





Inhibition of Microglial GSK3β Activity Is Common to Different Kinds of Antidepressants: A Proposal for an In Vitro Screen to Detect Novel Antidepressant Principles
Abstract
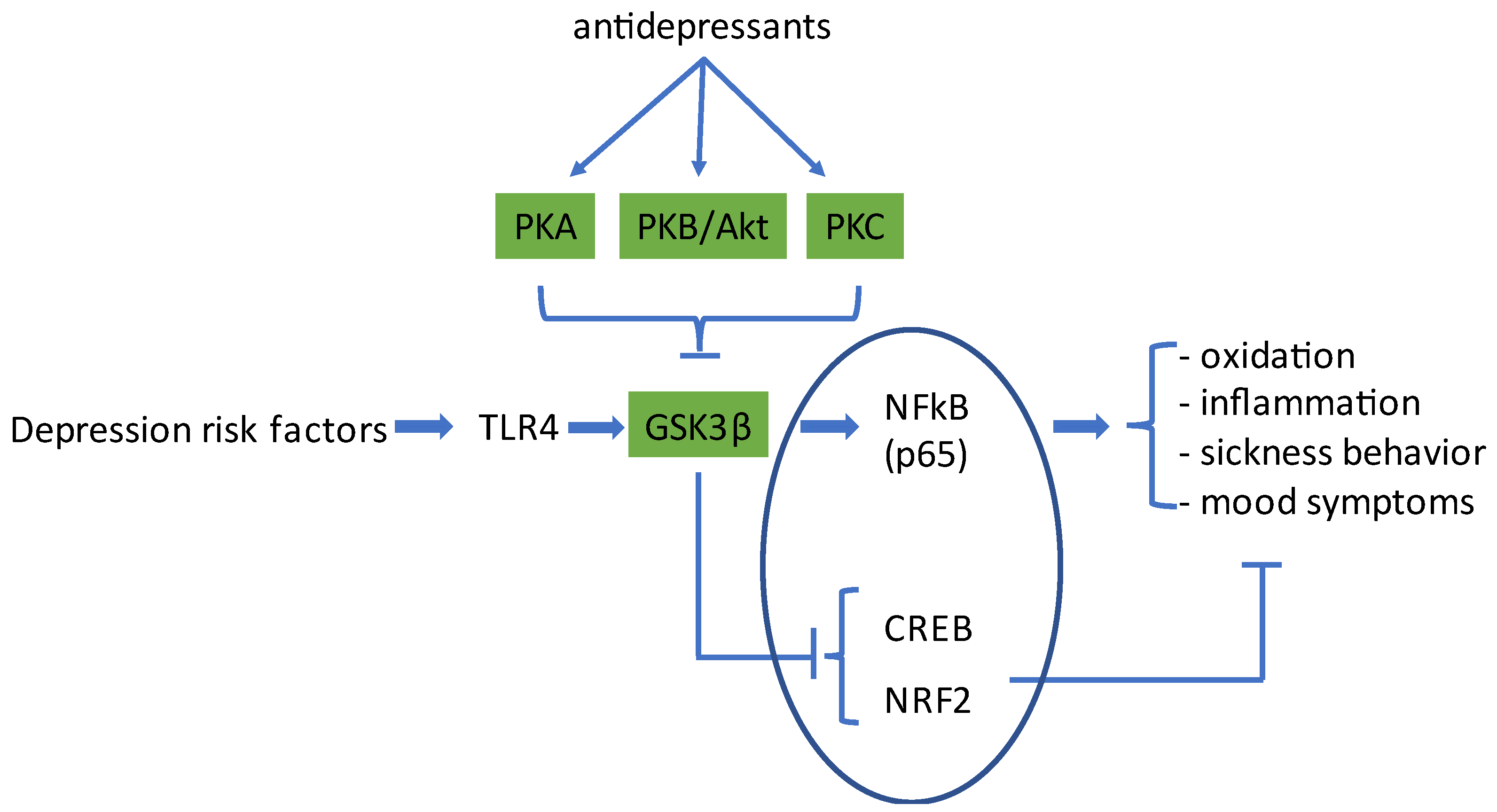
1. Introduction
2. Activation of Microglia Cells by Risk Factors for Depression
3. Classical Antidepressants Activate PKA to Inhibit GSK3β
4. Activation of PKB/Akt to Inhibit GSK3β
5. Gq-PLC-PKC-Induced GSK3β-Inhibition
5.1. Fatty Acids and Endocannabinoids
5.2. Opioid μ-Receptor Agonists
5.3. Hallucinogenic 5HT2 Agonists
6. Discussion
6.1. Ketamine
6.2. GSK3β Blockade by Antipsychotics
6.3. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Malhi, G.S.; Mann, J.J. Depression. Lancet 2018, 392, 2299–2312. [Google Scholar] [CrossRef]
- Schildkraut, J.J. The Catecholamine Hypothesis of Affective Disorders: A Review of Supporting Evidence. Am. J. Psychiatry 1965, 122, 509–522. [Google Scholar] [CrossRef]
- Capuron, L.; Miller, A.H. Immune system to brain signaling: Neuropsychopharmacological implications. Pharmacol. Ther. 2011, 130, 226–238. [Google Scholar] [CrossRef]
- Berk, M.; Williams, L.J.; Jacka, F.N.; O’Neil, A.; Pasco, J.A.; Moylan, S.; Allen, N.B.; Stuart, A.L.; Hayley, A.; Byrne, M.L.; et al. So depression is an inflammatory disease, but where does the inflammation come from? BMC Med. 2013, 11, 200. [Google Scholar] [CrossRef]
- Ögren, S.O.; Fuxe, K.; Agnati, L. The Importance of Brain Serotonergic Receptor Mechanisms for the Action of Antidepressant Drugs. Pharmacopsychiatry 1985, 18, 209–213. [Google Scholar] [CrossRef]
- Palazidou, E.; Beer, M.S.; Checkley, S.; Stahl, S.M. Pharmacologic exploitation of neurotransmitter receptors for the design of novel antidepressant drugs. Drug Des. Deliv. 1988, 2, 247–256. [Google Scholar]
- Broekkamp, C.L.E.; Leysen, D.; Peeters, B.W.; Pinder, R.M. Prospects for Improved Antidepressants. J. Med. Chem. 1995, 38, 4615–4633. [Google Scholar] [CrossRef]
- Kenis, G.; Maes, M. Effects of antidepressants on the production of cytokines. Int. J. Neuropsychopharmacol. 2002, 5, 401–412. [Google Scholar] [CrossRef]
- Haroon, E.; Raison, C.L.; Miller, A.H. Psychoneuroimmunology Meets Neuropsychopharmacology: Translational Implications of the Impact of Inflammation on Behavior. Neuropsychopharmacology 2011, 37, 137–162. [Google Scholar] [CrossRef]
- GBD 2019 Mental Disorders Collaborators. Global, regional, and national burden of 12 mental disorders in 204 countries and territories, 1990–2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet Psychiatry 2022, 9, 137–150. [Google Scholar] [CrossRef]
- COVID-19 Mental Disorders Collaborators. Global prevalence and burden of depressive and anxiety disorders in 204 countries and territories in 2020 due to the COVID-19 pandemic. Lancet 2021, 398, 1700–1712. [Google Scholar] [CrossRef]
- Lee, S.; Jeong, J.; Kwak, Y.; Park, S.K. Depression research: Where are we now? Mol. Brain 2010, 3, 8. [Google Scholar] [CrossRef]
- Koo, J.W.; Russo, S.J.; Ferguson, D.; Nestler, E.J.; Duman, R.S. Nuclear factor-κB is a critical mediator of stress-impaired neurogenesis and depressive behavior. Proc. Natl. Acad. Sci. USA 2010, 107, 2669–2674. [Google Scholar] [CrossRef]
- Cheng, Y.; Pardo, M.; Armini, R.D.S.; Martinez, A.; Mouhsine, H.; Zagury, J.-F.; Jope, R.S.; Beurel, E. Stress-induced neuroinflammation is mediated by GSK3-dependent TLR4 signaling that promotes susceptibility to depression-like behavior. Brain Behav. Immun. 2016, 53, 207–222. [Google Scholar] [CrossRef]
- Duda, P.; Hajka, D.; Wójcicka, O.; Rakus, D.; Gizak, A. GSK3β: A Master Player in Depressive Disorder Pathogenesis and Treatment Responsiveness. Cells 2020, 9, 727. [Google Scholar] [CrossRef]
- Krishnadas, R.; Cavanagh, J. Depression: An inflammatory illness? J. Neurol. Neurosurg. Psychiatry 2012, 83, 495–502. [Google Scholar] [CrossRef]
- Strawbridge, R.; Arnone, D.; Danese, A.; Papadopoulos, A.; Vives, A.H.; Cleare, A.J. Inflammation and clinical response to treatment in depression: A meta-analysis. Eur. Neuropsychopharmacol. 2015, 25, 1532–1543. [Google Scholar] [CrossRef]
- Benros, M.E.; Waltoft, B.L.; Nordentoft, M.; Østergaard, S.D.; Eaton, W.W.; Krogh, J.; Mortensen, P.B. Autoimmune Diseases and Severe Infections as Risk Factors for Mood Disorders: A nationwide study. JAMA Psychiatry 2013, 70, 812–820. [Google Scholar] [CrossRef]
- Bhandari, S.; Larson, M.E.; Kumar, N.; Stein, D. Association of Inflammatory Bowel Disease (IBD) with Depressive Symptoms in the United States Population and Independent Predictors of Depressive Symptoms in an IBD Population: A NHANES Study. Gut Liver 2017, 11, 512–519. [Google Scholar] [CrossRef]
- Beurel, E.; Toups, M.; Nemeroff, C.B. The Bidirectional Relationship of Depression and Inflammation: Double Trouble. Neuron 2020, 107, 234–256. [Google Scholar] [CrossRef]
- Kalkman, H.O. The Association Between Vascular Inflammation and Depressive Disorder. Causality, Biomarkers and Targeted Treatment. Pharmaceuticals 2020, 13, 92. [Google Scholar] [CrossRef] [PubMed]
- Vezzani, A. Epilepsy and Inflammation in the Brain: Overview and Pathophysiology. Epilepsy Curr. 2014, 14, 3–7. [Google Scholar] [CrossRef]
- Chung, Y.-C.; Ko, H.-W.; Bok, E.-G.; Park, E.-S.; Huh, S.-H.; Nam, J.-H.; Jin, B.-K. The role of neuroinflammation on the pathogenesis of Parkinson’s disease. BMB Rep. 2010, 43, 225–232. [Google Scholar] [CrossRef]
- Ouchi, Y.; Yagi, S.; Yokokura, M.; Sakamoto, M. Neuroinflammation in the living brain of Parkinson’s disease. Park. Relat. Disord. 2009, 15 (Suppl. S3), S200–S204. [Google Scholar] [CrossRef]
- Tan, Z.S.; Seshadri, S. Inflammation in the Alzheimer’s disease cascade: Culprit or innocent bystander? Alzheimer’s Res. Ther. 2010, 2, 6. [Google Scholar] [CrossRef]
- Sandberg, M.; Patil, J.; D’Angelo, B.; Weber, S.G.; Mallard, C. NRF2-regulation in brain health and disease: Implication of cerebral inflammation. Neuropharmacology 2014, 79, 298–306. [Google Scholar] [CrossRef]
- Sullivan, L.E.; Fiellin, D.A.; O’Connor, P.G. The prevalence and impact of alcohol problems in major depression: A systematic review. Am. J. Med. 2005, 118, 330–341. [Google Scholar] [CrossRef]
