S.W.; Hernandez, D.G.; et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat. Genet. 2009, 41, 1308–1312. [Google Scholar] [CrossRef]
- Piaceri, I.; Nacmias, B.; Sorbi, S. Genetics of familial and sporadic Alzheimer s disease. Front. Biosci. 2013, E5, 167–177. [Google Scholar] [CrossRef]
- Avazzadeh, S.; Baena, J.M.; Keighron, C.; Feller-Sanchez, Y.; Quinlan, L.R. Modelling Parkinson’s Disease: iPSCs towards Better Understanding of Human Pathology. Brain Sci. 2021, 11, 373. [Google Scholar] [CrossRef] [PubMed]
- Morgan, S.L.; Naderi, P.; Koler, K.; Pita-Juarez, Y.; Prokopenko, D.; Vlachos, I.S.; Tanzi, R.E.; Bertram, L.; Hide, W.A. Most pathways can be related to the pathogenesis of alzheimer’s disease. Front. Aging Neurosci. 2022, 14, 846902. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, K.; Baig, M.H.; Mushtaq, G.; Kamal, M.A.; Greig, N.H.; Choi, I. Commonalities in Biological Pathways, Genetics, and Cellular Mechanism between Alzheimer Disease and Other Neurodegenerative Diseases: An In Silico-Updated Overview. Curr. Alzheimer Res. 2017, 14, 1190–1197. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef]
- De Strooper, B.; Vassar, R.; Golde, T. The secretases: Enzymes with therapeutic potential in Alzheimer disease. Nat. Rev. Neurol. 2010, 6, 99–107. [Google Scholar] [CrossRef]
- Braak, H.; Alafuzoff, I.; Arzberger, T.; Kretzschmar, H.; Del Tredici, K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006, 112, 389–404. [Google Scholar] [CrossRef]
- Alonso, A.D.C.; Grundke-Iqbal, I.; Iqbal, K. Alzheimer’s disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nat. Med. 1996, 2, 783–787. [Google Scholar] [CrossRef]
- Wiersma, V.I.; Hoozemans, J.J.M.; Scheper, W. Untangling the origin and function of granulovacuolar degeneration bodies in neurodegenerative proteinopathies. Acta Neuropathol. Commun. 2020, 8, 153. [Google Scholar] [CrossRef]
- Castellani, R.J.; Gupta, Y.; Sheng, B.; Siedlak, S.L.; Harris, P.L.; Coller, J.M.; Perry, G.; Lee, H.-G.; Tabaton, M.; Smith, M.A.; et al. A novel origin for granulovacuolar degeneration in aging and Alzheimer’s disease: Parallels to stress granules. Lab. Investig. 2011, 91, 1777–1786. [Google Scholar] [CrossRef]
- Lang, A.E.; Lozano, A.M. Parkinson’s disease. First of two parts. N. Engl. J. Med. 1998, 339, 1044–1053. [Google Scholar] [CrossRef]
- Hoang, Q.Q. Pathway for Parkinson disease. Proc. Natl. Acad. Sci. USA 2014, 111, 2402–2403. [Google Scholar] [CrossRef] [PubMed]
- Shastry, B.S. Parkinson disease: Etiology, pathogenesis and future of gene therapy. Neurosci. Res. 2001, 41, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Corti, O.; Hampe, C.; Darios, F.; Ibanez, P.; Ruberg, M.; Brice, A. Parkinson’s disease: From causes to mechanisms. Comptes Rendus Biol. 2005, 328, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-H.; Burgess, J.D.; Faroqi, A.H.; DeMeo, N.N.; Fiesel, F.C.; Springer, W.; Delenclos, M.; McLean, P.J. Alpha-synuclein-induced mitochondrial dysfunction is mediated via a sirtuin 3-dependent pathway. Mol. Neurodegener. 2020, 15, 5. [Google Scholar] [CrossRef]
- Devi, L.; Raghavendran, V.; Prabhu, B.M.; Avadhani, N.G.; Anandatheerthavarada, H.K. Mitochondrial Import and Accumulation of alpha-Synuclein Impair Complex I in Human Dopaminergic Neuronal Cultures and Parkinson Disease Brain. J. Biol. Chem. 2008, 283, 9089–9100. [Google Scholar] [CrossRef] [PubMed]
- Darios, F.; Corti, O.; Hampe, C.; Muriel, M.-P.; Abbas, N.; Gu, W.-J.; Lücking, C.B.; Hirsch, E.C.; Rooney, T.; Ruberg, M.; et al. Parkin prevents mitochondrial swelling and cytochrome c release in mitochondria-dependent cell death. Hum. Mol. Genet. 2003, 12, 517–526. [Google Scholar] [CrossRef]
- Zhang, Y.; Gao, J.; Chung, K.K.K.; Huang, H.; Dawson, V.L.; Dawson, T.M. Parkin functions as an E2-dependent ubiquitin- protein ligase and promotes the degradation of the synaptic vesicle-associated protein, CDCrel-1. Proc. Natl. Acad. Sci. USA 2000, 97, 13354–13359. [Google Scholar] [CrossRef]
- Grimaldo, L.; Sandoval, A.; Garza-López, E.; Felix, R. Involvement of Parkin in the ubiquitin proteasome system-mediated degradation of N-type voltage-gated Ca2+ channels. PLoS ONE 2017, 12, e0185289. [Google Scholar] [CrossRef]
- Healy, D.G.; Abou-Sleiman, P.M.; Valente, E.M.; Gilks, W.P.; Bhatia, K.; Quinn, N.; Lees, A.J.; Wood, N.W. DJ-1 mutations in Parkinson’s disease. J. Med. Genet. 2004, 41, 248. [Google Scholar]
- Höglinger, G.U.; Carrard, G.; Michel, P.P.; Medja, F.; Lombès, A.; Ruberg, M.; Friguet, B.; Hirsch, E. Dysfunction of mitochondrial complex I and the proteasome: Interactions between two biochemical deficits in a cellular model of Parkinson’s disease. J. Neurochem. 2003, 86, 1297–1307. [Google Scholar] [CrossRef]
- McNaught, K.S.; Jenner, P. Proteasomal function is impaired in substantia nigra in Parkinson’s disease. Neurosci. Lett. 2001, 297, 191–194. [Google Scholar] [CrossRef]
- Abou-Sleiman, P.M.; Healy, D.G.; Quinn, N.; Lees, A.J.; Wood, N.W. The role of pathogenicDJ-1 mutations in Parkinson’s disease. Ann. Neurol. 2003, 54, 283–286. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Kim, K.S.; Iyirhiaro, G.O.; Marcogliese, P.C.; Callaghan, S.M.; Qu, D.; Kim, W.J.; Slack, R.S.; Park, D.S. DJ-1 modulates the unfolded protein response and cell death via upregulation of ATF4 following ER stress. Cell Death Dis. 2019, 10, 135. [Google Scholar] [CrossRef] [PubMed]
- Szeto, J.Y.; Lewis, S.J. Current Treatment Options for Alzheimer’s Disease and Parkinson’s Disease Dementia. Curr. Neuropharmacol. 2016, 14, 326–338. [Google Scholar] [CrossRef] [PubMed]
- Mehta, M.; Adem, A.; Sabbagh, M. New Acetylcholinesterase Inhibitors for Alzheimer’s Disease. Int. J. Alzheimers Dis. 2012, 2012, 728983. [Google Scholar] [CrossRef] [PubMed]
- Colović, M.B.; Krstić, D.Z.; Lazarević-Pašti, T.D.; Bondžić, A.M.; Vasić, V.M. Acetylcholinesterase inhibitors: Pharmacology and toxicology. Curr. Neuropharmacol. 2013, 11, 315–335. [Google Scholar] [CrossRef]
- Grossberg, G.T. Cholinesterase Inhibitors for the Treatment of Alzheimer’s Disease. Current Ther. Res. 2003, 64, 216–235. [Google Scholar]
