
Immortalized B Cells Transfected with mRNA of Antigen Fused to MITD (IBMAM): An Effective Tool for Antigen-Specific T-Cell Expansion and TCR Validation
 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Normal Healthy Blood, Patients, and Biospecimens
2.2. Cell Culture [B Cells, Feeder Cells, T Cells, Jurkat TCRαβ-KO (CD4+, CD8+)]
2.3. Plasmid DNA
2.4. Generation of Immortalized B Cells
2.5. In Vitro Transcription of Antigens mRNA and Electroporation of the mRNA to Immortalized B Cells
2.6. HLA Genotyping
2.7. Rapid Expansion Protocol (REP)
2.8. ELISA
2.9. Flow Cytometric Analysis
2.10. Immunofluorescence Staining
2.11. Jurkat T-Cell Activation Bioassay
2.12. IBMAM Stimulation of PBMCs
2.13. IBMAM Detection of Antigen-Specific T Cells (Cytokines Secretion Assay)
2.14. Expansion of Antigen-Specific T Cells
2.15. Statistical Analysis
3. Results
3.1. Characterization of Immortalized B Cells
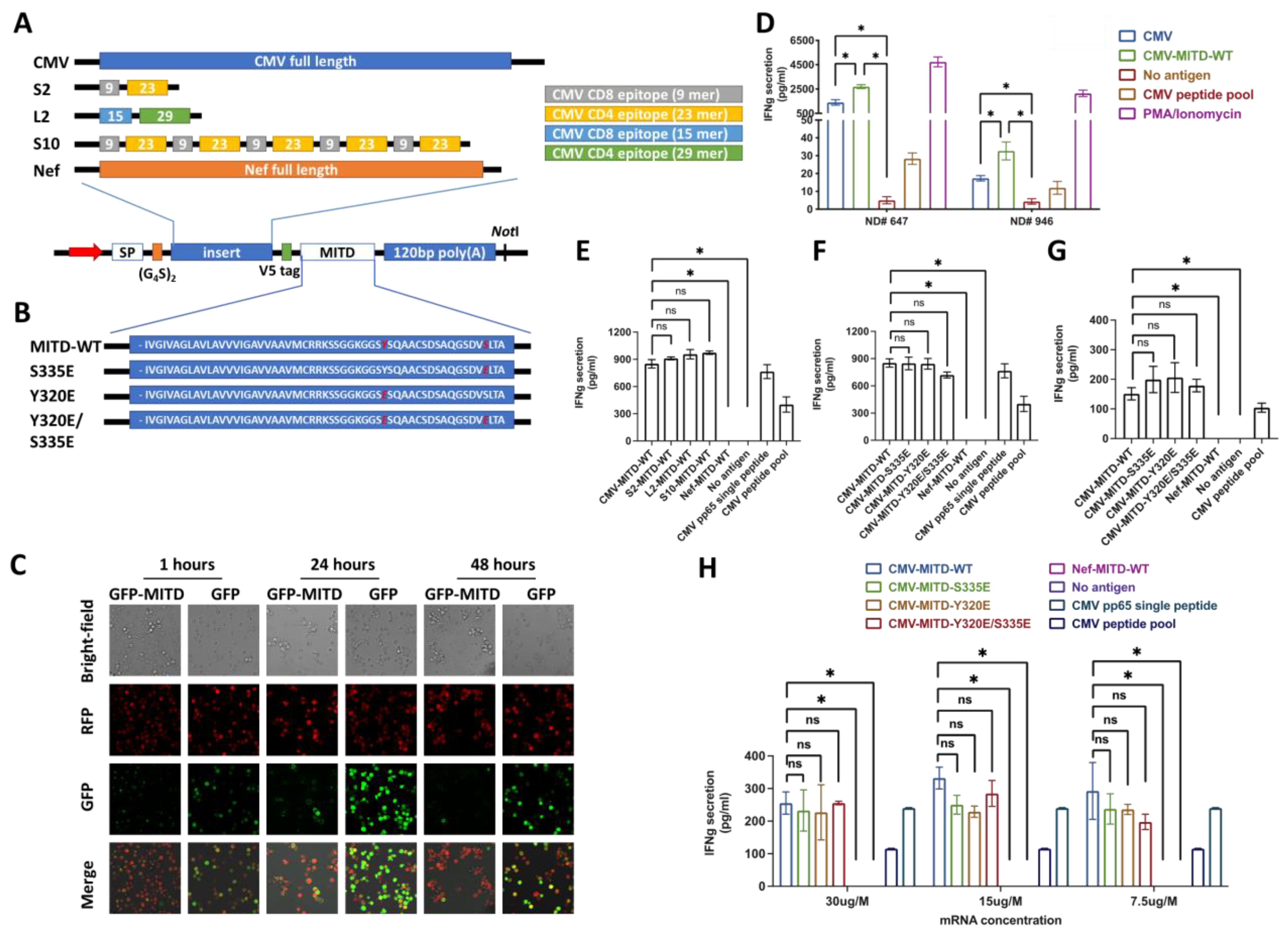
3.2. IBMAM: Antigen Presentation Efficiency Assessment with MITD/MITD Variants and Variable Epitope Arrangements
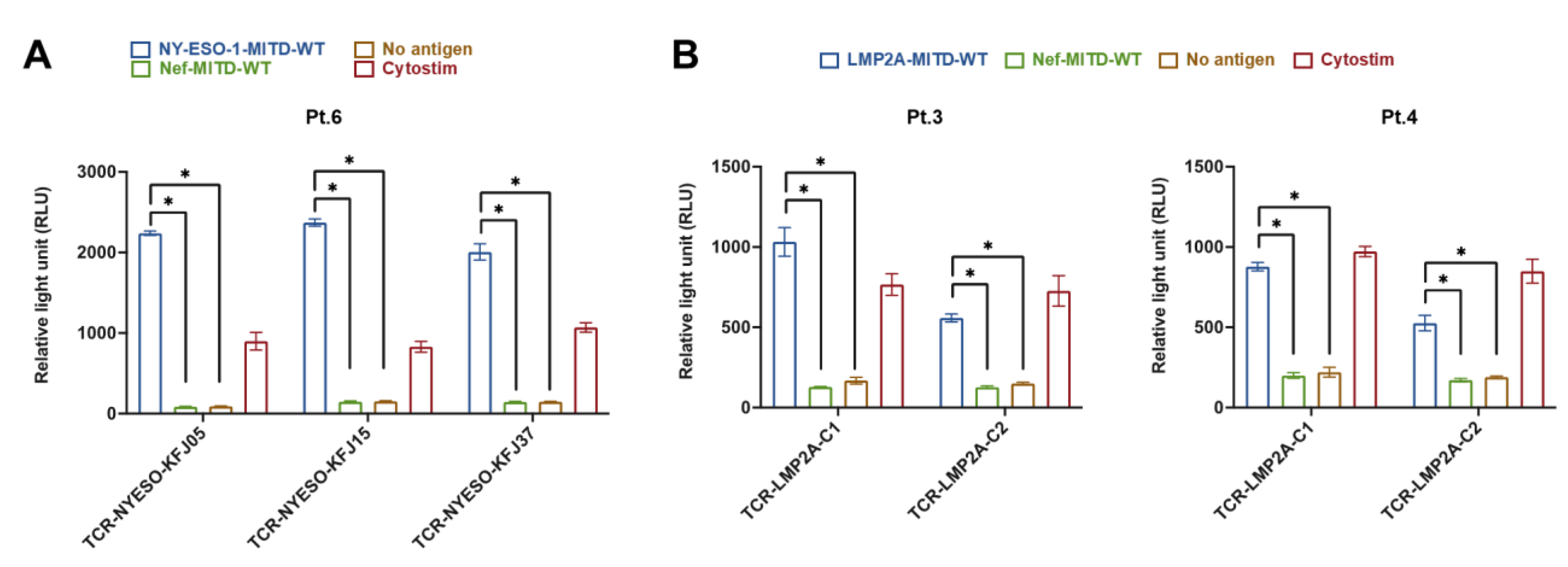
3.3. Assessment of the Interaction between Antigen-Specific T Cells and IBMAM
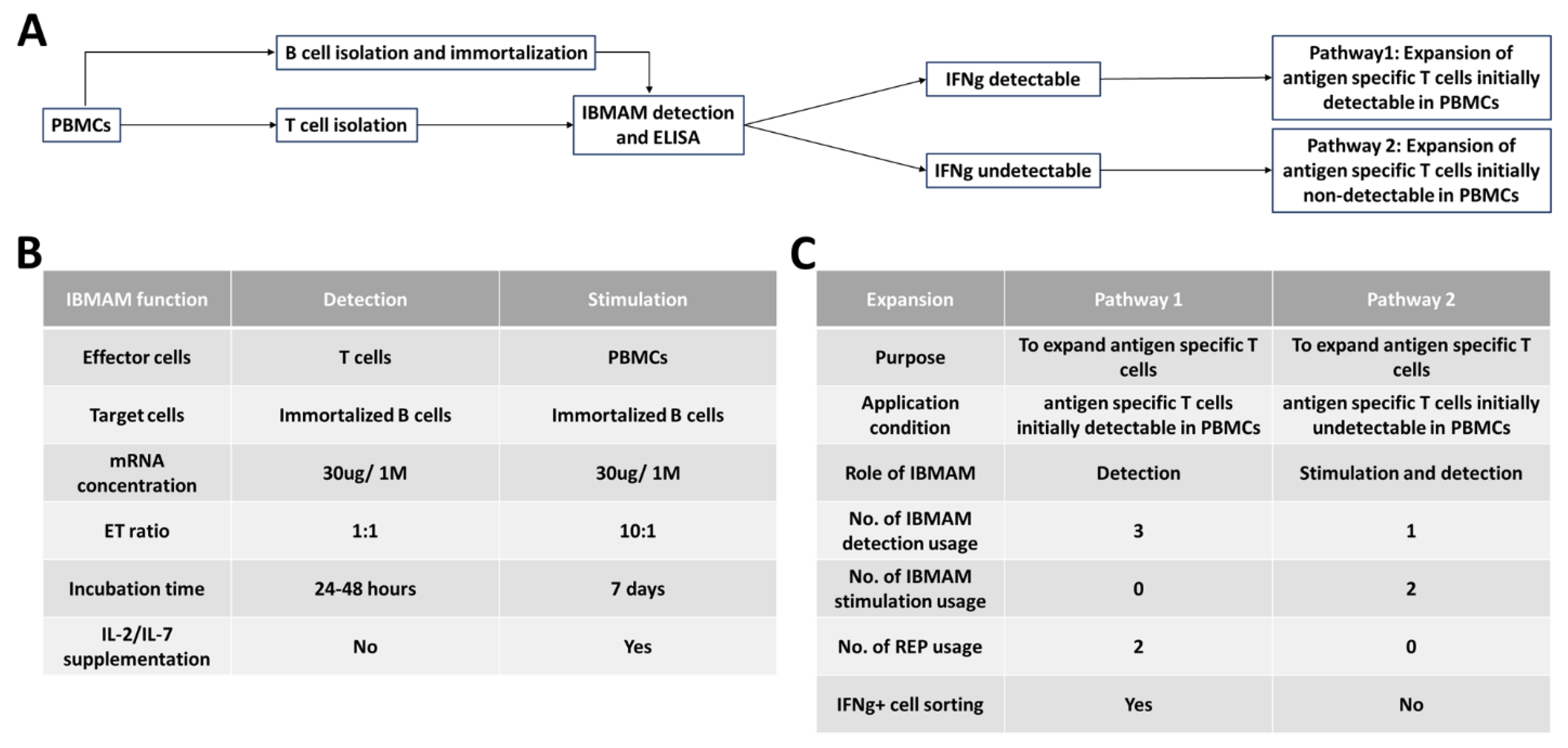
3.4. General Schema of IBMAM Application in Antigen-Specific T-Cells Expansion
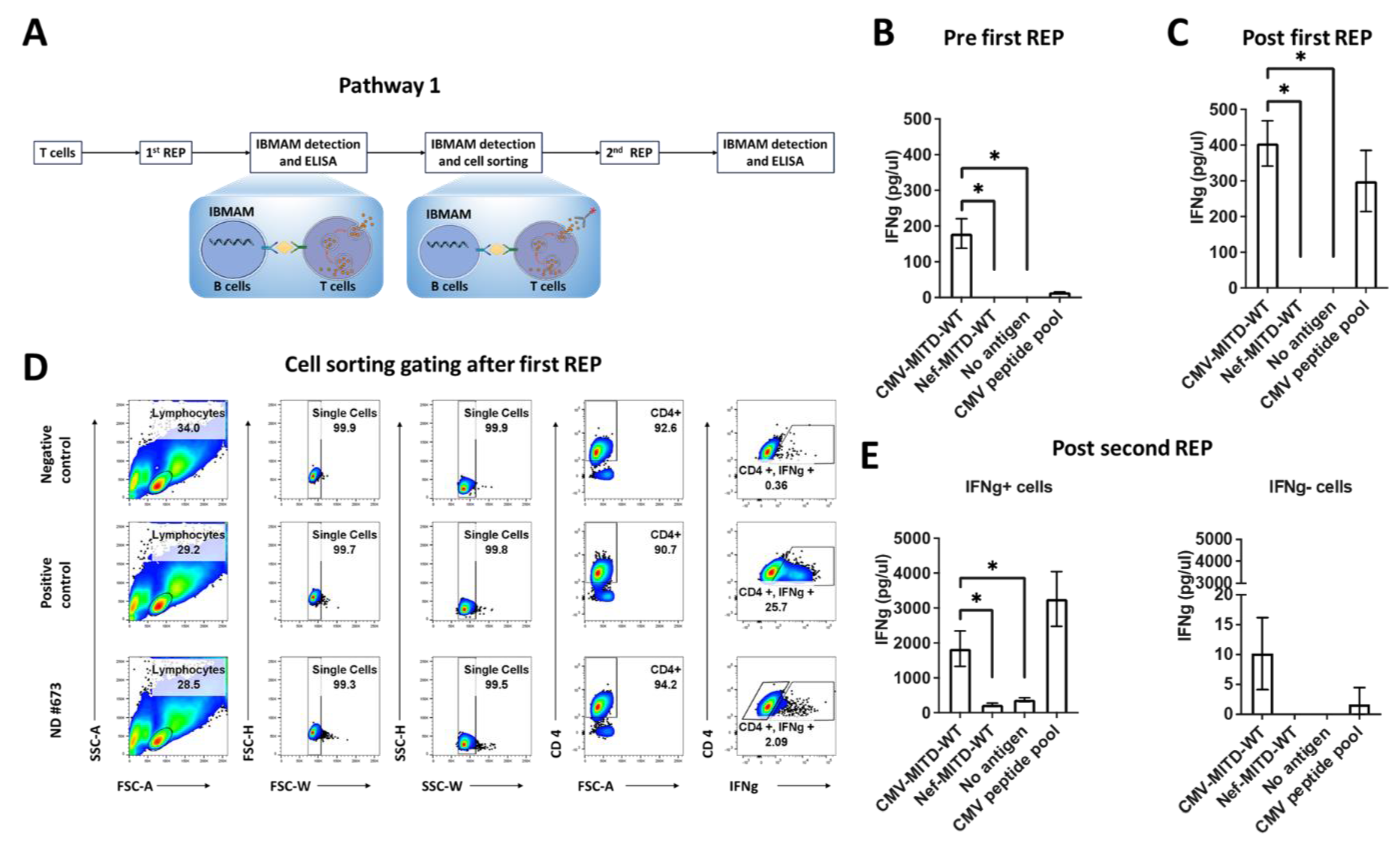
3.5. Pathway1: Expansion of Antigen-Specific T Cells Initially Detectable in PBMCs Using IBMAM
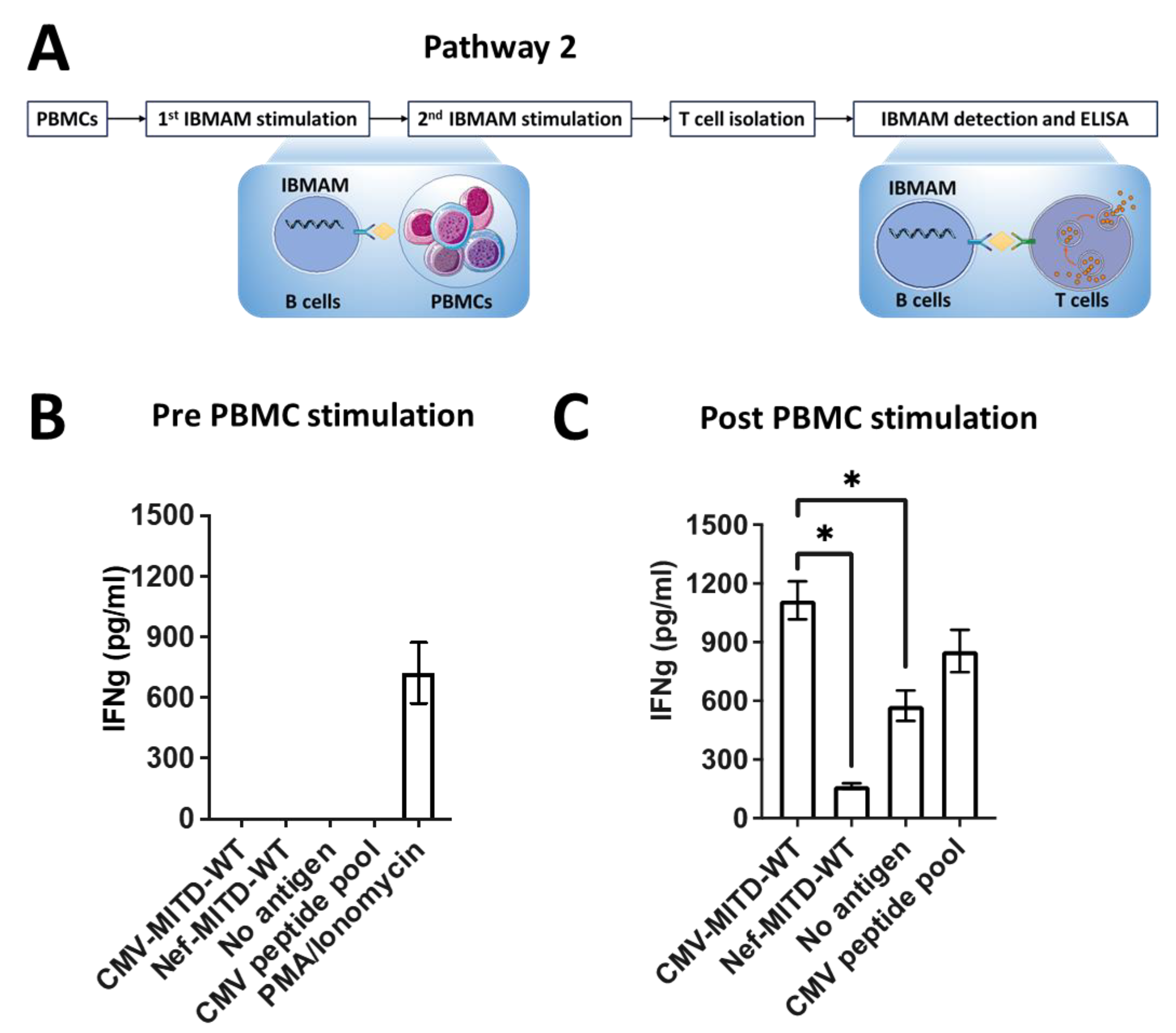
3.6. Pathway2: Expansion of Antigen-Specific T Cells Initially Undetectable in PBMCs Using IBMAM
3.7. IBMAM Platform Application to TCR Validation
4. Discussion
Limitation of the Study
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Di Blasi, D.; Claessen, I.; Turksma, A.W.; van Beek, J.; Ten Brinke, A. Guidelines for analysis of low-frequency antigen-specific T cell results: Dye-based proliferation assay vs (3)H-thymidine incorporation. J. Immunol. Methods 2020, 487, 112907. [Google Scholar] [CrossRef]