- Fergusson, D.M.; Boden, J.M.; Horwood, L.J. Tests of Causal Links Between Alcohol Abuse or Dependence and Major Depression. Arch. Gen. Psychiatry 2009, 66, 260–266. [Google Scholar] [CrossRef]
- Beurel, E.; Jope, R.S. Inflammation and lithium: Clues to mechanisms contributing to suicide-linked traits. Transl. Psychiatry 2014, 4, e488. [Google Scholar] [CrossRef]
- Mogensen, T.H. Pathogen Recognition and Inflammatory Signaling in Innate Immune Defenses. Clin. Microbiol. Rev. 2009, 22, 240–273. [Google Scholar] [CrossRef]
- Kaufmann, F.N.; Costa, A.P.; Ghisleni, G.; Diaz, A.P.; Rodrigues, A.L.S.; Peluffo, H.; Kaster, M.P. NLRP3 inflammasome-driven pathways in depression: Clinical and preclinical findings. Brain Behav. Immun. 2017, 64, 367–383. [Google Scholar] [CrossRef]
- Park, J.S.; Svetkauskaite, D.; He, Q.; Kim, J.Y.; Strassheim, D.; Ishizaka, A.; Abraham, E. Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J. Biol. Chem. 2004, 279, 7370–7377. [Google Scholar] [CrossRef]
- Kim, S.Y.; Son, M.; Lee, S.E.; Park, I.H.; Kwak, M.S.; Han, M.; Lee, H.S.; Kim, E.S.; Kim, J.-Y.; Lee, J.E.; et al. High-Mobility Group Box 1-Induced Complement Activation Causes Sterile Inflammation. Front. Immunol. 2018, 9, 705. [Google Scholar] [CrossRef]
- Alfonso-Loeches, S.; Pascual-Lucas, M.; Blanco, A.M.; Sanchez-Vera, I.; Guerri, C. Pivotal Role of TLR4 Receptors in Alcohol-Induced Neuroinflammation and Brain Damage. J. Neurosci. 2010, 30, 8285–8295. [Google Scholar] [CrossRef]
- Dantzer, R.; O’Connor, J.C.; Freund, G.G.; Johnson, R.W.; Kelley, K.W. From inflammation to sickness and depression: When the immune system subjugates the brain. Nat. Rev. Neurosci. 2008, 9, 46–56. [Google Scholar] [CrossRef]
- Maes, M.; Berk, M.; Goehler, L.; Song, C.; Anderson, G.; Gałecki, P.; Leonard, B. Depression and sickness behavior are Janus-faced responses to shared inflammatory pathways. BMC Med. 2012, 10, 66. [Google Scholar] [CrossRef]
- Kendler, K.S.; Hettema, J.M.; Butera, F.; Gardner, C.O.; Prescott, C.A. Life Event Dimensions of Loss, Humiliation, Entrapment, and Danger in the Prediction of Onsets of Major Depression and Generalized Anxiety. Arch. Gen. Psychiatry 2003, 60, 789–796. [Google Scholar] [CrossRef]
- Calcia, M.A.; Bonsall, D.R.; Bloomfield, P.S.; Selvaraj, S.; Barichello, T.; Howes, O.D. Stress and neuroinflammation: A systematic review of the effects of stress on microglia and the implications for mental illness. Psychopharmacology 2016, 233, 1637–1650. [Google Scholar] [CrossRef]
- Willner, P. The chronic mild stress (CMS) model of depression: History, evaluation and usage. Neurobiol. Stress 2017, 6, 78–93. [Google Scholar] [CrossRef]
- Rohleder, N. Stimulation of Systemic Low-Grade Inflammation by Psychosocial Stress. Psychosom. Med. 2014, 76, 181–189. [Google Scholar] [CrossRef]
- Yirmiya, R.; Rimmerman, N.; Reshef, R. Depression as a Microglial Disease. Trends Neurosci. 2015, 38, 637–658. [Google Scholar] [CrossRef] [PubMed]
- Kreisel, T.; Frank, M.G.; Licht, T.; Reshef, R.; Ben-Menachem-Zidon, O.; Baratta, M.V.; Maier, S.F.; Yirmiya, R. Dynamic microglial alterations underlie stress-induced depressive-like behavior and suppressed neurogenesis. Mol. Psychiatry 2014, 19, 699–709. [Google Scholar] [CrossRef] [PubMed]
- Beumer, W.; Gibney, S.M.; Drexhage, R.C.; Pont-Lezica, L.; Doorduin, J.; Klein, H.C.; Steiner, J.; Connor, T.J.; Harkin, A.; Versnel, M.A.; et al. The immune theory of psychiatric diseases: A key role for activated microglia and circulating monocytes. J. Leukoc. Biol. 2012, 92, 959–975. [Google Scholar] [CrossRef] [PubMed]
- Hinwood, M.; Morandini, J.; Day, T.A.; Walker, F.R. Evidence that Microglia Mediate the Neurobiological Effects of Chronic Psychological Stress on the Medial Prefrontal Cortex. Cereb. Cortex 2012, 22, 1442–1454. [Google Scholar] [CrossRef]
- Delpech, J.-C.; Madore, C.; Nadjar, A.; Joffre, C.; Wohleb, E.S.; Layé, S. Microglia in neuronal plasticity: Influence of stress. Neuropharmacology 2015, 96, 19–28. [Google Scholar] [CrossRef]
- Lehmann, M.L.; Cooper, H.A.; Maric, D.; Herkenham, M. Social defeat induces depressive-like states and microglial activation without involvement of peripheral macrophages. J. Neuroinflamm. 2016, 13, 224. [Google Scholar] [CrossRef]
- Lehmann, M.L.; Weigel, T.K.; Poffenberger, C.N.; Herkenham, M. The Behavioral Sequelae of Social Defeat Require Microglia and Are Driven by Oxidative Stress in Mice. J. Neurosci. 2019, 39, 5594–5605. [Google Scholar] [CrossRef]
- Tang, J.; Yu, W.; Chen, S.; Gao, Z.; Xiao, B. Microglia Polarization and Endoplasmic Reticulum Stress in Chronic Social Defeat Stress Induced Depression Mouse. Neurochem. Res. 2018, 43, 985–994. [Google Scholar] [CrossRef]
- McKim, D.B.; Niraula, A.; Tarr, A.J.; Wohleb, E.S.; Sheridan, J.F.; Godbout, J.P. Neuroinflammatory Dynamics Underlie Memory Impairments after Repeated Social Defeat. J. Neurosci. 2016, 36, 2590–2604. [Google Scholar] [CrossRef] [PubMed]
- Ohgidani, M.; Kato, T.A.; Sagata, N.; Hayakawa, K.; Shimokawa, N.; Sato-Kasai, M.; Kanba, S. TNF-α from hippocampal microglia induces working memory deficits by acute stress in mice. Brain Behav. Immun. 2016, 55, 17–24. [Google Scholar] [CrossRef]
- Wang, Y.-L.; Han, Q.-Q.; Gong, W.-Q.; Pan, D.-H.; Wang, L.-Z.; Hu, W.; Yang, M.; Li, B.; Yu, J.; Liu, Q. Microglial activation mediates chronic mild stress-induced depressive- and anxiety-like behavior in adult rats. J. Neuroinflamm. 2018, 15, 21. [Google Scholar] [CrossRef]
- Ramirez, K.; Sheridan, J.F. Antidepressant imipramine diminishes stress-induced inflammation in the periphery and central nervous system and related anxiety- and depressive- like behaviors. Brain Behav. Immun. 2016, 57, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zhang, Y.-P.; Li, Y.-Y.; Liu, B.-P.; Wang, H.-Y.; Li, K.-W.; Zhao, S.; Song, C. Minocycline ameliorates depressive behaviors and neuro-immune dysfunction induced by chronic unpredictable mild stress in the rat. Behav. Brain Res. 2019, 356, 348–357. [Google Scholar] [CrossRef]
- Ślusarczyk, J.; Trojan, E.; Głombik, K.; Budziszewska, B.; Kubera, M.; Lasoń, W.; Popiołek-Barczyk, K.; Mika, J.; Wędzony, K.; Basta-Kaim, A. Prenatal stress is a vulnerability factor for altered morphology and biological activity of microglia cells. Front. Cell. Neurosci. 2015, 9, 82. [Google Scholar] [CrossRef]