- Sharma, K. Cholinesterase inhibitors as Alzheimer’s therapeutics (Review). Mol. Med. Rep. 2019, 20, 1479–1487. [Google Scholar] [CrossRef]
- Olivares, D.; Deshpande, V.K.; Shi, Y.; Lahiri, D.K.; Greig, N.H.; Rogers, J.T.; Huang, X. N-methyl D-aspartate (NMDA) receptor antagonists and memantine treatment for Alzheimer’s disease, vascular dementia and Parkinson’s disease. Curr. Alzheimer Res. 2012, 9, 746–758. [Google Scholar] [CrossRef]
- Liu, J.; Chang, L.; Song, Y.; Li, H.; Wu, Y. The Role of NMDA Receptors in Alzheimer’s Disease. Front. Neurosci. 2019, 13, 43. [Google Scholar] [CrossRef]
- Laxton, A.W.; Stone, S.; Lozano, A.M. The Neurosurgical Treatment of Alzheimer’s Disease: A Review. Ster. Funct. Neurosurg. 2014, 92, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Adams, R.D.; Fisher, C.M.; Hakim, S.; Ojemann, R.G.; Sweet, W.H. Symptomatic occult hydrocephalus with “normal” cerebrospinal-fluid pressure. A treatable syndrome. N. Engl. J. Med. 1965, 273, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Hulstaert, F.; Blennow, K.; Ivanoiu, A.; Schoonderwaldt, H.C.; Riemenschneider, M.; De Deyn, P.P.; Bancher, C.; Cras, P.; Wiltfang, J.; Mehta, P.D.; et al. Improved discrimination of AD patients using beta-amyloid(1-42) and tau levels in CSF. Neurology 1999, 52, 1555–1562. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, H.S.; Wu, W.; Zhong, J.; Edgar, M. Omental transposition to the brain as a surgical method for treating Alzheimer’s disease. Neurol. Res. 2003, 25, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, H.S. A new approach to the treatment of Alzheimer’s disease: The need for a controlled study. J. Alzheimers Dis. 2011, 25, 209–212. [Google Scholar] [CrossRef]
- DeMaagd, G.; Philip, A. Parkinson’s disease and its management: Part 1: Disease entity, risk factors, pathophysiology, clinical presentation, and diagnosis. P T 2015, 40, 504–532. [Google Scholar] [PubMed]
- Jankovic, J.; Aguilar, L.G. Current approaches to the treatment of Parkinson’s disease. Neuropsychiatr. Dis. Treat. 2008, 4, 743–757. [Google Scholar] [CrossRef]
- van Laar, T.; De Deyn, P.P.; Aarsland, D.; Barone, P.; Galvin, J.E. Effects of cholinesterase inhibitors in Parkinson’s disease dementia: A review of clinical data. CNS Neurosci. Ther. 2011, 17, 428–441. [Google Scholar] [CrossRef]
- Tysnes, O.-B.; Storstein, A. Epidemiology of Parkinson’s disease. J. Neural. Transm. 2017, 124, 901–905. [Google Scholar] [CrossRef]
- Pagano, G.; Rengo, G.; Pasqualetti, G.; Femminella, G.D.; Monzani, F.; Ferrara, N.; Tagliati, M. Cholinesterase inhibitors for Parkinson’s disease: A systematic review and meta-analysis. J. Neurol. Neurosurg. Psychiatry 2014, 86, 767–773. [Google Scholar] [CrossRef]
- Zahoor, I.; Shafi, A.; Haq, E. Pharmacological treatment of parkinson’s disease. In Parkinson’s Disease: Pathogenesis and Clinical Aspects; Stoker, T.B., Greenl, J.C., Eds.; Codon Publications: Brisbane, Australia, 2018. [Google Scholar]
- Schapira, A.H.V. Present and future drug treatment for Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 2005, 76, 1472–1478. [Google Scholar] [CrossRef]
- Carrarini, C.; Russo, M.; Dono, F.; Di Pietro, M.; Rispoli, M.G.; Di Stefano, V.; Ferri, L.; Barbone, F.; Vitale, M.; Thomas, A.; et al. A Stage-Based Approach to Therapy in Parkinson’s Disease. Biomolecules 2019, 9, 388. [Google Scholar] [CrossRef] [PubMed]
- Antonini, A.; Odin, P.; Pahwa, R.; Aldred, J.; Alobaidi, A.; Jalundhwala, Y.J.; Kukreja, P.; Bergmann, L.; Inguva, S.; Bao, Y.; et al. The Long-Term Impact of Levodopa/Carbidopa Intestinal Gel on ‘Off’-time in Patients with Advanced Parkinson’s Disease: A Systematic Review. Adv. Ther. 2021, 38, 2854–2890. [Google Scholar] [CrossRef]
- Hurtig, H.I. Problems with Current Pharmacologic Treatment of Parkinson’s Disease. Exp. Neurol. 1997, 144, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Ahlskog, J.E. Treatment of early Parkinson’s disease: Are complicated strategies justified? Mayo Clin. Proc. 1996, 71, 659–670. [Google Scholar] [CrossRef] [PubMed]
- Mouchaileh, N.; Hughes, A.J. Pharmacological management of Parkinson’s disease in older people. J. Pharm. Pract. Res. 2020, 50, 445–454. [Google Scholar] [CrossRef]
- Companys-Alemany, J.; Turcu, A.L.; Bellver-Sanchis, A.; Loza, M.I.; Brea, J.M.; Canudas, A.M.; Leiva, R.; Vázquez, S.; Pallàs, M.; Griñán-Ferré, C. A Novel NMDA Receptor Antagonist Protects against Cognitive Decline Presented by Senescent Mice. Pharmaceutics 2020, 12, 284. [Google Scholar] [CrossRef]
- Bronstein, J.M.; Tagliati, M.; Alterman, R.L.; Lozano, A.M.; Volkmann, J.; Stefani, A.; Horak, F.B.; Okun, M.S.; Foote, K.D.; Krack, P.; et al. Deep brain stimulation for Parkinson disease: An expert consensus and review of key issues. Arch. Neurol. 2011, 68, 165. [Google Scholar] [CrossRef]
- Herzog, J.; Fietzek, U.; Hamel, W.; Morsnowski, A.; Steigerwald, F.; Schrader, B.; Weinert, D.M.; Pfister, G.; Müller, D.; Mehdorn, H.M.; et al. Most effective stimulation site in subthalamic deep brain stimulation for Parkinson’s disease. Mov. Disord. 2004, 19, 1050–1054. [Google Scholar] [CrossRef]
- Lozano, A.M.; Lipsman, N.; Bergman, H.; Brown, P.; Chabardes, S.; Chang, J.W.; Matthews, K.; McIntyre, C.C.; Schlaepfer, T.E.; Schulder, M.; et al. Deep brain stimulation: Current challenges and future directions. Nat. Rev. Neurol. 2019, 15, 148–160. [Google Scholar] [CrossRef]
- Luo, Y.; Sun, Y.; Tian, X.; Zheng, X.; Wang, X.; Li, W.; Wu, X.; Shu, B.; Hou, W. Deep Brain Stimulation for Alzheimer’s Disease: Stimulation Parameters and Potential Mechanisms of Action. Front. Aging Neurosci. 2021, 13, 619543. [Google Scholar] [CrossRef] [PubMed]
- Logsdon, R.G.; McCurry, S.M.; Teri, L. Evidence-Based Interventions to Improve Quality of Life for Individuals with Dementia. Alzheimers Care Today 2007, 8, 309–318. [Google Scholar] [PubMed]
- Vieregge, P.; Dethlefsen, J. Physical therapy and speech therapy in Parkinson syndrome—A status assessment. Fortschr. Neurol. Psychiatr. 1992, 60, 369–374. [Google Scholar] [CrossRef]