- Bullock, T.N.; Colella, T.A.; Engelhard, V.H. The density of peptides displayed by dendritic cells affects immune responses to human tyrosinase and gp100 in HLA-A2 transgenic mice. J. Immunol. 2000, 164, 2354–2361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.P.; Simon, P.; Lower, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrors, B.; et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Esprit, A.; de Mey, W.; Bahadur Shahi, R.; Thielemans, K.; Franceschini, L.; Breckpot, K. Neo-Antigen mRNA Vaccines. Vaccines 2020, 8, 776. [Google Scholar] [CrossRef] [PubMed]
- Lizee, G.; Basha, G.; Jefferies, W.A. Tails of wonder: Endocytic-sorting motifs key for exogenous antigen presentation. Trends Immunol. 2005, 26, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Basha, G.; Lizee, G.; Reinicke, A.T.; Seipp, R.P.; Omilusik, K.D.; Jefferies, W.A. MHC class I endosomal and lysosomal trafficking coincides with exogenous antigen loading in dendritic cells. PLoS ONE 2008, 3, e3247. [Google Scholar] [CrossRef] [PubMed]
- Kreiter, S.; Selmi, A.; Diken, M.; Sebastian, M.; Osterloh, P.; Schild, H.; Huber, C.; Tureci, O.; Sahin, U. Increased antigen presentation efficiency by coupling antigens to MHC class I trafficking signals. J. Immunol. 2008, 180, 309–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, M.; Foldvari, Z.; Giannakopoulou, E.; Boschen, M.L.; Stronen, E.; Yang, W.; Toebes, M.; Schubert, B.; Kohlbacher, O.; Schumacher, T.N.; et al. Induction of neoantigen-reactive T cells from healthy donors. Nat. Protoc. 2019, 14, 1926–1943. [Google Scholar] [CrossRef]
- Rossi, M.; Young, J.W. Human dendritic cells: Potent antigen-presenting cells at the crossroads of innate and adaptive immunity. J. Immunol. 2005, 175, 1373–1381. [Google Scholar] [CrossRef] [Green Version]