- Wohleb, E.S.; Hanke, M.L.; Corona, A.W.; Powell, N.D.; Stiner, L.M.; Bailey, M.T.; Nelson, R.J.; Godbout, J.P.; Sheridan, J.F. β-Adrenergic Receptor Antagonism Prevents Anxiety-Like Behavior and Microglial Reactivity Induced by Repeated Social Defeat. J. Neurosci. 2011, 31, 6277–6288. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.D.; Frank, M.G.; Tracey, K.J.; Watkins, L.R.; Maier, S.F. Stress Induces the Danger-Associated Molecular Pattern HMGB-1 in the Hippocampus of Male Sprague Dawley Rats: A Priming Stimulus of Microglia and the NLRP3 Inflammasome. J. Neurosci. 2015, 35, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.-L.; Chen, J.-G.; Wang, F. Microglia: A Central Player in Depression. Curr. Med. Sci. 2020, 40, 391–400. [Google Scholar] [CrossRef]
- Wang, H.; He, Y.; Sun, Z.; Ren, S.; Liu, M.; Wang, G.; Yang, J. Microglia in depression: An overview of microglia in the pathogenesis and treatment of depression. J. Neuroinflamm. 2022, 19, 132. [Google Scholar] [CrossRef]
- Rahimian, R.; Belliveau, C.; Chen, R.; Mechawar, N. Microglial Inflammatory-Metabolic Pathways and Their Potential Therapeutic Implication in Major Depressive Disorder. Front. Psychiatry 2022, 13, 871997. [Google Scholar] [CrossRef]
- Martin, M.; Rehani, K.; Jope, R.S.; Michalek, S.M. Toll-like receptor–mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat. Immunol. 2005, 6, 777–784. [Google Scholar] [CrossRef]
- Viatour, P.; Merville, M.-P.; Bours, V.; Chariot, A. Phosphorylation of NF-κB and IκB proteins: Implications in cancer and inflammation. Trends Biochem. Sci. 2005, 30, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Wen, A.Y.; Sakamoto, K.M.; Miller, L.S. The Role of the Transcription Factor CREB in Immune Function. J. Immunol. 2010, 185, 6413–6419. [Google Scholar] [CrossRef]
- Gill, A.J.; Kolson, D.L. Dimethyl Fumarate Modulation of Immune and Antioxidant Responses: Application to HIV Therapy. Crit. Rev. Immunol. 2013, 33, 307–359. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-W.; Lee, H.-K.; Shin, J.-H.; Lee, J.-K. Up-down Regulation of HO-1 and iNOS Gene Expressions by Ethyl Pyruvate via Recruiting p300 to Nrf2 and Depriving It from p65. Free. Radic. Biol. Med. 2013, 65, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.; Walker, A.J.; Walder, K.; Berk, M.; Marx, W.; Carvalho, A.F.; Maes, M.; Puri, B.K. Increasing Nrf2 Activity as a Treatment Approach in Neuropsychiatry. Mol. Neurobiol. 2021, 58, 2158–2182. [Google Scholar] [CrossRef]
- Zuo, C.; Cao, H.; Song, Y.; Gu, Z.; Huang, Y.; Yang, Y.; Miao, J.; Zhu, L.; Chen, J.; Jiang, Y.; et al. Nrf2: An all-rounder in depression. Redox Biol. 2022, 58, 102522. [Google Scholar] [CrossRef]
- Maes, M.; Fišar, Z.; Medina, M.; Scapagnini, G.; Nowak, G.; Berk, M. New drug targets in depression: Inflammatory, cell-mediated immune, oxidative and nitrosative stress, mitochondrial, antioxidant, and neuroprogressive pathways. And new drug candidates—Nrf2 activators and GSK-3 inhibitors. Inflammopharmacology 2012, 20, 127–150. [Google Scholar] [CrossRef]
- Rada, P.; Rojo, A.I.; Chowdhry, S.; McMahon, M.; Hayes, J.D.; Cuadrado, A. SCF/β-TrCP Promotes Glycogen Synthase Kinase 3-Dependent Degradation of the Nrf2 Transcription Factor in a Keap1-Independent Manner. Mol. Cell. Biol. 2011, 31, 1121–1133. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Kostov, R.V.; Kazantsev, A.G. The role of Nrf2 signaling in counteracting neurodegenerative diseases. FEBS J. 2018, 285, 3576–3590. [Google Scholar] [CrossRef]
- Kondoh, K.; Nishida, E. Regulation of MAP kinases by MAP kinase phosphatases. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2007, 1773, 1227–1237. [Google Scholar] [CrossRef]
- Kasuya, Y.; Umezawa, H.; Hatano, M. Stress-Activated Protein Kinases in Spinal Cord Injury: Focus on Roles of p38. Int. J. Mol. Sci. 2018, 19, 867. [Google Scholar] [CrossRef]
- Hoppstädter, J.; Ammit, A. Role of Dual-Specificity Phosphatase 1 in Glucocorticoid-Driven Anti-inflammatory Responses. Front. Immunol. 2019, 10, 1446. [Google Scholar] [CrossRef] [PubMed]
- Jope, R.S.; Cheng, Y.; Lowell, J.A.; Worthen, R.J.; Sitbon, Y.H.; Beurel, E. Stressed and Inflamed, Can GSK3 Be Blamed? Trends Biochem. Sci. 2017, 42, 180–192. [Google Scholar] [CrossRef] [PubMed]
- Beurel, E.; Michalek, S.M.; Jope, R.S. Innate and adaptive immune responses regulated by glycogen synthase kinase-3 (GSK3). Trends Immunol. 2010, 31, 24–31. [Google Scholar] [CrossRef]
- Engel, T.; Hernández, F.; Avila, J.; Lucas, J.J. Full Reversal of Alzheimer’s Disease-Like Phenotype in a Mouse Model with Conditional Overexpression of Glycogen Synthase Kinase-3. J. Neurosci. 2006, 26, 5083–5090. [Google Scholar] [CrossRef]
- Guo, H.; Callaway, J.B.; Ting, J.P.-Y. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef]
- Qin, L.; He, J.; Hanes, R.N.; Pluzarev, O.; Hong, J.-S.; Crews, F.T. Increased systemic and brain cytokine production and neuroinflammation by endotoxin following ethanol treatment. J. Neuroinflamm. 2008, 5, 10. [Google Scholar] [CrossRef]
- Forlenza, O.V.; Spink, J.M.; Dayanandan, R.; Anderton, B.H.; Olesen, O.F.; Lovestone, S. Muscarinic agonists reduce tau phosphorylation in non-neuronal cells via GSK-3β inhibition and in neurons. J. Neural Transm. 2000, 107, 1201–1212. [Google Scholar] [CrossRef]
- Pearce, L.R.; Komander, D.; Alessi, D.R. The nuts and bolts of AGC protein kinases. Nat. Rev. Mol. Cell Biol. 2010, 11, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Dwivedi, Y.; Pandey, G.N. Adenylyl cyclase-cyclicAMP signaling in mood disorders: Role of the crucial phosphorylating enzyme protein kinase A. Neuropsychiatr. Dis. Treat. 2008, 4, 161–176. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Richards, E.M.; Niciu, M.; Ionescu, D.F.; Zoghbi, S.S.; Hong, J.; Telu, S.; Hines, C.S.; Pike, V.W.; Zarate, C.A.; et al. cAMP signaling in brain is decreased in unmedicated depressed patients and increased by treatment with a selective serotonin reuptake inhibitor. Mol. Psychiatry 2017, 22, 754–759. [Google Scholar] [CrossRef] [PubMed]
- Schappi, J.M.; Rasenick, M.M. Gαs, adenylyl cyclase, and their relationship to the diagnosis and treatment of depression. Front. Pharmacol. 2022, 13, 1012778. [Google Scholar] [CrossRef]