- De la Rosa, A.; Olaso-Gonzalez, G.; Arc-Chagnaud, C.; Millan, F.; Salvador-Pascual, A.; García-Lucerga, C.; Blasco-Lafarga, C.; Garcia-Dominguez, E.; Carretero, A.; Correas, A.G.; et al. Physical exercise in the prevention and treatment of Alzheimer’s disease. J. Sport Health Sci. 2020, 9, 394–404. [Google Scholar] [CrossRef] [PubMed]
- Borrione, P.; Tranchita, E.; Sansone, P.; Parisi, A. Effects of physical activity in Parkinson’s disease: A new tool for rehabilitation. World J. Methodol. 2014, 4, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Duncan, T.; Valenzuela, M. Alzheimer’s disease, dementia, and stem cell therapy. Stem. Cell Res. Ther. 2017, 8, 111. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Cheung, H.-H. Stem Cell-Based Therapies for Parkinson Disease. Int. J. Mol. Sci. 2020, 21, 8060. [Google Scholar] [CrossRef]
- Liu, X.-Y.; Yang, L.-P.; Zhao, L. Stem cell therapy for Alzheimer’s disease. World J. Stem Cells 2020, 12, 787–802. [Google Scholar] [CrossRef]
- Gosselet, F.; Loiola, R.A.; Roig, A.; Rosell, A.; Culot, M. Central nervous system delivery of molecules across the blood-brain barrier. Neurochem. Int. 2021, 144, 104952. [Google Scholar] [CrossRef]
- Nguyen, D.D.; Lai, J.-Y. Synthesis, bioactive properties, and biomedical applications of intrinsically therapeutic nanoparticles for disease treatment. Chem. Eng. J. 2022, 435, 134970. [Google Scholar] [CrossRef]
- Veziroglu, E.M.; Mias, G.I. Characterizing Extracellular Vesicles and Their Diverse RNA Contents. Front. Genet. 2020, 11, 700. [Google Scholar] [CrossRef] [PubMed]
- van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Jain, S.K. Application potential of engineered liposomes in tumor targeting. In Multifunctional Systems for Combined Delivery, Biosensing and Diagnostics; Elsevier: Amsterdam, The Netherlands, 2017; pp. 171–191. [Google Scholar]
- Xie, J.; Bi, Y.; Zhang, H.; Dong, S.; Teng, L.; Lee, R.J.; Yang, Z. Cell-Penetrating Peptides in Diagnosis and Treatment of Human Diseases: From Preclinical Research to Clinical Application. Front. Pharmacol. 2020, 11, 697. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef]
- Vasudevan, S. Posttranscriptional Upregulation by MicroRNAs. Wiley Interdiscip. Rev. RNA 2012, 3, 311–330. [Google Scholar] [CrossRef]
- Robinson, D.G.; Ding, Y.; Jiang, L. Unconventional protein secretion in plants: A critical assessment. Protoplasma 2016, 253, 31–43. [Google Scholar] [CrossRef]
- Deatherage, B.L.; Cookson, B.T. Membrane Vesicle Release in Bacteria, Eukaryotes, and Archaea: A Conserved yet Underappreciated Aspect of Microbial Life. Infect. Immun. 2012, 80, 1948–1957. [Google Scholar] [CrossRef]
- Croese, T.; Furlan, R. Extracellular vesicles in neurodegenerative diseases. Mol. Asp. Med. 2018, 60, 52–61. [Google Scholar] [CrossRef]
- Hill, A.F. Extracellular Vesicles and Neurodegenerative Diseases. J. Neurosci. 2019, 39, 9269–9273. [Google Scholar] [CrossRef]
- Doyle, L.; Wang, M. Overview of Extracellular Vesicles, Their Origin, Composition, Purpose, and Methods for Exosome Isolation and Analysis. Cells 2019, 8, 727. [Google Scholar] [CrossRef]
- Kakarla, R.; Hur, J.; Kim, Y.J.; Kim, J.; Chwae, Y.-J. Apoptotic cell-derived exosomes: Messages from dying cells. Exp. Mol. Med. 2020, 52, 1–6. [Google Scholar] [CrossRef]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Transport into the Cell from the Plasma Membrane: Endocytosis; Garland Science: New York, NY, USA, 2002. [Google Scholar]
- Huotari, J.; Helenius, A. Endosome maturation. EMBO J. 2011, 30, 3481–3500. [Google Scholar] [CrossRef]
- Meldolesi, J. Exosomes and Ectosomes in Intercellular Communication. Curr. Biol. 2018, 28, R435–R444. [Google Scholar] [CrossRef] [PubMed]
- Abels, E.R.; Breakefield, X.O. Introduction to Extracellular Vesicles: Biogenesis, RNA Cargo Selection, Content, Release, and Uptake. Cell. Mol. Neurobiol. 2016, 36, 301–312. [Google Scholar] [CrossRef]
- Kalani, A.; Tyagi, A.; Tyagi, N. Exosomes: Mediators of Neurodegeneration, Neuroprotection and Therapeutics. Mol. Neurobiol. 2014, 49, 590–600. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhou, X.; Wei, M.; Gao, X.; Zhao, L.; Shi, R.; Sun, W.; Duan, Y.; Yang, G.; Yuan, L. In Vitro and in Vivo RNA Inhibition by CD9-HuR Functionalized Exosomes Encapsulated with miRNA or CRISPR/dCas9. Nano Lett. 2019, 19, 19–28. [Google Scholar] [CrossRef]
- Porro, C.; Panaro, M.A.; Lofrumento, D.D.; Hasalla, E.; Trotta, T. The multiple roles of exosomes in Parkinson’s disease: An overview. Immunopharmacol. Immunotoxicol. 2019, 41, 469–476. [Google Scholar] [CrossRef]
- Vogel, A.D.; Upadhya, R.; Shetty, A.K. Neural stem cell derived extracellular vesicles: Attributes and prospects for treating neurodegenerative disorders. Ebiomedicine 2018, 38, 273–282. [Google Scholar] [CrossRef]
- Danzer, K.M.; Kranich, L.R.; Ruf, W.P.; Cagsal-Getkin, O.; Winslow, A.R.; Zhu, L.; Vanderburg, C.R.; McLean, P.J. Exosomal cell-to-cell transmission of alpha synuclein oligomers. Mol. Neurodegener. 2012, 7, 42. [Google Scholar] [CrossRef]
- Yáñez-Mó, M.; Siljander, P.R.-M.; Andreu, Z.; Bedina Zavec, A.; Borràs, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef]
- Reed, S.L.; Escayg, A. Extracellular vesicles in the treatment of neurological disorders. Neurobiol. Dis. 2021, 157, 105445. [Google Scholar] [CrossRef]
- Wang, N.; Mi, X.; Gao, B.; Gu, J.; Wang, W.; Zhang, Y.; Wang, X. Minocycline inhibits brain inflammation and attenuates spontaneous recurrent seizures following pilocarpine-induced status epilepticus. Neuroscience 2015, 287, 144–156. [Google Scholar] [CrossRef]
- Ding, M.; Shen, Y.; Wang, P.; Xie, Z.; Xu, S.; Zhu, Z.; Wang, Y.; Lyu, Y.; Wang, D.; Xu, L.; et al. Exosomes Isolated from Human Umbilical Cord Mesenchymal Stem Cells Alleviate Neuroinflammation and Reduce Amyloid-Beta Deposition by Modulating Microglial Activation in Alzheimer’s Disease. Neurochem. Res. 2018, 43, 2165–2177. [Google Scholar] [CrossRef]