- Kwakkenbos, M.J.; Diehl, S.A.; Yasuda, E.; Bakker, A.Q.; van Geelen, C.M.; Lukens, M.V.; van Bleek, G.M.; Widjojoatmodjo, M.N.; Bogers, W.M.; Mei, H.; et al. Generation of stable monoclonal antibody-producing B cell receptor-positive human memory B cells by genetic programming. Nat. Med. 2010, 16, 123–128. [Google Scholar] [CrossRef]
- Caeser, R.; Gao, J.; Di Re, M.; Gong, C.; Hodson, D.J. Genetic manipulation and immortalized culture of ex vivo primary human germinal center B cells. Nat. Protoc. 2021, 16, 2499–2519. [Google Scholar] [CrossRef] [PubMed]
- Scheper, W.; Kelderman, S.; Fanchi, L.F.; Linnemann, C.; Bendle, G.; de Rooij, M.A.J.; Hirt, C.; Mezzadra, R.; Slagter, M.; Dijkstra, K.; et al. Low and variable tumor reactivity of the intratumoral TCR repertoire in human cancers. Nat. Med. 2019, 25, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Lapointe, R.; Bellemare-Pelletier, A.; Housseau, F.; Thibodeau, J.; Hwu, P. CD40-stimulated B lymphocytes pulsed with tumor antigens are effective antigen-presenting cells that can generate specific T cells. Cancer Res. 2003, 63, 2836–2843. [Google Scholar] [PubMed]
- Holtkamp, S.; Kreiter, S.; Selmi, A.; Simon, P.; Koslowski, M.; Huber, C.; Tureci, O.; Sahin, U. Modification of antigen-encoding RNA increases stability, translational efficacy, and T-cell stimulatory capacity of dendritic cells. Blood 2006, 108, 4009–4017. [Google Scholar] [CrossRef]
- Simon, P.; Omokoko, T.A.; Breitkreuz, A.; Hebich, L.; Kreiter, S.; Attig, S.; Konur, A.; Britten, C.M.; Paret, C.; Dhaene, K.; et al. Functional TCR retrieval from single antigen-specific human T cells reveals multiple novel epitopes. Cancer Immunol. Res. 2014, 2, 1230–1244. [Google Scholar] [CrossRef] [Green Version]
- Scheeren, F.A.; Naspetti, M.; Diehl, S.; Schotte, R.; Nagasawa, M.; Wijnands, E.; Gimeno, R.; Vyth-Dreese, F.A.; Blom, B.; Spits, H. STAT5 regulates the self-renewal capacity and differentiation of human memory B cells and controls Bcl-6 expression. Nat. Immunol. 2005, 6, 303–313. [Google Scholar] [CrossRef]