- Karege, F.; Perroud, N.; Burkhardt, S.; Fernandez, R.; Ballmann, E.; La Harpe, R.; Malafosse, A. Protein levels of β-catenin and activation state of glycogen synthase kinase-3β in major depression. A study with postmortem prefrontal cortex. J. Affect. Disord. 2012, 136, 185–188. [Google Scholar] [CrossRef]
- Bansal, Y.; Singh, R.; Parhar, I.; Kuhad, A.; Soga, T. Quinolinic Acid and Nuclear Factor Erythroid 2-Related Factor 2 in Depression: Role in Neuroprogression. Front. Pharmacol. 2019, 10, 452. [Google Scholar] [CrossRef] [PubMed]
- Forlenza, M.J.; Miller, G.E. Increased Serum Levels of 8-Hydroxy-2′-Deoxyguanosine in Clinical Depression. Psychosom. Med. 2006, 68, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Savage, K.; Gogarty, L.; Lea, A.; Deleuil, S.; Nolidin, K.; Croft, K.; Stough, C. The Relationship between F2-Isoprostanes Plasma Levels and Depression Symptoms in Healthy Older Adults. Antioxidants 2022, 11, 822. [Google Scholar] [CrossRef]
- Yan, Y.; Jiang, W.; Liu, L.; Wang, X.; Ding, C.; Tian, Z.; Zhou, R. Dopamine Controls Systemic Inflammation through Inhibition of NLRP3 Inflammasome. Cell 2015, 160, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Chen, T.; Li, G.; Jia, Y.; Wang, J.; Xue, L.; Chen, Y. Dopamine Alters Lipopolysaccharide-Induced Nitric Oxide Production in Microglial Cells via Activation of D1-Like Receptors. Neurochem. Res. 2019, 44, 947–958. [Google Scholar] [CrossRef]
- Perreault, M.L.; Jones-Tabah, J.; O’Dowd, B.F.; George, S.R. A physiological role for the dopamine D5 receptor as a regulator of BDNF and Akt signalling in rodent prefrontal cortex. Int. J. Neuropsychopharmacol. 2013, 16, 477–483. [Google Scholar] [CrossRef]
- Fujita, H.; Tanaka, J.; Maeda, N.; Sakanaka, M. Adrenergic agonists suppress the proliferation of microglia through β2-adrenergic receptor. Neurosci. Lett. 1998, 242, 37–40. [Google Scholar] [CrossRef]
- Russo, C.D.; Boullerne, A.I.; Gavrilyuk, V.; Feinstein, D.L. Inhibition of microglial inflammatory responses by norepinephrine: Effects on nitric oxide and interleukin-1β production. J. Neuroinflamm. 2004, 1, 9. [Google Scholar] [CrossRef] [PubMed]
- Mahé, C.; Loetscher, E.; Dev, K.K.; Bobirnac, I.; Otten, U.; Schoeffter, P. Serotonin 5-HT7 receptors coupled to induction of interleukin-6 in human microglial MC-3 cells. Neuropharmacology 2005, 49, 40–47. [Google Scholar] [CrossRef]
- Nakahashi, T.; Fujimura, H.; Altar, C.; Li, J.; Kambayashi, J.-I.; Tandon, N.N.; Sun, B. Vascular endothelial cells synthesize and secrete brain-derived neurotrophic factor. FEBS Lett. 2000, 470, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.; Xu, Y.; Pearse, D.D. Cyclic AMP is a key regulator of M1 to M2a phenotypic conversion of microglia in the presence of Th2 cytokines. J. Neuroinflamm. 2016, 13, 9. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, M.; Nishida, A.; Zensho, H.; Yamawaki, S. Chronic antidepressant exposure enhances 5-hydroxytryptamine7 receptor-mediated cyclic adenosine monophosphate accumulation in rat frontocortical astrocytes. J. Pharmacol. Exp. Ther. 1996, 279, 1551–1558. [Google Scholar]
- Tynan, R.J.; Weidenhofer, J.; Hinwood, M.; Cairns, M.J.; Day, T.A.; Walker, F.R. A comparative examination of the anti-inflammatory effects of SSRI and SNRI antidepressants on LPS stimulated microglia. Brain Behav. Immun. 2012, 26, 469–479. [Google Scholar] [CrossRef]
- Hashioka, S.; Klegeris, A.; Monji, A.; Kato, T.; Sawada, M.; McGeer, P.L.; Kanba, S. Antidepressants inhibit interferon-γ-induced microglial production of IL-6 and nitric oxide. Exp. Neurol. 2007, 206, 33–42. [Google Scholar] [CrossRef]
- Bielecka, A.M.; Paul-Samojedny, M.; Obuchowicz, E. Moclobemide exerts anti-inflammatory effect in lipopolysaccharide-activated primary mixed glial cell culture. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2010, 382, 409–417. [Google Scholar] [CrossRef]
- Wang, L.; Wang, R.; Liu, L.; Qiao, D.; Baldwin, D.S.; Hou, R. Effects of SSRIs on peripheral inflammatory markers in patients with major depressive disorder: A systematic review and meta-analysis. Brain Behav. Immun. 2019, 79, 24–38. [Google Scholar] [CrossRef]
- Dionisie, V.; Filip, G.A.; Manea, M.C.; Manea, M.; Riga, S. The anti-inflammatory role of SSRI and SNRI in the treatment of depression: A review of human and rodent research studies. Inflammopharmacology 2021, 29, 75–90. [Google Scholar] [CrossRef]
- Krabbe, G.; Matyash, V.; Pannasch, U.; Mamer, L.; Boddeke, H.W.; Kettenmann, H. Activation of serotonin receptors promotes microglial injury-induced motility but attenuates phagocytic activity. Brain Behav. Immun. 2012, 26, 419–428. [Google Scholar] [CrossRef] [PubMed]
- de las Casas-Engel, M.; Domínguez-Soto, A.; Sierra-Filardi, E.; Bragado, R.; Nieto, C.; Puig-Kroger, A.; Samaniego, R.; Loza, M.; Corcuera, M.T.; Gómez-Aguado, F.; et al. Serotonin Skews Human Macrophage Polarization through HTR2B and HTR7. J. Immunol. 2013, 190, 2301–2310. [Google Scholar] [CrossRef]
- Kettenmann, H.; Hanisch, U.-K.; Noda, M.; Verkhratsky, A. Physiology of Microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef] [PubMed]
- Harry, G.J. Microglia during development and aging. Pharmacol. Ther. 2013, 139, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Rush, A.J.; Sackeim, H.A.; Marangell, L.B.; George, M.S.; Brannan, S.K.; Davis, S.M.; Lavori, P.; Howland, R.; Kling, M.A.; Rittberg, B.; et al. Effects of 12 Months of Vagus Nerve Stimulation in Treatment-Resistant Depression: A Naturalistic Study. Biol. Psychiatry 2005, 58, 355–363. [Google Scholar] [CrossRef]
- Bajbouj, M.; Merkl, A.; Schlaepfer, T.; Frick, C.; Zobel, A.; Maier, W.; O’Keane, V.; Corcoran, C.; Adolfsson, R.; Trimble, M.; et al. Two-Year Outcome of Vagus Nerve Stimulation in Treatment-Resistant Depression. J. Clin. Psychopharmacol. 2010, 30, 273–281. [Google Scholar] [CrossRef]
- Howland, R.H. Vagus Nerve Stimulation. Curr. Behav. Neurosci. Rep. 2014, 1, 64–73. [Google Scholar] [CrossRef]
- Bonaz, B.; Sinniger, V.; Hoffmann, D.; Clarençon, D.; Mathieu, N.; Dantzer, C.; Vercueil, L.; Picq, C.; Trocmé, C.; Faure, P.; et al. Chronic vagus nerve stimulation in Crohn’s disease: A 6-month follow-up pilot study. Neurogastroenterol. Motil. 2016, 28, 948–953. [Google Scholar] [CrossRef]