- Calabria, E.; Scambi, I.; Bonafede, R.; Schiaffino, L.; Peroni, D.; Potrich, V.; Capelli, C.; Schena, F.; Mariotti, R. ASCs-Exosomes Recover Coupling Efficiency and Mitochondrial Membrane Potential in an in vitro Model of ALS. Front. Neurosci. 2019, 13, 1070. [Google Scholar] [CrossRef]
- Yang, L.; Zhai, Y.; Hao, Y.; Zhu, Z.; Cheng, G. The Regulatory Functionality of Exosomes Derived from hUMSCs in 3D Culture for Alzheimer’s Disease Therapy. Small 2020, 16, e1906273. [Google Scholar] [CrossRef]
- Riazifar, M.; Pone, E.J.; Lötvall, J.; Zhao, W. Stem Cell Extracellular Vesicles: Extended Messages of Regeneration. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 125–154. [Google Scholar] [CrossRef]
- Mead, B.; Tomarev, S. Bone Marrow-Derived Mesenchymal Stem Cells-Derived Exosomes Promote Survival of Retinal Ganglion Cells Through miRNA-Dependent Mechanisms. Stem. Cells Transl. Med. 2017, 6, 1273–1285. [Google Scholar] [CrossRef]
- Elia, C.A.; Tamborini, M.; Rasile, M.; Desiato, G.; Marchetti, S.; Swuec, P.; Mazzitelli, S.; Clemente, F.; Anselmo, A.; Matteoli, M.; et al. Intracerebral Injection of Extracellular Vesicles from Mesenchymal Stem Cells Exerts Reduced Aβ Plaque Burden in Early Stages of a Preclinical Model of Alzheimer’s Disease. Cells 2019, 8, 1059. [Google Scholar] [CrossRef]
- Canales-Aguirre, A.; Reza-Zaldivar, E.; Hernández-Sapiéns, M.; Gutiérrez-Mercado, Y.K.; Sandoval-Ávila, S.; Gomez-Pinedo, U.; Márquez-Aguirre, A.L.; Vázquez-Méndez, E.; Padilla-Camberos, E. Mesenchymal stem cell-derived exosomes promote neurogenesis and cognitive function recovery in a mouse model of Alzheimer’s disease. Neural Regen. Res. 2019, 14, 1626–1634. [Google Scholar] [CrossRef]
- Chen, C.C.; Liu, L.; Ma, F.; Wong, C.W.; Guo, X.E.; Chacko, J.V.; Farhoodi, H.P.; Zhang, S.X.; Zimak, J.; Ségaliny, A.; et al. Elucidation of Exosome Migration Across the Blood–Brain Barrier Model In Vitro. Cell. Mol. Bioeng. 2016, 9, 509–529. [Google Scholar] [CrossRef]
- Liu, S.; Fan, M.; Xu, J.-X.; Yang, L.-J.; Qi, C.-C.; Xia, Q.-R.; Ge, J.-F. Exosomes derived from bone-marrow mesenchymal stem cells alleviate cognitive decline in AD-like mice by improving BDNF-related neuropathology. J. Neuroinflamm. 2022, 19, 35. [Google Scholar] [CrossRef]
- Kwok, Z.H.; Wang, C.; Jin, Y. Extracellular Vesicle Transportation and Uptake by Recipient Cells: A Critical Process to Regulate Human Diseases. Processes 2021, 9, 273. [Google Scholar] [CrossRef] [PubMed]
- Jurgielewicz, B.; Yao, Y.; Stice, S.L. Kinetics and Specificity of HEK293T Extracellular Vesicle Uptake using Imaging Flow Cytometry. Nanoscale Res. Lett. 2020, 15, 170. [Google Scholar] [CrossRef] [PubMed]
- Rana, S.; Yue, S.; Stadel, D.; Zöller, M. Toward tailored exosomes: The exosomal tetraspanin web contributes to target cell selection. Int. J. Biochem. Cell Biol. 2012, 44, 1574–1584. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Molina, C.; Sandoval, M.; Henzi, R.; Ramírez, J.P.; Varas-Godoy, M.; Luarte, A.; Lafourcade, C.A.; Lopez-Verrilli, A.; Smalla, K.-H.; Kaehne, T.; et al. Small Extracellular Vesicles in Rat Serum Contain Astrocyte-Derived Protein Biomarkers of Repetitive Stress. Int. J. Neuropsychopharmacol. 2019, 22, 232–246. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Wang, L.; Zeng, X.; Schwarz, H.; Nanda, H.S.; Peng, X.; Zhou, Y. Exosomes, a New Star for Targeted Delivery. Front. Cell Dev. Biol. 2021, 9, 751079. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Takahashi, Y.; Takakura, Y. Possibility of Exosome-Based Therapeutics and Challenges in Production of Exosomes Eligible for Therapeutic Application. Biol. Pharm. Bull. 2018, 41, 835–842. [Google Scholar] [CrossRef]
- Jarmalavičiūtė, A.; Tunaitis, V.; Pivoraitė, U.; Venalis, A.; Pivoriūnas, A. Exosomes from dental pulp stem cells rescue human dopaminergic neurons from 6-hydroxy-dopamine–induced apoptosis. Cytotherapy 2015, 17, 932–939. [Google Scholar] [CrossRef]
- Fernandes, A.; Ribeiro, A.R.; Monteiro, M.; Garcia, G.; Vaz, A.R.; Brites, D. Secretome from SH-SY5Y APPSwe cells trigger time-dependent CHME3 microglia activation phenotypes, ultimately leading to miR-21 exosome shuttling. Biochimie 2018, 155, 67–82. [Google Scholar] [CrossRef]
- Loch-Neckel, G.; Matos, A.T.; Vaz, A.R.; Brites, D. Challenges in the Development of Drug Delivery Systems Based on Small Extracellular Vesicles for Therapy of Brain Diseases. Front. Pharmacol. 2022, 13, 839790. [Google Scholar] [CrossRef]
- Strimpakos, A.; Sharma, R.A. Curcumin: Preventive and Therapeutic Properties in Laboratory Studies and Clinical Trials. Antioxid. Redox Signal. 2008, 10, 511–546. [Google Scholar] [CrossRef]
- Sun, D.; Zhuang, X.; Xiang, X.; Liu, Y.; Zhang, S.; Liu, C.; Barnes, S.; Grizzle, W.; Miller, D.; Zhang, H.-G. A Novel Nanoparticle Drug Delivery System: The Anti-inflammatory Activity of Curcumin Is Enhanced When Encapsulated in Exosomes. Mol. Ther. 2010, 18, 1606–1614. [Google Scholar] [CrossRef]
- Hu, S.; Maiti, P.; Ma, Q.; Zuo, X.; Jones, M.R.; Cole, G.M.; Frautschy, S.A. Clinical development of curcumin in neurodegenerative disease. Expert Rev. Neurother. 2015, 15, 629–637. [Google Scholar] [CrossRef]
- Qu, M.; Lin, Q.; Huang, L.; Fu, Y.; Wang, L.; He, S.; Fu, Y.; Yang, S.; Zhang, Z.; Zhang, L.; et al. Dopamine-loaded blood exosomes targeted to brain for better treatment of Parkinson’s disease. J. Control. Release 2018, 287, 156–166. [Google Scholar] [CrossRef]
- Haney, M.J.; Klyachko, N.L.; Zhao, Y.; Gupta, R.; Plotnikova, E.G.; He, Z.; Patel, T.; Piroyan, A.; Sokolsky, M.; Kabanov, A.V.; et al. Exosomes as drug delivery vehicles for Parkinson’s disease therapy. J. Control. Release 2015, 207, 18–30. [Google Scholar] [CrossRef]
- Zhao, Y.; Haney, M.J.; Gupta, R.; Bohnsack, J.P.; He, Z.; Kabanov, A.V.; Batrakova, E.V. GDNF-Transfected Macrophages Produce Potent Neuroprotective Effects in Parkinson’s Disease Mouse Model. PLoS ONE 2014, 9, e106867. [Google Scholar] [CrossRef]
- Lai, R.C.; Yeo, R.W.Y.; Tan, K.H.; Lim, S.K. Exosomes for drug delivery—A novel application for the mesenchymal stem cell. Biotechnol. Adv. 2013, 31, 543–551. [Google Scholar] [CrossRef]