- Zhong, C.; Gragert, L.; Maiers, M.; Hill, B.T.; Garcia-Gomez, J.; Gendzekhadze, K.; Senitzer, D.; Song, J.; Weisenburger, D.; Goldstein, L.; et al. The association between HLA and non-Hodgkin lymphoma subtypes, among a transplant-indicated population. Leuk. Lymphoma 2019, 60, 2899–2908. [Google Scholar] [CrossRef] [Green Version]
- Dudley, M.E.; Wunderlich, J.R.; Shelton, T.E.; Even, J.; Rosenberg, S.A. Generation of tumor-infiltrating lymphocyte cultures for use in adoptive transfer therapy for melanoma patients. J. Immunother. 2003, 26, 332–342. [Google Scholar] [CrossRef]
- Kusam, S.; Vasanwala, F.H.; Dent, A.L. Transcriptional repressor BCL-6 immortalizes germinal center-like B cells in the absence of p53 function. Oncogene 2004, 23, 839–844. [Google Scholar] [CrossRef]
- Kwakkenbos, M.J.; van Helden, P.M.; Beaumont, T.; Spits, H. Stable long-term cultures of self-renewing B cells and their applications. Immunol. Rev. 2016, 270, 65–77. [Google Scholar] [CrossRef] [Green Version]
- Zotos, D.; Coquet, J.M.; Zhang, Y.; Light, A.; D’Costa, K.; Kallies, A.; Corcoran, L.M.; Godfrey, D.I.; Toellner, K.M.; Smyth, M.J.; et al. IL-21 regulates germinal center B cell differentiation and proliferation through a B cell-intrinsic mechanism. J. Exp. Med. 2010, 207, 365–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Cruz, T.G.; Liu, S.; Khalili, J.S.; Whittington, M.; Zhang, M.; Overwijk, W.; Lizee, G. Natural splice variant of MHC class I cytoplasmic tail enhances dendritic cell-induced CD8+ T-cell responses and boosts anti-tumor immunity. PLoS ONE 2011, 6, e22939. [Google Scholar] [CrossRef] [Green Version]
- Qiao, Y.; Molina, H.; Pandey, A.; Zhang, J.; Cole, P.A. Chemical rescue of a mutant enzyme in living cells. Science 2006, 311, 1293–1297. [Google Scholar] [CrossRef] [PubMed]
