
NEK1-Mediated Phosphorylation of YAP1 Is Key to Prostate Cancer Progression
 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plasmids and Antibodies
2.2. Cell Treatment
2.3. RNA Extraction
2.4. Realtime Quantitative PCR (RT-qPCR) for p73, TEAD Targets, AR and EMT Genes
2.5. Scratch-Wound Repair Assay
2.6. Immunohistochemistry
2.7. Animal Maintenance and Procedures
2.8. Ethics Approval and Consent to Participate
2.9. Statistical Analysis
3. Results
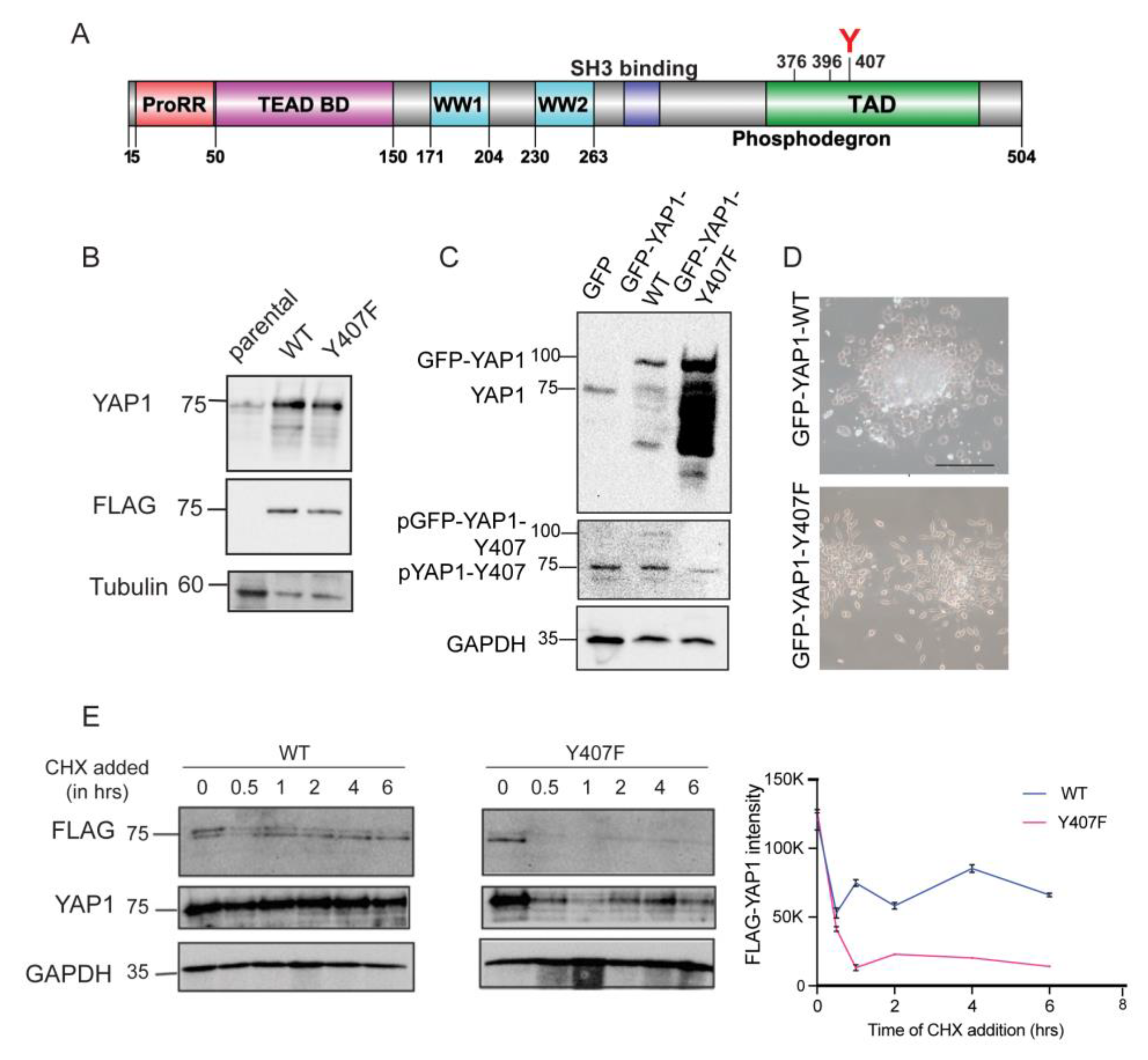
3.1. NEK1 Phosphorylates YAP1 at Y407 and Increases Its Stability
3.2. NEK1-Dependent Phosphorylation of YAP1 Leads to Increased YAP1 Transcriptional Activity upon Mitomycin C (MMC)-Induced DNA Damage
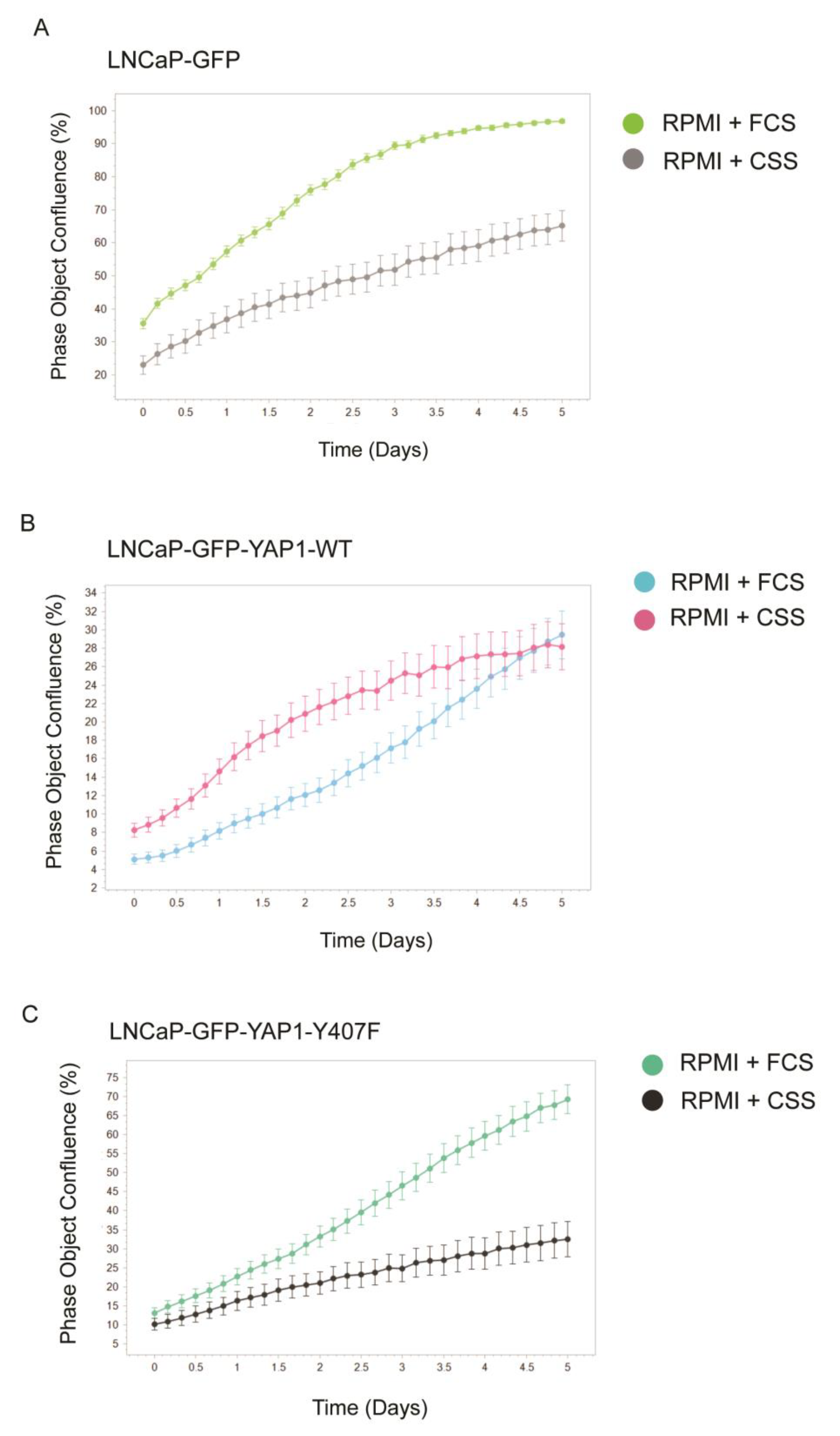
3.3. YAP1 Overexpression, but Not the Y407F Hypoactive/Unstable Mutant, Transforms LNCaP Cells to Androgen-Independent Growth
3.4. Analysis of Transcriptomic Changes in LNCaP Cells Overexpressing GFP-YAP
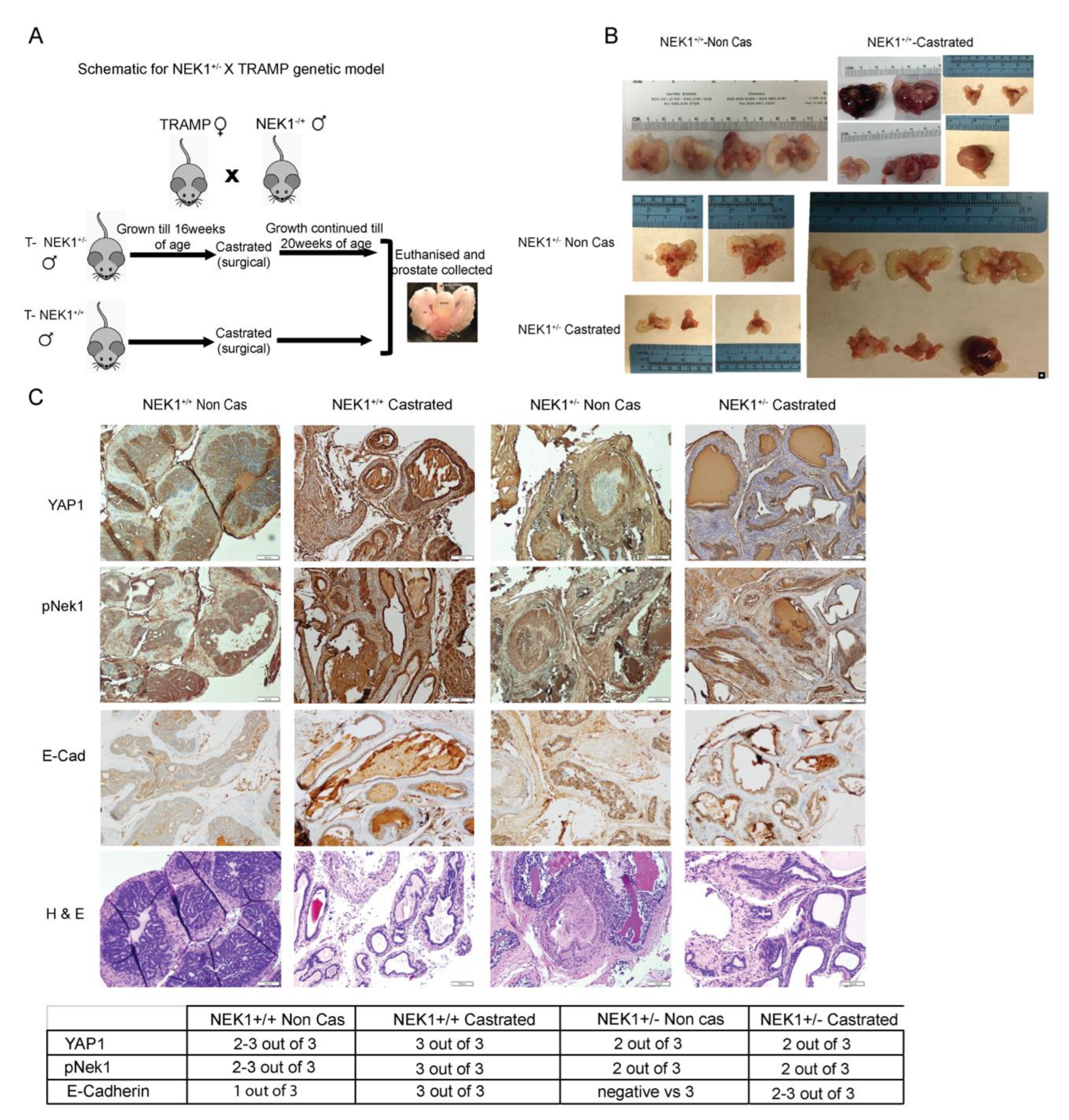
3.5. YAP1 Expression Is NEK1-Dependent in a Castrated TRAMP Mouse Model
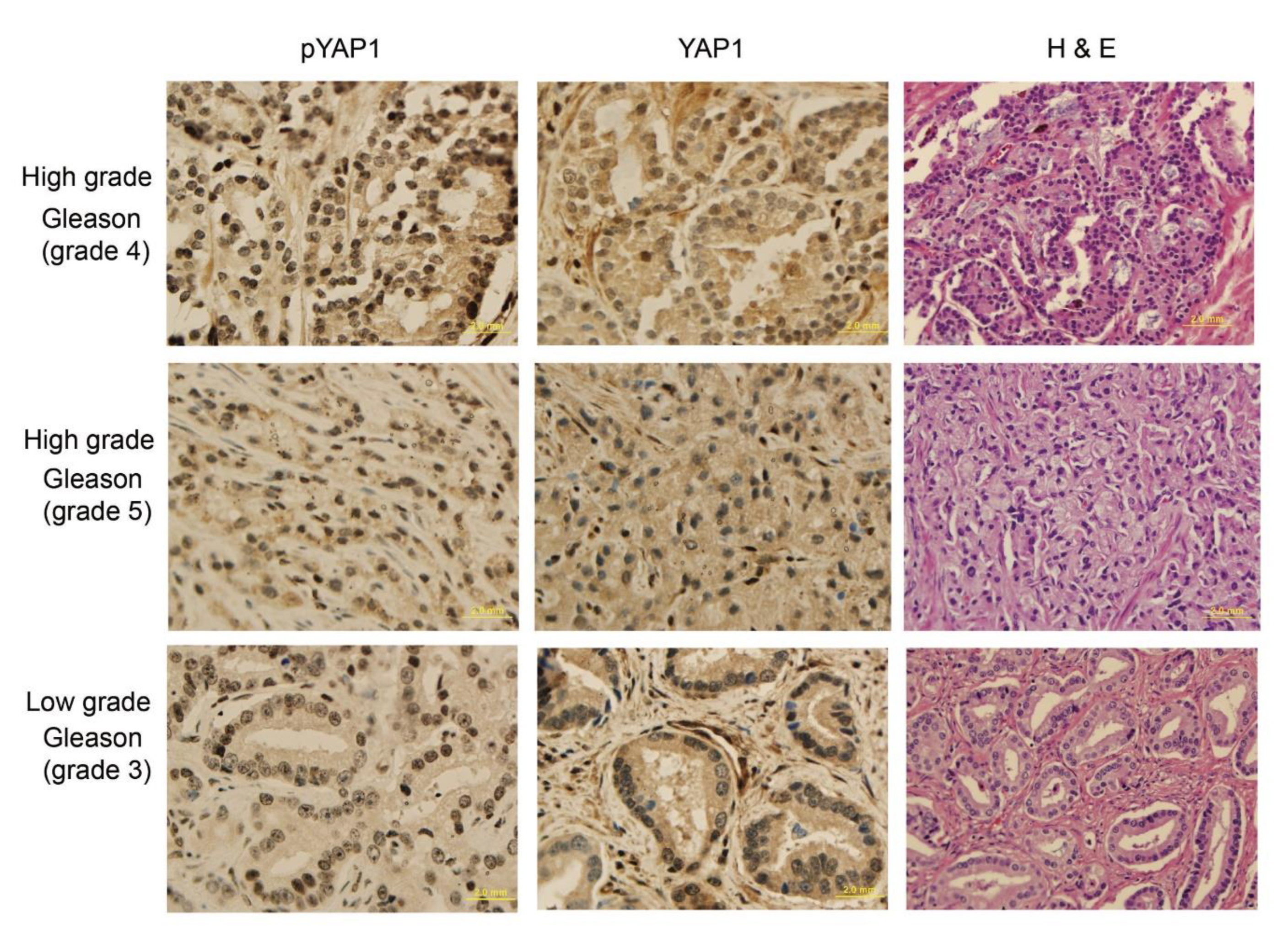
3.6. Analysis of YAP1 and pYAP1-Y407 in Pca and Correlation with Gleason Score (GS)
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AD/I | Androgen-dependent or -independent |
| ADT | Androgen Deprivation Therapy |
| AR | Androgen Receptor |
| ATR | Ataxia Telangiectasia Related |
| BAX | Bcl2 Associate Apoptosis protein X |
| CSS | Charcoal-Stripped Serum |
| EMT/MET | Epithelial to Mesenchymal Transition and vice versa |
| FKBP5 | FK506 binding protein 5 (an immunophilin) |
| GFP | Green Fluorescent Protein |
| GS | Gleason Score |
| H&E | Hematoxylin-Eosin stain |
| mCRPC | Metastatic Castrate Resistant Prostate Cancer |