- Haney, M.J.; Zhao, Y.; Fallon, J.K.; Yue, W.; Li, S.M.; Lentz, E.E.; Erie, D.; Smith, P.C.; Batrakova, E.V. Extracellular Vesicles as Drug Delivery System for the Treatment of Neurodegenerative Disorders: Optimization of the Cell Source. Adv. NanoBiomed Res. 2021, 1, 2100064. [Google Scholar] [CrossRef]
- Henning-Knechtel, A.; Kumar, S.; Wallin, C.; Król, S.; Wärmländer, S.K.; Jarvet, J.; Esposito, G.; Kirmizialtin, S.; Gräslund, A.; Hamilton, A.D.; et al. Designed Cell-Penetrating Peptide Inhibitors of Amyloid-beta Aggregation and Cytotoxicity. Cell Rep. Phys. Sci. 2020, 1, 100014. [Google Scholar] [CrossRef]
- Dietz, G.P.H.; Bähr, M. Synthesis of cell-penetrating peptides and their application in neurobiology. Methods Mol. Biol. 2007, 399, 181–198. [Google Scholar]
- Magzoub, M.; Gräslund, A. Cell-penetrating peptides: [corrected] from inception to application. Q. Rev. Biophys. 2004, 37, 147–195. [Google Scholar] [CrossRef] [PubMed]
- Ruseska, I.; Zimmer, A. Internalization mechanisms of cell-penetrating peptides. Beilstein J. Nanotechnol. 2020, 11, 101–123. [Google Scholar] [CrossRef] [PubMed]
- Madani, F.; Lindberg, S.; Langel, Ü.; Futaki, S.; Gräslund, A. Mechanisms of Cellular Uptake of Cell-Penetrating Peptides. J. Biophys. 2011, 2011, 414729. [Google Scholar] [CrossRef]
- Langel, Ü. Cell-Penetrating Peptides and Transportan. Pharmaceutics 2021, 13, 987. [Google Scholar] [CrossRef] [PubMed]
- Gerbal-Chaloin, S.; Gondeau, C.; Aldrian-Herrada, G.; Heitz, F.; Gauthier-Rouviere, C.; Divita, G. First step of the cell-penetrating peptide mechanism involves Rac1 GTPase-dependent actin-network remodelling. Biol. Cell 2007, 99, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.Q.; Hölscher, C. GIP has neuroprotective effects in Alzheimer and Parkinson’s disease models. Peptides 2020, 125, 170184. [Google Scholar] [CrossRef] [PubMed]
- Athauda, D.; Foltynie, T. The glucagon-like peptide 1 (GLP) receptor as a therapeutic target in Parkinson’s disease: Mechanisms of action. Drug Discov. Today 2016, 21, 802–818. [Google Scholar] [CrossRef] [PubMed]
- Hölscher, C. Protective properties of GLP-1 and associated peptide hormones in neurodegenerative disorders. Br. J. Pharmacol. 2022, 179, 695–714. [Google Scholar] [CrossRef]
- Magzoub, M. Combating Proteins with Proteins: Engineering Cell-Penetrating Peptide Antagonists of Amyloid-β Aggregation and Associated Neurotoxicity. DNA Cell Biol. 2020, 39, 920–925. [Google Scholar] [CrossRef]
- Liu, M.; Fang, X.; Yang, Y.; Wang, C. Peptide-Enabled Targeted Delivery Systems for Therapeutic Applications. Front. Bioeng. Biotechnol. 2021, 9, 701504. [Google Scholar] [CrossRef]
- Foged, C.; Nielsen, H.M. Cell-penetrating peptides for drug delivery across membrane barriers. Expert Opin. Drug Deliv. 2008, 5, 105–117. [Google Scholar] [CrossRef]
- El-Andaloussi, S.; Järver, P.; Johansson, H.J.; Langel, U. Cargo-dependent cytotoxicity and delivery efficacy of cell-penetrating peptides: A comparative study. Biochem. J. 2007, 407, 285–292. [Google Scholar] [CrossRef]
- Singh, T.; Murthy, A.S.N.; Yang, H.-J.; Im, J. Versatility of cell-penetrating peptides for intracellular delivery of siRNA. Drug Deliv. 2018, 25, 1996–2006. [Google Scholar] [CrossRef]
- Kardani, K.; Milani, A.; Shabani, S.H.; Bolhassani, A. Cell penetrating peptides: The potent multi-cargo intracellular carriers. Expert Opin. Drug Deliv. 2019, 16, 1227–1258. [Google Scholar] [CrossRef]
- Fonseca, S.B.; Pereira, M.P.; Kelley, S.O. Recent advances in the use of cell-penetrating peptides for medical and biological applications. Adv. Drug Deliv. Rev. 2009, 61, 953–964. [Google Scholar] [CrossRef]
- Ramsey, J.D.; Flynn, N.H. Cell-penetrating peptides transport therapeutics into cells. Pharmacol. Ther. 2015, 154, 78–86. [Google Scholar] [CrossRef]
- Li, H.; Tsui, T.Y.; Ma, W. Intracellular Delivery of Molecular Cargo Using Cell-Penetrating Peptides and the Combination Strategies. Int. J. Mol. Sci. 2015, 16, 19518–19536. [Google Scholar] [CrossRef]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Wightman, B.; Ha, I.; Ruvkun, G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell 1993, 75, 855–862. [Google Scholar] [CrossRef]
- Ha, M.; Kim, V.N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef]
- Friedländer, M.R.; Lizano, E.; Houben, A.J.S.; Bezdan, D.; Báñez-Coronel, M.; Kudla, G.; Mateu-Huertas, E.; Kagerbauer, B.; González, J.; Chen, K.C.; et al. Evidence for the biogenesis of more than 1,000 novel human microRNAs. Genome Biol. 2014, 15, R57. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-C.; Chan, W.-C.; Hu, L.-Y.; Lai, C.-H.; Hsu, C.-N.; Lin, W.-C. Identification of homologous microRNAs in 56 animal genomes. Genomics 2010, 96, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Davis-Dusenbery, B.N.; Hata, A. Mechanisms of control of microRNA biogenesis. J. Biochem. 2010, 148, 381–392. [Google Scholar]
- Pasquinelli, A.E.; Reinhart, B.J.; Slack, F.; Martindale, M.Q.; Kuroda, M.I.; Maller, B.; Hayward, D.C.; Ball, E.E.; Degnan, B.; Müller, P.; et al. Conservation of the sequence and temporal expression of let-7 heterochronic regulatory RNA. Nature 2000, 408, 86–89. [Google Scholar] [CrossRef] [PubMed]
- Broughton, J.P.; Lovci, M.T.; Huang, J.L.; Yeo, G.W.; Pasquinelli, A.E. Pairing beyond the Seed Supports MicroRNA Targeting Specificity. Mol. Cell 2016, 64, 320–333. [Google Scholar] [CrossRef] [PubMed]
- Coller, H.A.; Sang, L.; Roberts, J.M. A New Description of Cellular Quiescence. PLoS Biol. 2006, 4, e83. [Google Scholar] [CrossRef] [PubMed]
- Maciotta, S.; Meregalli, M.; Torrente, Y. The involvement of microRNAs in neurodegenerative diseases. Front. Cell Neurosci. 2013, 7, 265. [Google Scholar] [CrossRef]
- Friedman, R.C.; Farh, K.K.-H.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef]
- Sadlon, A.; Takousis, P.; Alexopoulos, P.; Evangelou, E.; Prokopenko, I.; Perneczky, R. miRNAs Identify Shared Pathways in Alzheimer’s and Parkinson’s Diseases. Trends Mol. Med. 2019, 25, 662–672. [Google Scholar] [CrossRef]