- Wills, M.R.; Carmichael, A.J.; Mynard, K.; Jin, X.; Weekes, M.P.; Plachter, B.; Sissons, J.G. The human cytotoxic T-lymphocyte (CTL) response to cytomegalovirus is dominated by structural protein pp65: Frequency, specificity, and T-cell receptor usage of pp65-specific CTL. J. Virol. 1996, 70, 7569–7579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nastke, M.D.; Herrgen, L.; Walter, S.; Wernet, D.; Rammensee, H.G.; Stevanovic, S. Major contribution of codominant CD8 and CD4 T cell epitopes to the human cytomegalovirus-specific T cell repertoire. Cell Mol. Life Sci. 2005, 62, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.K.; Cha, S.C.; Smith, D.L.; Kim, K.H.; Parshottam, S.R.; Rao, S.; Popescu, M.; Lee, V.Y.; Neelapu, S.S.; Kwak, L.W. Phase I study of an active immunotherapy for asymptomatic phase Lymphoplasmacytic lymphoma with DNA vaccines encoding antigen-chemokine fusion: Study protocol. BMC Cancer 2018, 18, 187. [Google Scholar] [CrossRef] [Green Version]
- Friedman, K.M.; Prieto, P.A.; Devillier, L.E.; Gross, C.A.; Yang, J.C.; Wunderlich, J.R.; Rosenberg, S.A.; Dudley, M.E. Tumor-specific CD4+ melanoma tumor-infiltrating lymphocytes. J. Immunother. 2012, 35, 400–408. [Google Scholar] [CrossRef]
- Kitano, S.; Tsuji, T.; Liu, C.; Hirschhorn-Cymerman, D.; Kyi, C.; Mu, Z.; Allison, J.P.; Gnjatic, S.; Yuan, J.D.; Wolchok, J.D. Enhancement of tumor-reactive cytotoxic CD4+ T cell responses after ipilimumab treatment in four advanced melanoma patients. Cancer Immunol. Res. 2013, 1, 235–244. [Google Scholar] [CrossRef] [Green Version]
- Baskar, S.; Kobrin, C.B.; Kwak, L.W. Autologous lymphoma vaccines induce human T cell responses against multiple, unique epitopes. J. Clin. Investig. 2004, 113, 1498–1510. [Google Scholar] [CrossRef] [PubMed]