- Borovikova, L.V.; Ivanova, S.; Zhang, M.; Yang, H.; Botchkina, G.I.; Watkins, L.R.; Wang, H.; Abumrad, N.; Eaton, J.W.; Tracey, K.J. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 2000, 405, 458–462. [Google Scholar] [CrossRef] [PubMed]
- De Jonge, W.J.; Van Der Zanden, E.P.; The, F.O.; Bijlsma, M.F.; Van Westerloo, D.J.; Bennink, R.J.; Berthoud, H.-R.; Uematsu, S.; Akira, S.; van den Wijngaard, R.M.; et al. Stimulation of the vagus nerve attenuates macrophage activation by activating the Jak2-STAT3 signaling pathway. Nat. Immunol. 2005, 6, 844–851. [Google Scholar] [CrossRef]
- Bencherif, M.; Lippiello, P.M.; Lucas, R.; Marrero, M.B. Alpha7 nicotinic receptors as novel therapeutic targets for inflammation-based diseases. Cell. Mol. Life Sci. 2011, 68, 931–949. [Google Scholar] [CrossRef] [PubMed]
- Rehani, K.; Scott, D.A.; Renaud, D.; Hamza, H.; Williams, L.R.; Wang, H.; Martin, M. Cotinine-induced convergence of the cholinergic and PI3 kinase-dependent anti-inflammatory pathways in innate immune cells. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2008, 1783, 375–382. [Google Scholar] [CrossRef]
- Bitner, R.S.; Nikkel, A.L.; Markosyan, S.; Otte, S.; Puttfarcken, P.; Gopalakrishnan, M. Selective α7 nicotinic acetylcholine receptor activation regulates glycogen synthase kinase3β and decreases tau phosphorylation in vivo. Brain Res. 2009, 1265, 65–74. [Google Scholar] [CrossRef]
- Echeverria, V.; Zeitlin, R.; Burgess, S.; Patel, S.; Barman, A.; Thakur, G.; Mamcarz, M.; Wang, L.; Sattelle, D.B.; Kirschner, D.A.; et al. Cotinine Reduces Amyloid-β Aggregation and Improves Memory in Alzheimer’s Disease Mice. J. Alzheimer’s Dis. 2011, 24, 817–835. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, D.; Lee, C.-H.L.; Flood, D.; Marger, F.; Donnelly-Roberts, D. Therapeutic Potential of α7 Nicotinic Acetylcholine Receptors. Pharmacol. Rev. 2015, 67, 1025–1073. [Google Scholar] [CrossRef] [PubMed]
- Egea, J.; Buendia, I.; Parada, E.; Navarro, E.; León, R.; Lopez, M.G. Anti-inflammatory role of microglial alpha7 nAChRs and its role in neuroprotection. Biochem. Pharmacol. 2015, 97, 463–472. [Google Scholar] [CrossRef]
- Parada, E.; Egea, J.; Buendia, I.; Negredo, P.; Cunha, A.C.; Cardoso, S.; Soares, M.P.; López, M.G. The Microglial α7-Acetylcholine Nicotinic Receptor Is a Key Element in Promoting Neuroprotection by Inducing Heme Oxygenase-1 via Nuclear Factor Erythroid-2-Related Factor 2. Antioxid. Redox Signal. 2013, 19, 1135–1148. [Google Scholar] [CrossRef]
- Navarro, E.; Gonzalez-Lafuente, L.; Pérez-Liébana, I.; Buendia, I.; López-Bernardo, E.; Sánchez-Ramos, C.; Prieto, I.; Cuadrado, A.; Satrustegui, J.; Cadenas, S.; et al. Heme-Oxygenase I and PCG-1α Regulate Mitochondrial Biogenesis via Microglial Activation of Alpha7 Nicotinic Acetylcholine Receptors Using PNU282987. Antioxidants Redox Signal. 2017, 27, 93–105. [Google Scholar] [CrossRef]
- Zhao, D.; Xu, X.; Pan, L.; Zhu, W.; Fu, X.; Guo, L.; Lu, Q.; Wang, J. Pharmacologic activation of cholinergic alpha7 nicotinic receptors mitigates depressive-like behavior in a mouse model of chronic stress. J. Neuroinflamm. 2017, 14, 234. [Google Scholar] [CrossRef] [PubMed]
- Kerschensteiner, M.; Gallmeier, E.; Behrens, L.; Leal, V.V.; Misgeld, T.; Klinkert, W.E.F.; Kolbeck, R.; Hoppe, E.; Oropeza-Wekerle, R.-L.; Bartke, I.; et al. Activated Human T Cells, B Cells, and Monocytes Produce Brain-derived Neurotrophic Factor In Vitro and in Inflammatory Brain Lesions: A Neuroprotective Role of Inflammation? J. Exp. Med. 1999, 189, 865–870. [Google Scholar] [CrossRef]
- Yasuda, S.; Liang, M.-H.; Marinova, Z.I.; Yahyavi, A.; Chuang, D.-M. The mood stabilizers lithium and valproate selectively activate the promoter IV of brain-derived neurotrophic factor in neurons. Mol. Psychiatry 2009, 14, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Ghosal, S.; Bang, E.; Yue, W.; Hare, B.; Lepack, A.E.; Girgenti, M.J.; Duman, R.S. Activity-Dependent Brain-Derived Neurotrophic Factor Release Is Required for the Rapid Antidepressant Actions of Scopolamine. Biol. Psychiatry 2018, 83, 29–37. [Google Scholar] [CrossRef]
- Wiese, S.; Karus, M.; Faissner, A. Astrocytes as a Source for Extracellular Matrix Molecules and Cytokines. Front. Pharmacol. 2012, 3, 120. [Google Scholar] [CrossRef] [PubMed]
- Wray, N.H.; Schappi, J.M.; Singh, H.; Senese, N.B.; Rasenick, M.M. NMDAR-independent, cAMP-dependent antidepressant actions of ketamine. Mol. Psychiatry 2019, 24, 1833–1843. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.-S.; Peng, G.-S.; Li, G.; Yang, S.; Wu, X.; Wang, C.-C.; Wilson, B.A.; Lu, R.-B.; Gean, P.-W.; Chuang, D.-M.; et al. Valproate protects dopaminergic neurons in midbrain neuron/glia cultures by stimulating the release of neurotrophic factors from astrocytes. Mol. Psychiatry 2006, 11, 1116–1125. [Google Scholar] [CrossRef]
- Cherry, J.D.; Olschowka, J.A.; O’Banion, M.K. Neuroinflammation and M2 microglia: The good, the bad, and the inflamed. J. Neuroinflamm. 2014, 11, 98. [Google Scholar] [CrossRef]
- Nakajima, K.; Kikuchi, Y.; Ikoma, E.; Honda, S.; Ishikawa, M.; Liu, Y.; Kohsaka, S. Neurotrophins regulate the function of cultured microglia. Glia 1998, 24, 272–289. [Google Scholar] [CrossRef]
- Zhang, J.; Geula, C.; Lu, C.; Koziel, H.; Hatcher, L.M.; Roisen, F.J. Neurotrophins regulate proliferation and survival of two microglial cell lines in vitro. Exp. Neurol. 2003, 183, 469–481. [Google Scholar] [CrossRef]
- Parkhurst, C.N.; Yang, G.; Ninan, I.; Savas, J.N.; Yates, J.R., 3rd; Lafaille, J.J.; Hempstead, B.L.; Littman, D.R.; Gan, W.-B. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 2013, 155, 1596–1609. [Google Scholar] [CrossRef]
- Gomes, C.; Ferreira, R.; George, J.; Sanches, R.; Rodrigues, D.I.; Gonçalves, N.; A Cunha, R. Activation of microglial cells triggers a release of brain-derived neurotrophic factor (BDNF) inducing their proliferation in an adenosine A2A receptor-dependent manner: A2A receptor blockade prevents BDNF release and proliferation of microglia. J. Neuroinflamm. 2013, 10, 16. [Google Scholar] [CrossRef]