- Siedlecki-Wullich, D.; Miñano-Molina, A.; Rodríguez-Álvarez, J. microRNAs as Early Biomarkers of Alzheimer’s Disease: A Synaptic Perspective. Cells 2021, 10, 113. [Google Scholar] [CrossRef]
- Roser, A.E.; Gomes, L.C.; Schünemann, J.; Maass, F.; Lingor, P. Circulating miRNAs as Diagnostic Biomarkers for Parkinson’s Disease. Front. Neurosci. 2018, 12, 625. [Google Scholar] [CrossRef] [PubMed]
- Barbagallo, C.; Mostile, G.; Baglieri, G.; Giunta, F.; Luca, A.; Raciti, L.; Zappia, M.; Purrello, M.; Ragusa, M.; Nicoletti, A. Specific Signatures of Serum miRNAs as Potential Biomarkers to Discriminate Clinically Similar Neurodegenerative and Vascular-Related Diseases. Cell. Mol. Neurobiol. 2020, 40, 531–546. [Google Scholar] [CrossRef] [PubMed]
- Kiko, T.; Nakagawa, K.; Tsuduki, T.; Furukawa, K.; Arai, H.; Miyazawa, T. MicroRNAs in Plasma and Cerebrospinal Fluid as Potential Markers for Alzheimer’s Disease. J. Alzheimers Dis. 2014, 39, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Schwienbacher, C.; Foco, L.; Picard, A.; Corradi, E.; Serafin, A.; Panzer, J.; Zanigni, S.; Blankenburg, H.; Facheris, M.F.; Giannini, G.; et al. Plasma and White Blood Cells Show Different miRNA Expression Profiles in Parkinson’s Disease. J. Mol. Neurosci. 2017, 62, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Huang, Z.; Chen, M.; Wang, C.; Chen, X.; Chen, J.; Zhang, J. Identification of a panel of five serum miRNAs as a biomarker for Parkinson’s disease. Park. Relat. Disord. 2016, 22, 68–73. [Google Scholar] [CrossRef]
- Hou, T.-Y.; Zhou, Y.; Zhu, L.-S.; Wang, X.; Pang, P.; Wang, D.-Q.; Liuyang, Z.-Y.; Man, H.; Lu, Y.; Zhu, L.-Q.; et al. Correcting abnormalities in miR-124/PTPN1 signaling rescues tau pathology in Alzheimer’s disease. J. Neurochem. 2020, 154, 441–457. [Google Scholar] [CrossRef]
- Basak, I.; Patil, K.S.; Alves, G.; Larsen, J.P.; Møller, S.G. microRNAs as neuroregulators, biomarkers and therapeutic agents in neurodegenerative diseases. Cell. Mol. Life Sci. 2016, 73, 811–827. [Google Scholar] [CrossRef]
- Miñones-Moyano, E.; Porta, S.; Escaramís, G.; Rabionet, R.; Iraola, S.; Kagerbauer, B.; Espinosa-Parrilla, Y.; Ferrer, I.; Estivill, X.; Martí, E. MicroRNA profiling of Parkinson’s disease brains identifies early downregulation of miR-34b/c which modulate mitochondrial function. Hum. Mol. Genet. 2011, 20, 3067–3078. [Google Scholar] [CrossRef]
- Esteves, M.; Abreu, R.; Fernandes, H.; Serra-Almeida, C.; Martins, P.A.; Barão, M.; Cristóvão, A.C.; Saraiva, C.; Ferreira, R.; Ferreira, L.; et al. MicroRNA-124-3p-enriched small extracellular vesicles as a therapeutic approach for Parkinson’s disease. Mol. Ther. 2022, 30, 3176–3192. [Google Scholar] [CrossRef]
- Recasens, A.; Perier, C.; Sue, C.M. Role of microRNAs in the Regulation of alpha-Synuclein Expression: A Systematic Review. Front. Mol. Neurosci. 2016, 9, 128. [Google Scholar] [CrossRef]
- Guy, R.; Offen, D. Promising Opportunities for Treating Neurodegenerative Diseases with Mesenchymal Stem Cell-Derived Exosomes. Biomolecules 2020, 10, 1320. [Google Scholar] [CrossRef] [PubMed]
- Elsharkasy, O.; Nordin, J.; Hagey, D.; de Jong, O.; Schiffelers, R.; El Andaloussi, S.; Vader, P. Extracellular vesicles as drug delivery systems: Why and how? Adv. Drug Deliv. Rev. 2020, 159, 332–343. [Google Scholar] [CrossRef] [PubMed]
- Pedrioli, G.; Piovesana, E.; Vacchi, E.; Balbi, C. Extracellular Vesicles as Promising Carriers in Drug Delivery: Considerations from a Cell Biologist’s Perspective. Biology 2021, 10, 376. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, I.K.; Wood, M.J.A.; Fuhrmann, G. Extracellular vesicles as a next-generation drug delivery platform. Nat. Nanotechnol. 2021, 16, 748–759. [Google Scholar] [CrossRef] [PubMed]
- Lalatsa, A.; Schatzlein, A.G.; Uchegbu, I.F. Strategies to Deliver Peptide Drugs to the Brain. Mol. Pharm. 2014, 11, 1081–1093. [Google Scholar] [CrossRef] [PubMed]
- Upadhya, R.; Shetty, A.K. Extracellular Vesicles for the Diagnosis and Treatment of Parkinson’s Disease. Aging Dis. 2021, 12, 1438–1450. [Google Scholar] [CrossRef]
- Vandendriessche, C.; Bruggeman, A.; Van Cauwenberghe, C.; Vandenbroucke, R. Extracellular Vesicles in Alzheimer’s and Parkinson’s Disease: Small Entities with Large Consequences. Cells 2020, 9, 2485. [Google Scholar] [CrossRef]
- Berumen Sánchez, G.; Bunn, K.E.; Pua, H.H.; Rafat, M. Extracellular vesicles: Mediators of intercellular communication in tissue injury and disease. Cell Commun. Signal 2021, 19, 104. [Google Scholar] [CrossRef]
- Coleman, B.M.; Hill, A.F. Extracellular vesicles—Their role in the packaging and spread of misfolded proteins associated with neurodegenerative diseases. Semin. Cell Dev. Biol. 2015, 40, 89–96. [Google Scholar] [CrossRef]
- Mathieu, M.; Martin-Jaular, L.; Lavieu, G.; Théry, C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat. Cell Biol. 2019, 21, 9–17. [Google Scholar] [CrossRef]
- Hoppstädter, J.; Dembek, A.; Linnenberger, R.; Dahlem, C.; Barghash, A.; Fecher-Trost, C.; Fuhrmann, G.; Koch, M.; Kraegeloh, A.; Huwer, H.; et al. Toll-Like Receptor 2 Release by Macrophages: An Anti-inflammatory Program Induced by Glucocorticoids and Lipopolysaccharide. Front. Immunol. 2019, 10, 1634. [Google Scholar] [CrossRef] [PubMed]
- Szepesi, Z.; Manouchehrian, O.; Bachiller, S.; Deierborg, T. Bidirectional Microglia–Neuron Communication in Health and Disease. Front. Cell. Neurosci. 2018, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M.J.A. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011, 29, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Valadi, H.; Ekström, K.; Bossios, A.; Sjöstrand, M.; Lee, J.J.; Lötvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Delpech, J.-C.; Herron, S.; Botros, M.B.; Ikezu, T. Neuroimmune Crosstalk through Extracellular Vesicles in Health and Disease. Trends Neurosci. 2019, 42, 361–372. [Google Scholar] [CrossRef]
- Sun, P.; Liu, D.Z.; Jickling, G.C.; Sharp, F.R.; Yin, K.-J. MicroRNA-based therapeutics in central nervous system injuries. J. Cereb. Blood Flow Metab. 2018, 38, 1125–1148. [Google Scholar] [CrossRef]