- Biragyn, A.; Surenhu, M.; Yang, D.; Ruffini, P.A.; Haines, B.A.; Klyushnenkova, E.; Oppenheim, J.J.; Kwak, L.W. Mediators of innate immunity that target immature, but not mature, dendritic cells induce antitumor immunity when genetically fused with nonimmunogenic tumor antigens. J. Immunol. 2001, 167, 6644–6653. [Google Scholar] [CrossRef] [Green Version]
- Bendandi, M.; Gocke, C.D.; Kobrin, C.B.; Benko, F.A.; Sternas, L.A.; Pennington, R.; Watson, T.M.; Reynolds, C.W.; Gause, B.L.; Duffey, P.L.; et al. Complete molecular remissions induced by patient-specific vaccination plus granulocyte-monocyte colony-stimulating factor against lymphoma. Nat. Med. 1999, 5, 1171–1177. [Google Scholar] [CrossRef]
- Khodadoust, M.S.; Olsson, N.; Wagar, L.E.; Haabeth, O.A.; Chen, B.; Swaminathan, K.; Rawson, K.; Liu, C.L.; Steiner, D.; Lund, P.; et al. Antigen presentation profiling reveals recognition of lymphoma immunoglobulin neoantigens. Nature 2017, 543, 723–727. [Google Scholar] [CrossRef] [Green Version]
- Lu, T.L.; Pugach, O.; Somerville, R.; Rosenberg, S.A.; Kochenderfer, J.N.; Better, M.; Feldman, S.A. A Rapid Cell Expansion Process for Production of Engineered Autologous CAR-T Cell Therapies. Hum. Gene Ther. Methods 2016, 27, 209–218. [Google Scholar] [CrossRef]
- Chan, K.F.; Gully, B.S.; Gras, S.; Beringer, D.X.; Kjer-Nielsen, L.; Cebon, J.; McCluskey, J.; Chen, W.; Rossjohn, J. Divergent T-cell receptor recognition modes of a HLA-I restricted extended tumour-associated peptide. Nat. Commun. 2018, 9, 1026. [Google Scholar] [CrossRef] [Green Version]
- Huisman, W.; Gille, I.; van der Maarel, L.E.; Hageman, L.; Morton, L.T.; de Jong, R.C.M.; Heemskerk, M.H.M.; Amsen, D.; Falkenburg, J.H.F.; Jedema, I. Identification of Functional HLA-A*01: 01-Restricted EBV-LMP2-Specific T-cell Receptors. J. Infect. Dis. 2020, 226, 833–842. [Google Scholar] [CrossRef]