- Jiang, Y.; Wei, N.; Zhu, J.; Lu, T.; Chen, Z.; Xu, G.; Liu, X. Effects of Brain-Derived Neurotrophic Factor on Local Inflammation in Experimental Stroke of Rat. Mediat. Inflamm. 2010, 2010, 372423. [Google Scholar] [CrossRef]
- Porcher, C.; Medina, I.; Gaiarsa, J.-L. Mechanism of BDNF Modulation in GABAergic Synaptic Transmission in Healthy and Disease Brains. Front. Cell. Neurosci. 2018, 12, 273. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Frye, M.A.; Shelton, R.C. Review of Pharmacological Treatment in Mood Disorders and Future Directions for Drug Development. Neuropsychopharmacology 2012, 37, 77–101. [Google Scholar] [CrossRef] [PubMed]
- Tao, X.; Finkbeiner, S.; Arnold, D.B.; Shaywitz, A.J.; Greenberg, M.E. Ca2+ Influx Regulates BDNF Transcription by a CREB Family Transcription Factor-Dependent Mechanism. Neuron 1998, 20, 709–726. [Google Scholar] [CrossRef]
- Krishnan, V.; Nestler, E.J. The molecular neurobiology of depression. Nature 2008, 455, 894–902. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.-H.; Kim, Y.-K. BDNF mRNA expression of peripheral blood mononuclear cells was decreased in depressive patients who had or had not recently attempted suicide. J. Affect. Disord. 2010, 125, 369–373. [Google Scholar] [CrossRef]
- McEwen, B.S.; Chattarji, S.; Diamond, D.M.; Jay, T.M.; Reagan, L.P.; Svenningsson, P.; Fuchs, E. The neurobiological properties of tianeptine (Stablon): From monoamine hypothesis to glutamatergic modulation. Mol. Psychiatry 2010, 15, 237–249. [Google Scholar] [CrossRef]
- Schmidt, H.D.; Shelton, R.C.; Duman, R.S. Functional Biomarkers of Depression: Diagnosis, Treatment, and Pathophysiology. Neuropsychopharmacology 2011, 36, 2375–2394. [Google Scholar] [CrossRef]
- Björkholm, C.; Monteggia, L.M. BDNF—A Key Transducer of Antidepressant Effects. Neuropharmacology 2016, 102, 72–79. [Google Scholar] [CrossRef]
- Hait, N.C.; Wise, L.; Allegood, J.C.; O’Brien, M.; Avni, D.; Reeves, T.M.; Knapp, P.; Lu, J.; Luo, C.; Miles, M.F.; et al. Active, phosphorylated fingolimod inhibits histone deacetylases and facilitates fear extinction memory. Nat. Neurosci. 2014, 17, 971–980. [Google Scholar] [CrossRef]
- di Nuzzo, L.; Orlando, R.; Tognoli, C.; Di Pietro, P.; Bertini, G.; Miele, J.; Bucci, D.; Motolese, M.; Scaccianoce, S.; Caruso, A.; et al. Antidepressant activity of fingolimod in mice. Pharmacol. Res. Perspect. 2015, 3, e00135. [Google Scholar] [CrossRef] [PubMed]
- Covington, H.E., 3rd; Vialou, V.F.; LaPlant, Q.; Ohnishi, Y.N.; Nestler, E.J. Hippocampal-dependent antidepressant-like activity of histone deacetylase inhibition. Neurosci. Lett. 2011, 493, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Lima, I.V.D.A.; Almeida-Santos, A.F.; Ferreira-Vieira, T.H.; Aguiar, D.C.; Ribeiro, F.M.; Campos, A.C.; de Oliveira, A.C.P. Antidepressant-like effect of valproic acid—Possible involvement of PI3K/Akt/mTOR pathway. Behav. Brain Res. 2017, 329, 166–171. [Google Scholar] [CrossRef] [PubMed]
- Boorman, E.; Zajkowska, Z.; Ahmed, R.; Pariante, C.M.; Zunszain, P.A. Crosstalk between endocannabinoid and immune systems: A potential dysregulation in depression? Psychopharmacology 2016, 233, 1591–1604. [Google Scholar] [CrossRef]
- Biringer, R.G. Endocannabinoid signaling pathways: Beyond CB1R and CB2R. J. Cell Commun. Signal. 2021, 15, 335–360. [Google Scholar] [CrossRef] [PubMed]
- Núñez, E.; Benito, C.; Pazos, M.R.; Barbachano, A.; Fajardo, O.; González, S.; Tolón, R.M.; Romero, J. Cannabinoid CB2 receptors are expressed by perivascular microglial cells in the human brain: An immunohistochemical study. Synapse 2004, 53, 208–213. [Google Scholar] [CrossRef] [PubMed]
- Turcotte, C.; Blanchet, M.-R.; LaViolette, M.; Flamand, N. The CB2 receptor and its role as a regulator of inflammation. Cell. Mol. Life Sci. 2016, 73, 4449–4470. [Google Scholar] [CrossRef]
- Benyó, Z.; Ruisanchez, É.; Leszl-Ishiguro, M.; Sándor, P.; Pacher, P. Endocannabinoids in cerebrovascular regulation. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H785–H801. [Google Scholar] [CrossRef] [PubMed]
- Roche, M.; Finn, D.P. Brain CB2 Receptors: Implications for Neuropsychiatric Disorders. Pharmaceuticals 2010, 3, 2517–2553. [Google Scholar] [CrossRef]
- Chiang, N.; de la Rosa, X.; Libreros, S.; Serhan, C.N. Novel Resolvin D2 Receptor Axis in Infectious Inflammation. J. Immunol. 2017, 198, 842–851. [Google Scholar] [CrossRef]
- Chiang, N.; Dalli, J.; Colas, R.A.; Serhan, C.N. Identification of resolvin D2 receptor mediating resolution of infections and organ protection. J. Exp. Med. 2015, 212, 1203–1217. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Li, L.; Jiang, B.; Feng, Z.; Yang, L.; Tang, J.; Chen, Q.; Zhang, J.; Tan, Q.; Feng, H.; et al. Cannabinoid receptor-2 stimulation suppresses neuroinflammation by regulating microglial M1/M2 polarization through the cAMP/PKA pathway in an experimental GMH rat model. Brain Behav. Immun. 2016, 58, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Bie, B.; Wu, J.; Foss, J.F.; Naguib, M. An overview of the cannabinoid type 2 receptor system and its therapeutic potential. Curr. Opin. Anaesthesiol. 2018, 31, 407–414. [Google Scholar] [CrossRef]
- Gutiérrez, G.; Pérez-Ortiz, J.; Gutiérrez-Adán, A.; Manzanares, J. Depression-resistant endophenotype in mice overexpressing cannabinoid CB 2 receptors. Br. J. Pharmacol. 2010, 160, 1773–1784. [Google Scholar] [CrossRef]
- Meijerink, J.; Balvers, M.; Witkamp, R. N-acyl amines of docosahexaenoic acid and other n-3 polyunsatured fatty acids—From fishy endocannabinoids to potential leads. Br. J. Pharmacol. 2013, 169, 772–783. [Google Scholar] [CrossRef] [PubMed]
- Park, T.; Chen, H.; Kevala, K.; Lee, J.-W.; Kim, H.-Y. N-Docosahexaenoylethanolamine ameliorates LPS-induced neuroinflammation via cAMP/PKA-dependent signaling. J. Neuroinflamm. 2016, 13, 284. [Google Scholar] [CrossRef]
- Park, T.; Chen, H.; Kim, H.-Y. GPR110 (ADGRF1) mediates anti-inflammatory effects of N-docosahexaenoylethanolamine. J. Neuroinflamm. 2019, 16, 225. [Google Scholar] [CrossRef]
- Tyrtyshnaia, A.; Konovalova, S.; Bondar, A.; Ermolenko, E.; Sultanov, R.; Manzhulo, I. Anti-Inflammatory Activity of N-Docosahexaenoylethanolamine and N-Eicosapentaenoylethanolamine in a Mouse Model of Lipopolysaccharide-Induced Neuroinflammation. Int. J. Mol. Sci. 2021, 22, 10728. [Google Scholar] [CrossRef]