| MMC | Mitomycin C |
| NEK1 | NIMA Related Kinase 1 |
| NEPC | Neuroendocrine Prostate Cancer |
| PCa | Prostate Cancer |
| PIN | Prostate Intraepithelial Neoplasia |
| PRAD | Prostate Adenocarcinoma |
| TAD | Transcription Activation Domain |
| TRAMP mice | Transgenic Adenocarcinoma of the Mouse Prostate |
References
- Lei, Q.Y.; Zhang, H.; Zhao, B.; Zha, Z.Y.; Bai, F.; Pei, X.H.; Zhao, S.; Xiong, Y.; Guan, K.L. TAZ promotes cell proliferation and epithelial-mesenchymal transition and is inhibited by the hippo pathway. Mol. Cell Biol. 2008, 28, 2426–2436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, F.X.; Zhao, B.; Guan, K.L. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell 2015, 163, 811–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, B.; Li, L.; Lei, Q.; Guan, K.L. The Hippo-YAP pathway in organ size control and tumorigenesis: An updated version. Genes Dev. 2010, 24, 862–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salem, O.; Hansen, C.G. The Hippo Pathway in Prostate Cancer. Cells 2019, 8, 370. [Google Scholar] [CrossRef] [Green Version]
- Kuser-Abali, G.; Alptekin, A.; Lewis, M.; Garraway, I.P.; Cinar, B. YAP1 and AR interactions contribute to the switch from androgen-dependent to castration-resistant growth in prostate cancer. Nat. Commun. 2015, 6, 8126. [Google Scholar] [CrossRef] [Green Version]
- Noh, M.-G.; Kim, S.S.; Hwang, E.C.; Kwon, D.D.; Choi, C. Yes-Associated Protein Expression Is Correlated to the Differentiation of Prostate Adenocarcinoma. J. Pathol. Transl. Med. 2017, 51, 365–373. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Yang, S.; Chen, X.; Stauffer, S.; Yu, F.; Lele, S.M.; Fu, K.; Datta, K.; Palermo, N.; Chen, Y.; et al. The hippo pathway effector YAP regulates motility, invasion, and castration-resistant growth of prostate cancer cells. Mol. Cell. Biol. 2015, 35, 1350–1362. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Xie, P.; Wang, X.; Pan, P.; Wang, Y.; Zhang, H.; Dong, Y.; Shi, Y.; Jiang, Y.; Yu, R.; et al. YAP Promotes Migration and Invasion of Human Glioma Cells. J. of Mol. Neurosci. 2018, 64, 262–272. [Google Scholar] [CrossRef]
- Khalil, M.I.; Ghosh, I.; Singh, V.; Chen, J.; Zhu, H.; De Benedetti, A. NEK1 Phosphorylation of YAP Promotes Its Stabilization and Transcriptional Output. Cancers 2020, 12, 3666. [Google Scholar] [CrossRef]
- Jang, E.J.; Jeong, H.; Han, K.H.; Kwon, H.M.; Hong, J.H.; Hwang, E.S. TAZ suppresses NFAT5 activity through tyrosine phosphorylation. Mol Cell Biol. 2012, 32, 4925–4932. [Google Scholar] [CrossRef] [Green Version]
- Sudol, M.; Shields, D.C.; Farooq, A. Structures of YAP protein domains reveal promising targets for development of new cancer drugs. Semin Cell Dev. Biol. 2012, 23, 827–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van de Kooij, B.; Creixell, P.; van Vlimmeren, A.; Joughin, B.A.; Miller, C.J.; Haider, N.; Simpson, C.D.; Linding, R.; Stambolic, V.; Turk, B.E.; et al. Comprehensive substrate specificity profiling of the human Nek kinome reveals unexpected signaling outputs. Elife 2019, 8, e44635. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Bhoir, S.; Chikhale, R.V.; Hussain, J.; Dwyer, D.; Bryce, R.A.; Kirubakaran, S.; De Benedetti, A. Generation of phenothiazine with potent anti-TLK1 activity for prostate cancer therapy. Iscience 2020, 23, 101474. [Google Scholar] [CrossRef]