- Michlewski, G.; Cáceres, J.F. Post-transcriptional control of miRNA biogenesis. RNA 2019, 25, 1–16. [Google Scholar] [CrossRef]
- Bernardo, B.C.; Ooi, J.Y.Y.; Lin, R.C.Y.; McMullen, J.R. miRNA therapeutics: A new class of drugs with potential therapeutic applications in the heart. Future Med. Chem. 2015, 7, 1771–1792. [Google Scholar] [CrossRef]
- Sonntag, K.-C. MicroRNAs and deregulated gene expression networks in neurodegeneration. Brain Res. 2010, 1338, 48–57. [Google Scholar] [CrossRef]
- Faghihi, M.A.; Modarresi, F.; Khalil, A.M.; Wood, D.E.; Sahagan, B.G.; Morgan, T.E.; Finch, C.E.; Laurent, G.S.; Kenny, P.J.; Wahlestedt, C. Expression of a noncoding RNA is elevated in Alzheimer’s disease and drives rapid feed-forward regulation of beta-secretase. Nat. Med. 2008, 14, 723–730. [Google Scholar] [CrossRef]
- Cogswell, J.P.; Ward, J.; Taylor, I.A.; Waters, M.; Shi, Y.; Cannon, B.; Kelnar, K.; Kemppainen, J.; Brown, D.; Chen, C.; et al. Identification of miRNA changes in Alzheimer’s disease brain and CSF yields putative biomarkers and insights into disease pathways. J. Alzheimers Dis. 2008, 14, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Goh, S.Y.; Chao, Y.X.; Dheen, S.T.; Tan, E.-K.; Tay, S.S.-W. Role of micrornas in parkinson’s disease. Int. J. Mol. Sci. 2019, 20, 5649. [Google Scholar] [CrossRef] [PubMed]
- Junn, E.; Mouradian, M.M. MicroRNAs in neurodegenerative diseases and their therapeutic potential. Pharmacol. Ther. 2012, 133, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.C.; Chae, Y.-J.; Kabaria, S.; Chaudhuri, A.D.; Jain, M.R.; Li, H.; Mouradian, M.M.; Junn, E. MicroRNA-7 Protects against 1-Methyl-4-Phenylpyridinium-Induced Cell Death by Targeting RelA. J. Neurosci. 2014, 34, 12725–12737. [Google Scholar] [CrossRef]
- Leggio, L.; Vivarelli, S.; L’Episcopo, F.; Tirolo, C.; Caniglia, S.; Testa, N.; Marchetti, B.; Iraci, N. microRNAs in Parkinson’s Disease: From Pathogenesis to Novel Diagnostic and Therapeutic Approaches. Int. J. Mol. Sci. 2017, 18, 2698. [Google Scholar] [CrossRef]
- Wiedrick, J.T.; Phillips, J.I.; Lusardi, T.A.; McFarland, T.J.; Lind, B.; Sandau, U.S.; Harrington, C.A.; Lapidus, J.A.; Galasko, D.R.; Quinn, J.F.; et al. Validation of MicroRNA Biomarkers for Alzheimer’s Disease in Human Cerebrospinal Fluid. J. Alzheimers Dis. 2019, 67, 875–891. [Google Scholar] [CrossRef]
- Takousis, P.; Sadlon, A.; Schulz, J.; Wohlers, I.; Dobricic, V.; Middleton, L.; Lill, C.M.; Perneczky, R.; Bertram, L. Differential expression of microRNAs in Alzheimer’s disease brain, blood, and cerebrospinal fluid. Alzheimers Dement. 2019, 15, 1468–1477. [Google Scholar] [CrossRef]
- Swarbrick, S.; Wragg, N.; Ghosh, S.; Stolzing, A. Systematic Review of miRNA as Biomarkers in Alzheimer’s Disease. Mol. Neurobiol. 2019, 56, 6156–6167. [Google Scholar] [CrossRef]
- Keller, A.; Backes, C.; Haas, J.; Leidinger, P.; Maetzler, W.; Deuschle, C.; Berg, D.; Ruschil, C.; Galata, V.; Ruprecht, K.; et al. Validating Alzheimer’s disease micro RNAs using next-generation sequencing. Alzheimers Dement. 2016, 12, 565–576. [Google Scholar] [CrossRef]
- Higaki, S.; Muramatsu, M.; Matsuda, A.; Matsumoto, K.; Satoh, J.-I.; Michikawa, M.; Niida, S. Defensive effect of microRNA-200b/c against amyloid-beta peptide-induced toxicity in Alzheimer’s disease models. PLoS ONE 2018, 13, e0196929. [Google Scholar] [CrossRef]
- Munir, J.; Yoon, J.K.; Ryu, S. Therapeutic miRNA-Enriched Extracellular Vesicles: Current Approaches and Future Prospects. Cells 2020, 9, 2271. [Google Scholar] [CrossRef] [PubMed]
- Berry, C. Intracellular delivery of nanoparticles via the HIV-1 tat peptide. Nanomedicine 2008, 3, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Rizzuti, M.; Nizzardo, M.; Zanetta, C.; Ramirez, A.; Corti, S. Therapeutic applications of the cell-penetrating HIV-1 Tat peptide. Drug Discov. Today 2015, 20, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Reissmann, S. Cell penetration: Scope and limitations by the application of cell-penetrating peptides. J. Pept. Sci. 2014, 20, 760–784. [Google Scholar] [CrossRef]
- Nakase, I.; Noguchi, K.; Aoki, A.; Takatani-Nakase, T.; Fujii, I.; Futaki, S. Arginine-rich cell-penetrating peptide-modified extracellular vesicles for active macropinocytosis induction and efficient intracellular delivery. Sci. Rep. 2017, 7, 1991. [Google Scholar] [CrossRef]
- Nakase, I. Biofunctional Peptide-Modified Extracellular Vesicles Enable Effective Intracellular Delivery via the Induction of Macropinocytosis. Processes 2021, 9, 224. [Google Scholar] [CrossRef]
- Hirase, S.; Aoki, A.; Hattori, Y.; Morimoto, K.; Noguchi, K.; Fujii, I.; Takatani-Nakase, T.; Futaki, S.; Kirihata, M.; Nakase, I. Dodecaborate-Encapsulated Extracellular Vesicles with Modification of Cell-Penetrating Peptides for Enhancing Macropinocytotic Cellular Uptake and Biological Activity in Boron Neutron Capture Therapy. Mol. Pharm. 2022, 19, 1135–1145. [Google Scholar] [CrossRef]
- Mashouri, L.; Yousefi, H.; Aref, A.R.; Ahadi, A.M.; Molaei, F.; Alahari, S.K. Exosomes: Composition, biogenesis, and mechanisms in cancer metastasis and drug resistance. Mol. Cancer 2019, 18, 75. [Google Scholar] [CrossRef]
- García-Manrique, P.; Matos, M.; Gutiérrez, G.; Pazos, C.; Blanco-López, M.C. Therapeutic biomaterials based on extracellular vesicles: Classification of bio-engineering and mimetic preparation routes. J. Extracell. Vesicles 2018, 7, 1422676. [Google Scholar] [CrossRef]
- Pascucci, L.; Coccè, V.; Bonomi, A.; Ami, D.; Ceccarelli, P.; Ciusani, E.; Viganò, L.; Locatelli, A.; Sisto, F.; Doglia, S.M.; et al. Paclitaxel is incorporated by mesenchymal stromal cells and released in exosomes that inhibit in vitro tumor growth: A new approach for drug delivery. J. Control. Release 2014, 192, 262–270. [Google Scholar] [CrossRef]
- Li, S.-P.; Lin, Z.-X.; Jiang, X.-Y.; Yu, X.-Y. Exosomal cargo-loading and synthetic exosome-mimics as potential therapeutic tools. Acta Pharmacol. Sin. 2018, 39, 542–551. [Google Scholar] [CrossRef]