- Lee, J.-W.; Huang, B.X.; Kwon, H.; Rashid, A.; Kharebava, G.; Desai, A.; Patnaik, S.; Marugan, J.; Kim, H.-Y. Orphan GPR110 (ADGRF1) targeted by N-docosahexaenoylethanolamine in development of neurons and cognitive function. Nat. Commun. 2016, 7, 13123. [Google Scholar] [CrossRef]
- McDougle, D.R.; Watson, J.E.; Abdeen, A.A.; Adili, R.; Caputo, M.P.; Krapf, J.E.; Johnson, R.W.; Kilian, K.A.; Holinstat, M.; Das, A. Anti-inflammatory ω-3 endocannabinoid epoxides. Proc. Natl. Acad. Sci. USA 2017, 114, E6034–E6043. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Jia, J.; Liu, X.; Bai, F.; Wang, Q.; Xiong, L. Activation of murine microglial N9 cells is attenuated through cannabinoid receptor CB2 signaling. Biochem. Biophys. Res. Commun. 2015, 458, 92–97. [Google Scholar] [CrossRef]
- Yang, B.; Lin, L.; Bazinet, R.P.; Chien, Y.-C.; Chang, J.P.-C.; Satyanarayanan, S.K.; Su, H.; Su, K.-P. Clinical Efficacy and Biological Regulations of ω–3 PUFA-Derived Endocannabinoids in Major Depressive Disorder. Psychother. Psychosom. 2019, 88, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Ninković, J.; Roy, S. Role of the mu-opioid receptor in opioid modulation of immune function. Amino Acids 2013, 45, 9–24. [Google Scholar] [CrossRef]
- Lee, J.W.; Joshi, S.; Chan, J.S.; Wong, Y.H. Differential coupling of mu-, delta-, and kappa-opioid receptors to G alpha16-mediated stimulation of phospholipase C. J. Neurochem. 1998, 70, 2203–2211. [Google Scholar] [CrossRef]
- Ślusarczyk, J.; Trojan, E.; Głombik, K.; Piotrowska, A.; Budziszewska, B.; Kubera, M.; Popiołek-Barczyk, K.; Lasoń, W.; Mika, J.; Basta-Kaim, A. Targeting the NLRP3 Inflammasome-Related Pathways via Tianeptine Treatment-Suppressed Microglia Polarization to the M1 Phenotype in Lipopolysaccharide-Stimulated Cultures. Int. J. Mol. Sci. 2018, 19, 1965. [Google Scholar] [CrossRef] [PubMed]
- Emrich, H.M.; Vogt, P.; Herz, A. Possible Antidepressive Effects of Opioids: Action of Buprenorphine. Ann. N. Y. Acad. Sci. 1982, 398, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Kasper, S.; McEwen, B.S. Neurobiological and Clinical Effects of the Antidepressant Tianeptine. CNS Drugs 2008, 22, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Peciña, M.; Karp, J.F.; Mathew, S.; Todtenkopf, M.S.; Ehrich, E.W.; Zubieta, J.-K. Endogenous opioid system dysregulation in depression: Implications for new therapeutic approaches. Mol. Psychiatry 2019, 24, 576–587. [Google Scholar] [CrossRef]
- Loric, S.; Maroteaux, L.; Kellermann, O.; Launay, J.M. Functional serotonin-2B receptors are expressed by a teratocarcinoma-derived cell line during serotoninergic differentiation. Mol. Pharmacol. 1995, 47, 458–466. [Google Scholar]
- Cox, D.A.; Cohen, M.L. 5-HT2B receptor signaling in the rat stomach fundus: Dependence on calcium influx, calcium release and protein kinase C. Behav. Brain Res. 1996, 73, 289–292. [Google Scholar] [CrossRef] [PubMed]
- Naito, K.; Tanaka, C.; Mitsuhashi, M.; Moteki, H.; Kimura, M.; Natsume, H.; Ogihara, M. Signal Transduction Mechanism for Serotonin 5-HT2B Receptor-Mediated DNA Synthesis and Proliferation in Primary Cultures of Adult Rat Hepatocytes. Biol. Pharm. Bull. 2016, 39, 121–129. [Google Scholar] [CrossRef]
- Carhart-Harris, R.; Giribaldi, B.; Watts, R.; Baker-Jones, M.; Murphy-Beiner, A.; Murphy, R.; Martell, J.; Blemings, A.; Erritzoe, D.; Nutt, D.J. Trial of Psilocybin versus Escitalopram for Depression. N. Engl. J. Med. 2021, 384, 1402–1411. [Google Scholar] [CrossRef] [PubMed]
- Carhart-Harris, R.L.; Bolstridge, M.; Rucker, J.; Day, C.M.J.; Erritzoe, D.; Kaelen, M.; Bloomfield, M.; Rickard, J.A.; Forbes, B.; Feilding, A.; et al. Psilocybin with psychological support for treatment-resistant depression: An open-label feasibility study. Lancet Psychiatry 2016, 3, 619–627. [Google Scholar] [CrossRef] [PubMed]
- Gukasyan, N.; Davis, A.K.; Barrett, F.S.; Cosimano, M.P.; Sepeda, N.D.; Johnson, M.W.; Griffiths, R.R. Efficacy and safety of psilocybin-assisted treatment for major depressive disorder: Prospective 12-month follow-up. J. Psychopharmacol. 2022, 36, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Yovell, Y.; Bar, G.; Mashiah, M.; Baruch, Y.; Briskman, I.; Asherov, J.; Lotan, A.; Rigbi, A.; Panksepp, J. Ultra-Low-Dose Buprenorphine as a Time-Limited Treatment for Severe Suicidal Ideation: A Randomized Controlled Trial. Am. J. Psychiatry 2016, 173, 491–498. [Google Scholar] [CrossRef]
- Samuels, B.A.; Nautiyal, K.M.; Kruegel, A.C.; Levinstein, M.R.; Magalong, V.M.; Gassaway, M.M.; Grinnell, S.G.; Han, J.; Ansonoff, M.A.; E Pintar, J.; et al. The Behavioral Effects of the Antidepressant Tianeptine Require the Mu-Opioid Receptor. Neuropsychopharmacology 2017, 42, 2052–2063. [Google Scholar] [CrossRef]
- Serafini, G.; Adavastro, G.; Canepa, G.; De Berardis, D.; Valchera, A.; Pompili, M.; Nasrallah, H.; Amore, M. The Efficacy of Buprenorphine in Major Depression, Treatment-Resistant Depression and Suicidal Behavior: A Systematic Review. Int. J. Mol. Sci. 2018, 19, 2410. [Google Scholar] [CrossRef]
- Nelson, C.J.; Schneider, G.M.; Lysle, D.T. Involvement of Central μ- but Not δ- or κ-Opioid Receptors in Immunomodulation. Brain Behav. Immun. 2000, 14, 170–184. [Google Scholar] [CrossRef]
- Nummenmaa, L.; Karjalainen, T.; Isojärvi, J.; Kantonen, T.; Tuisku, J.; Kaasinen, V.; Joutsa, J.; Nuutila, P.; Kalliokoski, K.; Hirvonen, J.; et al. Lowered endogenous mu-opioid receptor availability in subclinical depression and anxiety. Neuropsychopharmacology 2020, 45, 1953–1959. [Google Scholar] [CrossRef]
- Gassaway, M.M.; Rives, M.-L.; Kruegel, A.C.; Javitch, J.A.; Sames, D. The atypical antidepressant and neurorestorative agent tianeptine is a μ-opioid receptor agonist. Transl. Psychiatry 2014, 4, e411. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Loram, L.C.; Ramos, K.; de Jesus, A.J.; Thomas, J.; Cheng, K.; Reddy, A.; Somogyi, A.A.; Hutchinson, M.R.; Watkins, L.R.; et al. Morphine activates neuroinflammation in a manner parallel to endotoxin. Proc. Natl. Acad. Sci. USA 2012, 109, 6325–6330. [Google Scholar] [CrossRef] [PubMed]