- Khalil, M.I.; De Benedetti, A. Tousled-like kinase 1: A novel factor with multifaceted role in mCRPC progression and development of therapy resistance. Cancer Drug Resist. 2022, 5, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Khalil, M.I.; Singh, V.; King, J.; De Benedetti, A. TLK1-mediated MK5-S354 phosphorylation drives prostate cancer cell motility and may signify distinct pathologies. Mol. Oncol. 2022, 16, 2537–2557. [Google Scholar] [CrossRef]
- Polci, R.; Peng, A.; Chen, P.L.; Riley, D.J.; Chen, Y. NIMA-related protein kinase 1 is involved early in the ionizing radiation-induced DNA damage response. Cancer Res. 2004, 64, 8800–8803. [Google Scholar] [CrossRef] [Green Version]
- Watts, P.L.; Plumb, J.A.; Courtney, J.M.; Scott, R. Sensitivity of cell lines to mitomycin C. Brit. J. of Urolog. 1996, 77, 363–366. [Google Scholar] [CrossRef]
- Levy, D.; Adamovich, Y.; Reuven, N.; Shaul, Y. The Yes-associated protein 1 stabilizes p73 by preventing Itch-mediated ubiquitination of p73. Cell Death Differ. 2007, 14, 743–751. [Google Scholar] [CrossRef] [Green Version]
- Levy, D.; Adamovich, Y.; Reuven, N.; Shaul, Y. Yap1 phosphorylation by c-Abl is a critical step in selective activation of proapoptotic genes in response to DNA damage. Mol. Cell 2008, 29, 350–361. [Google Scholar] [CrossRef]
- Lapi, E.; Di Agostino, S.; Donzelli, S.; Gal, H.; Domany, E.; Rechavi, G.; Pandolfi, P.P.; Givol, D.; Strano, S.; Lu, X.; et al. PML, YAP, and p73 Are Components of a Proapoptotic Autoregulatory Feedback Loop. Mol. Cell 2008, 32, 803–814. [Google Scholar] [CrossRef]
- Walsh, L.; Schmuckli-Maurer, J.; Billinton, N.; Barker, M.G.; Heyer, W.D.; Walmsley, R.M. DNA-damage induction of RAD54 can be regulated independently of the RAD9- and DDC1-dependent checkpoints that regulate RNR2. Curr. Genet 2002, 41, 232–240. [Google Scholar] [CrossRef]