- Good, K.L.; Bryant, V.L.; Tangye, S.G. Kinetics of human B cell behavior and amplification of proliferative responses following stimulation with IL-21. J. Immunol. 2006, 177, 5236–5247. [Google Scholar] [CrossRef] [Green Version]
- Bonehill, A.; Heirman, C.; Thielemans, K. Genetic approaches for the induction of a CD4+ T cell response in cancer immunotherapy. J. Gene Med. 2005, 7, 686–695. [Google Scholar] [CrossRef]
- Rowell, J.F.; Ruff, A.L.; Guarnieri, F.G.; Staveley-O’Carroll, K.; Lin, X.; Tang, J.; August, J.T.; Siliciano, R.F. Lysosome-associated membrane protein-1-mediated targeting of the HIV-1 envelope protein to an endosomal/lysosomal compartment enhances its presentation to MHC class II-restricted T cells. J. Immunol. 1995, 155, 1818–1828. [Google Scholar] [CrossRef]
- Livingston, B.D.; Newman, M.; Crimi, C.; McKinney, D.; Chesnut, R.; Sette, A. Optimization of epitope processing enhances immunogenicity of multiepitope DNA vaccines. Vaccine 2001, 19, 4652–4660. [Google Scholar] [CrossRef]
- Guild, B.C.; Strominger, J.L. Human and murine class I MHC antigens share conserved serine 335, the site of HLA phosphorylation in vivo. J. Biol. Chem. 1984, 259, 9235–9240. [Google Scholar] [CrossRef]
- Loube, S.R.; Owen, M.J.; Crumpton, M.J. Human class I histocompatibility antigens (HLA-A,B,C). A small proportion only is phosphorylated. Biochem. J. 1983, 210, 79–87. [Google Scholar] [CrossRef] [Green Version]
- Capps, G.G.; Zuniga, M.C. Phosphorylation of class I MHC molecules in the absence of phorbol esters is an intracellular event and may be characteristic of trafficking molecules. Mol. Immunol. 2000, 37, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Santos, S.G.; Powis, S.J.; Arosa, F.A. Misfolding of major histocompatibility complex class I molecules in activated T cells allows cis-interactions with receptors and signaling molecules and is associated with tyrosine phosphorylation. J. Biol. Chem. 2004, 279, 53062–53070. [Google Scholar] [CrossRef] [Green Version]