- van der Meel, R.; Fens, M.H.A.M.; Vader, P.; van Solinge, W.W.; Eniola-Adefeso, O.; Schiffelers, R.M. Extracellular vesicles as drug delivery systems: Lessons from the liposome field. J. Control. Release 2014, 195, 72–85. [Google Scholar] [CrossRef]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T Immunotherapy for Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef]
- Steinman, R.M. Decisions About Dendritic Cells: Past, Present, and Future. Annu. Rev. Immunol. 2012, 30, 1–22. [Google Scholar] [CrossRef]
- Li, L.; Lu, S.; Liang, X.; Cao, B.; Wang, S.; Jiang, J.; Luo, H.; He, S.; Lang, J.; Zhu, G. γδTDEs: An Efficient Delivery System for miR-138 with Anti-tumoral and Immunostimulatory Roles on Oral Squamous Cell Carcinoma. Mol. Ther. Nucleic Acids 2019, 14, 101–113. [Google Scholar] [CrossRef]
- Lee, J.; Kim, J.; Jeong, M.; Lee, H.; Goh, U.; Kim, H.; Kim, B.; Park, J.-H. Liposome-Based Engineering of Cells to Package Hydrophobic Compounds in Membrane Vesicles for Tumor Penetration. Nano Lett. 2015, 15, 2938–2944. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Wang, Y.; Huang, L. Exosomes from M1-Polarized Macrophages Potentiate the Cancer Vaccine by Creating a Pro-inflammatory Microenvironment in the Lymph Node. Mol. Ther. 2017, 25, 1665–1675. [Google Scholar] [CrossRef] [PubMed]
- Pessina, A.; Bonomi, A.; Coccè, V.; Invernici, G.; Navone, S.; Cavicchini, L.; Sisto, F.; Ferrari, M.; Viganò, L.; Locatelli, A.; et al. Mesenchymal Stromal Cells Primed with Paclitaxel Provide a New Approach for Cancer Therapy. PLoS ONE 2011, 6, e28321. [Google Scholar] [CrossRef] [PubMed]
- Meng, W.; He, C.; Hao, Y.; Wang, L.; Li, L.; Zhu, G. Prospects and challenges of extracellular vesicle-based drug delivery system: Considering cell source. Drug Deliv. 2020, 27, 585–598. [Google Scholar] [CrossRef] [PubMed]
- Vergauwen, G.; Dhondt, B.; Van Deun, J.; De Smedt, E.; Berx, G.; Timmerman, E.; Gevaert, K.; Miinalainen, I.; Cocquyt, V.; Braems, G.; et al. Confounding factors of ultrafiltration and protein analysis in extracellular vesicle research. Sci. Rep. 2017, 7, 2704. [Google Scholar] [CrossRef]
- Stranska, R.; Gysbrechts, L.; Wouters, J.; Vermeersch, P.; Bloch, K.; Dierickx, D.; Andrei, G.; Snoeck, R. Comparison of membrane affinity-based method with size-exclusion chromatography for isolation of exosome-like vesicles from human plasma. J. Transl. Med. 2018, 16, 1. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [PubMed]
- Botchway, B.O.; Okoye, F.C.; Chen, Y.; Arthur, W.E.; Fang, M. Alzheimer Disease: Recent Updates on Apolipoprotein E and Gut Microbiome Mediation of Oxidative Stress, and Prospective Interventional Agents. Aging Dis. 2022, 13, 87–102. [Google Scholar] [CrossRef] [PubMed]
- Brandebura, A.N.; Paumier, A.; Onur, T.S.; Allen, N.J. Astrocyte contribution to dysfunction, risk and progression in neurodegenerative disorders. Nat. Rev. Neurosci. 2023, 24, 23–39. [Google Scholar] [CrossRef] [PubMed]
- Pachler, K.; Lener, T.; Streif, D.; Dunai, Z.A.; Desgeorges, A.; Feichtner, M.; Öller, M.; Schallmoser, K.; Rohde, E.; Gimona, M. A Good Manufacturing Practice–grade standard protocol for exclusively human mesenchymal stromal cell–derived extracellular vesicles. Cytotherapy 2017, 19, 458–472. [Google Scholar] [CrossRef]
- Reissmann, S.; Filatova, M.P. New generation of cell-penetrating peptides: Functionality and potential clinical application. J. Pept. Sci. 2021, 27, e3300. [Google Scholar] [CrossRef]
- Palm, C.; Jayamanne, M.; Kjellander, M.; Hällbrink, M. Peptide degradation is a critical determinant for cell-penetrating peptide uptake. Biochim. Biophys. Acta 2007, 1768, 1769–1776. [Google Scholar] [CrossRef]
- Hirose, H.; Takeuchi, T.; Osakada, H.; Pujals, S.; Katayama, S.; Nakase, I.; Kobayashi, S.; Haraguchi, T.; Futaki, S. Transient Focal Membrane Deformation Induced by Arginine-rich Peptides Leads to Their Direct Penetration into Cells. Mol. Ther. 2012, 20, 984–993. [Google Scholar] [CrossRef]
- Seisel, Q.; Pelletier, F.; Deshayes, S.; Boisguerin, P. How to evaluate the cellular uptake of CPPs with fluorescence techniques: Dissecting methodological pitfalls associated to tryptophan-rich peptides. Biochim. Biophys. Acta Biomembr. 2019, 1861, 1533–1545. [Google Scholar] [CrossRef]
- Kristensen, M.; Birch, D.; Nielsen, H.M. Applications and Challenges for Use of Cell-Penetrating Peptides as Delivery Vectors for Peptide and Protein Cargos. Int. J. Mol. Sci. 2016, 17, 185. [Google Scholar] [CrossRef]
- Rennert, R.; Wespe, C.; Beck-Sickinger, A.G.; Neundorf, I. Developing novel hCT derived cell-penetrating peptides with improved metabolic stability. Biochim. Biophys. Acta 2006, 1758, 347–354. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Keighron, C.N.; Avazzadeh, S.; Goljanek-Whysall, K.; McDonagh, B.; Howard, L.; Ritter, T.; Quinlan, L.R. Extracellular Vesicles, Cell-Penetrating Peptides and miRNAs as Future Novel Therapeutic Interventions for Parkinson’s and Alzheimer’s Disease. Biomedicines 2023, 11, 728. https://doi.org/10.3390/biomedicines11030728
Keighron CN, Avazzadeh S, Goljanek-Whysall K, McDonagh B, Howard L, Ritter T, Quinlan LR. Extracellular Vesicles, Cell-Penetrating Peptides and miRNAs as Future Novel Therapeutic Interventions for Parkinson’s and Alzheimer’s Disease. Biomedicines. 2023; 11(3):728. https://doi.org/10.3390/biomedicines11030728
Chicago/Turabian StyleKeighron, Cameron Noah, Sahar Avazzadeh, Katarzyna Goljanek-Whysall, Brian McDonagh, Linda Howard, Thomas Ritter, and Leo R. Quinlan. 2023. "Extracellular Vesicles, Cell-Penetrating Peptides and miRNAs as Future Novel Therapeutic Interventions for Parkinson’s and Alzheimer’s Disease" Biomedicines 11, no. 3: 728. https://doi.org/10.3390/biomedicines11030728
APA StyleKeighron, C. N., Avazzadeh, S., Goljanek-Whysall, K., McDonagh, B., Howard, L., Ritter, T., & Quinlan, L. R. (2023). Extracellular Vesicles, Cell-Penetrating Peptides and miRNAs as Future Novel Therapeutic Interventions for Parkinson’s and Alzheimer’s Disease. Biomedicines, 11(3), 728. https://doi.org/10.3390/biomedicines11030728