- Singh, V.; Connelly, Z.M.; Shen, X.; De Benedetti, A. Identification of the proteome complement of humanTLK1 reveals it binds and phosphorylates NEK1 regulating its activity. Cell. Cycle 2017, 16, 915–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Liu, J.; Ying, X.; Lin, P.C.; Zhou, B.P. Twist-mediated Epithelial-mesenchymal Transition Promotes Breast Tumor Cell Invasion via Inhibition of Hippo Pathway. Sci. Rep. 2016, 6, 24606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, M.; Domsch, K.; Bartle-Schultheis, J.; Reim, I.; Schaub, C. Twist regulates Yorkie activity to guide lineage reprogramming of syncytial alary muscles. Cell Rep. 2022, 38, 110295. [Google Scholar] [CrossRef] [PubMed]
- Dave, N.; Guaita-Esteruelas, S.; Gutarra, S.; Frias, À.; Beltran, M.; Peiró, S.; de Herreros, A.G. Functional cooperation between Snail1 and twist in the regulation of ZEB1 expression during epithelial to mesenchymal transition. J. Biol. Chem. 2011, 286, 12024–12032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, N.L.; Nagle, R.B.; Cress, A.E.; Heimark, R.L. N-Cadherin expression in human prostate carcinoma cell lines. An epithelial-mesenchymal transformation mediating adhesion withStromal cells. Am. J. Pathol. 1999, 155, 787–798. [Google Scholar] [CrossRef]
- Totaro, A.; Panciera, T.; Piccolo, S. YAP/TAZ upstream signals and downstream responses. Nat. Cell Biol. 2018, 20, 888–899. [Google Scholar] [CrossRef]