- Guild, B.C.; Erikson, R.L.; Strominger, J.L. HLA-A2 and HLA-B7 antigens are phosphorylated in vitro by rous sarcoma virus kinase (pp60v-src) at a tyrosine residue encoded in a highly conserved exon of the intracellular domain. Proc. Natl. Acad. Sci. USA 1983, 80, 2894–2898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuniga, E.I.; Harker, J.A. T-cell exhaustion due to persistent antigen: Quantity not quality? Eur. J. Immunol. 2012, 42, 2285–2289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parkhurst, M.R.; Robbins, P.F.; Tran, E.; Prickett, T.D.; Gartner, J.J.; Jia, L.; Ivey, G.; Li, Y.F.; El-Gamil, M.; Lalani, A.; et al. Unique Neoantigens Arise from Somatic Mutations in Patients with Gastrointestinal Cancers. Cancer Discov. 2019, 9, 1022–1035. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Z.; Zhang, T.; Anderson, A.; Lee, V.; Szymura, S.; Dong, Z.; Kuang, B.; Oh, E.; Liu, J.; Neelapu, S.S.; et al. Immortalized B Cells Transfected with mRNA of Antigen Fused to MITD (IBMAM): An Effective Tool for Antigen-Specific T-Cell Expansion and TCR Validation. Biomedicines 2023, 11, 796. https://doi.org/10.3390/biomedicines11030796
Wang Z, Zhang T, Anderson A, Lee V, Szymura S, Dong Z, Kuang B, Oh E, Liu J, Neelapu SS, et al. Immortalized B Cells Transfected with mRNA of Antigen Fused to MITD (IBMAM): An Effective Tool for Antigen-Specific T-Cell Expansion and TCR Validation. Biomedicines. 2023; 11(3):796. https://doi.org/10.3390/biomedicines11030796
Chicago/Turabian StyleWang, Zhe, Tiantian Zhang, Aaron Anderson, Vincent Lee, Szymon Szymura, Zhenyuan Dong, Benjamin Kuang, Elizabeth Oh, Jingwei Liu, Sattva S. Neelapu, and et al. 2023. "Immortalized B Cells Transfected with mRNA of Antigen Fused to MITD (IBMAM): An Effective Tool for Antigen-Specific T-Cell Expansion and TCR Validation" Biomedicines 11, no. 3: 796. https://doi.org/10.3390/biomedicines11030796