- Zhang, Y.; Donaher, J.L.; Das, S.; Li, X.; Reinhardt, F.; Krall, J.A.; Lambert, A.W.; Thiru, P.; Keys, H.R.; Khan, M.; et al. Genome-wide CRISPR screen identifies PRC2 and KMT2D-COMPASS as regulators of distinct EMT trajectories that contribute differentially to metastasis. Nat. Cell Biol. 2022, 24, 554–564. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Tretiakova, M.S.; Silvis, M.R.; Lucas, J.; Klezovitch, O.; Coleman, I.; Bolouri, H.; Kutyavin, V.I.; Morrissey, C.; True, L.D.; et al. ERG Activates the YAP1 Transcriptional Program and Induces the Development of Age-Related Prostate Tumors. Cancer Cell. 2015, 27, 797–808. [Google Scholar] [CrossRef] [Green Version]
- Febbo, P.G.; Lowenberg, M.; Thorner, A.R.; Brown, M.; Loda, M.; Golub, T.R. Androgen mediated regulation and functional implications of fkbp51 expression in prostate cancer. J. Urol. 2005, 173, 1772–1777. [Google Scholar] [CrossRef]
- Singh, V.; Jaiswal, P.K.; Ghosh, I.; Koul, H.K.; Yu, X.; De Benedetti, A. The TLK1-Nek1 axis promotes prostate cancer progression. Cancer Lett. 2019, 453, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Zou, R.; Xu, Y.; Feng, Y.; Shen, M.; Yuan, F.; Yuan, Y. YAP nuclear-cytoplasmic translocation is regulated by mechanical signaling, protein modification, and metabolism. Cell Biol. Int. 2020, 44, 1416–1425. [Google Scholar] [CrossRef] [PubMed]
- Calses, P.C.; Crawford, J.J.; Lill, J.R.; Dey, A. Hippo Pathway in Cancer: Aberrant Regulation and Therapeutic Opportunities. Trends Cancer 2019, 5, 297–307. [Google Scholar] [CrossRef] [Green Version]
- Basu, S.; Totty, N.F.; Irwin, M.S.; Sudol, M.; Downward, J. Akt phosphorylates the Yes-associated protein, YAP, to induce interaction with 14-3-3 and attenuation of p73-mediated apoptosis. Mol. Cell. 2003, 11, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, S.; Dupont, S.; Cordenonsi, M. The biology of YAP/TAZ: Hippo signaling and beyond. Physiol. Rev. 2014, 94, 1287–1312. [Google Scholar] [CrossRef] [PubMed]
- Raghubir, M.; Azeem, S.M.; Hasnat, R.; Rahman, C.N.; Wong, L.; Yan, S.; Huang, Y.Q.; Zhagui, R.; Blyufer, A.; Tariq, I.; et al. Riluzole-induced apoptosis in osteosarcoma is mediated through Yes-associated protein upon phosphorylation by c-Abl Kinase. Sci. Rep. 2021, 11, 20974. [Google Scholar] [CrossRef] [PubMed]
- Nardone, G.; Oliver-De La Cruz, J.; Vrbsky, J.; Martini, C.; Pribyl, J.; Skládal, P.; Pešl, M.; Caluori, G.; Pagliari, S.; Martino, F.; et al. YAP regulates cell mechanics by controlling focal adhesion assembly. Nat. Commun. 2017, 8, 15321. [Google Scholar] [CrossRef] [Green Version]
- Feige, E.; Shalom, O.; Tsuriel, S.; Yissachar, N.; Motro, B. Nek1 shares structural and functional similarities with NIMA kinase. Biochim. Biophys. Acta. 2006, 1763, 272–281. [Google Scholar] [CrossRef] [Green Version]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghosh, I.; Khalil, M.I.; Mirza, R.; King, J.; Olatunde, D.; De Benedetti, A. NEK1-Mediated Phosphorylation of YAP1 Is Key to Prostate Cancer Progression. Biomedicines 2023, 11, 734. https://doi.org/10.3390/biomedicines11030734
Ghosh I, Khalil MI, Mirza R, King J, Olatunde D, De Benedetti A. NEK1-Mediated Phosphorylation of YAP1 Is Key to Prostate Cancer Progression. Biomedicines. 2023; 11(3):734. https://doi.org/10.3390/biomedicines11030734
Chicago/Turabian StyleGhosh, Ishita, Md Imtiaz Khalil, Rusella Mirza, Judy King, Damilola Olatunde, and Arrigo De Benedetti. 2023. "NEK1-Mediated Phosphorylation of YAP1 Is Key to Prostate Cancer Progression" Biomedicines 11, no. 3: 734. https://doi.org/10.3390/biomedicines11030734