- Lutz, P.-E.; Kieffer, B.L. Opioid receptors: Distinct roles in mood disorders. Trends Neurosci. 2013, 36, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Halberstadt, A.L.; Geyer, M.A. Multiple receptors contribute to the behavioral effects of indoleamine hallucinogens. Neuropharmacology 2011, 61, 364–381. [Google Scholar] [CrossRef]
- Ling, S.; Ceban, F.; Lui, L.M.W.; Lee, Y.; Teopiz, K.M.; Rodrigues, N.B.; Lipsitz, O.; Gill, H.; Subramaniapillai, M.; Mansur, R.B.; et al. Molecular Mechanisms of Psilocybin and Implications for the Treatment of Depression. CNS Drugs 2022, 36, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Desouza, L.A.; Benekareddy, M.; Fanibunda, S.E.; Mohammad, F.; Janakiraman, B.; Ghai, U.; Gur, T.; Blendy, J.A.; Vaidya, V.A. The Hallucinogenic Serotonin2A Receptor Agonist, 2,5-Dimethoxy-4-Iodoamphetamine, Promotes cAMP Response Element Binding Protein-Dependent Gene Expression of Specific Plasticity-Associated Genes in the Rodent Neocortex. Front. Mol. Neurosci. 2021, 14, 790213. [Google Scholar] [CrossRef] [PubMed]
- Glennon, R.A.; Rosecrans, J.A. Speculations on the mechanism of action of hallucinogenic indolealkylamines. Neurosci. Biobehav. Rev. 1981, 5, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Szabo, A.; Gogolak, P.; Koncz, G.; Foldvari, Z.; Pazmandi, K.; Miltner, N.; Poliska, S.; Bacsi, A.; Djurovic, S.; Rajnavolgyi, E. Immunomodulatory capacity of the serotonin receptor 5-HT2B in a subset of human dendritic cells. Sci. Rep. 2018, 8, 1765. [Google Scholar] [CrossRef]
- Bierhaus, A.; Wolf, J.; Andrassy, M.; Rohleder, N.; Humpert, P.M.; Petrov, D.; Ferstl, R.; von Eynatten, M.; Wendt, T.; Rudofsky, G.; et al. A mechanism converting psychosocial stress into mononuclear cell activation. Proc. Natl. Acad. Sci. USA 2003, 100, 1920–1925. [Google Scholar] [CrossRef]
- Bailey, M.T.; Engler, H.; Powell, N.D.; Padgett, D.A.; Sheridan, J.F. Repeated social defeat increases the bactericidal activity of splenic macrophages through a Toll-like receptor-dependent pathway. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 293, R1180–R1190. [Google Scholar] [CrossRef]
- Diz-Chaves, Y.; Pernía, O.; Carrero, P.; Garcia-Segura, L.M. Prenatal stress causes alterations in the morphology of microglia and the inflammatory response of the hippocampus of adult female mice. J. Neuroinflamm. 2012, 9, 71. [Google Scholar] [CrossRef]
- Lindqvist, D.; Dhabhar, F.S.; James, S.J.; Hough, C.M.; Jain, F.A.; Bersani, F.S.; Reus, V.I.; Verhoeven, J.E.; Epel, E.S.; Mahan, L.; et al. Oxidative stress, inflammation and treatment response in major depression. Psychoneuroendocrinology 2017, 76, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Vilhardt, F.; Haslund-Vinding, J.; Jaquet, V.; McBean, G. Microglia antioxidant systems and redox signalling. Br. J. Pharmacol. 2017, 174, 1719–1732. [Google Scholar] [CrossRef]
- Bellezza, I.; Giambanco, I.; Minelli, A.; Donato, R. Nrf2-Keap1 signaling in oxidative and reductive stress. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2018, 1865, 721–733. [Google Scholar] [CrossRef] [PubMed]
- Martín-Hernández, D.; Caso, J.R.; Meana, J.J.; Callado, L.F.; Madrigal, J.L.M.; García-Bueno, B.; Leza, J.C. Intracellular inflammatory and antioxidant pathways in postmortem frontal cortex of subjects with major depression: Effect of antidepressants. J. Neuroinflamm. 2018, 15, 251. [Google Scholar] [CrossRef] [PubMed]
- Nadjar, A.; Leyrolle, Q.; Joffre, C.; Laye, S. Bioactive lipids as new class of microglial modulators: When nutrition meets neuroimunology. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2017, 79, 19–26. [Google Scholar] [CrossRef]
- Hsiao, C.-C.; Sankowski, R.; Prinz, M.; Smolders, J.; Huitinga, I.; Hamann, J. GPCRomics of Homeostatic and Disease-Associated Human Microglia. Front. Immunol. 2021, 12, 674189. [Google Scholar] [CrossRef]
- Wang, Q.; Lv, C.; Sun, Y.; Han, X.; Wang, S.; Mao, Z.; Xin, Y.; Zhang, B. The Role of Alpha-Lipoic Acid in the Pathomechanism of Acute Ischemic Stroke. Cell. Physiol. Biochem. 2018, 48, 42–53. [Google Scholar] [CrossRef]
- Xu, J.; Drew, P.D. 9-Cis-retinoic acid suppresses inflammatory responses of microglia and astrocytes. J. Neuroimmunol. 2006, 171, 135–144. [Google Scholar] [CrossRef]
- Suh, H.-S.; Zhao, M.-L.; Derico, L.; Choi, N.; Lee, S.C. Insulin-like growth factor 1 and 2 (IGF1, IGF2) expression in human microglia: Differential regulation by inflammatory mediators. J. Neuroinflamm. 2013, 10, 37. [Google Scholar] [CrossRef]
- Zhang, B.; Yang, L.; Konishi, Y.; Maeda, N.; Sakanaka, M.; Tanaka, J. Suppressive effects of phosphodiesterase type IV inhibitors on rat cultured microglial cells: Comparison with other types of cAMP-elevating agents. Neuropharmacology 2002, 42, 262–269. [Google Scholar] [CrossRef]
- Dinarello, C.A. Anti-inflammatory Agents: Present and Future. Cell 2010, 140, 935–950. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, Y.; Moriyama, M.; Kawabe, K.; Satoh, H.; Takano, K.; Azuma, Y.-T.; Nakamura, Y. Lauric Acid Alleviates Neuroinflammatory Responses by Activated Microglia: Involvement of the GPR40-Dependent Pathway. Neurochem. Res. 2018, 43, 1723–1735. [Google Scholar] [CrossRef]
- Wang, Y.-P.; Wu, Y.; Li, L.-Y.; Zheng, J.; Liu, R.-G.; Zhou, J.-P.; Yuan, S.-Y.; Shang, Y.; Yao, S.-L. Aspirin-triggered lipoxin A4attenuates LPS-induced pro-inflammatory responses by inhibiting activation of NF-κB and MAPKs in BV-2 microglial cells. J. Neuroinflamm. 2011, 8, 95. [Google Scholar] [CrossRef] [PubMed]
- Rey, C.; Nadjar, A.; Buaud, B.; Vaysse, C.; Aubert, A.; Pallet, V.; Layé, S.; Joffre, C. Resolvin D1 and E1 promote resolution of inflammation in microglial cells in vitro. Brain Behav. Immun. 2016, 55, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Lamont, G.J.; Lamont, R.J.; Uriarte, S.M.; Wang, H.; Scott, D.A. Resolvin D1, resolvin D2 and maresin 1 activate the GSK3β anti-inflammatory axis in TLR4-engaged human monocytes. J. Endotoxin Res. 2016, 22, 186–195. [Google Scholar] [CrossRef]
- Tylek, K.; Trojan, E.; Leśkiewicz, M.; Regulska, M.; Bryniarska, N.; Curzytek, K.; Lacivita, E.; Leopoldo, M.; Basta-Kaim, A. Time-Dependent Protective and Pro-Resolving Effects of FPR2 Agonists on Lipopolysaccharide-Exposed Microglia Cells Involve Inhibition of NF-κB and MAPKs Pathways. Cells 2021, 10, 2373. [Google Scholar] [CrossRef] [PubMed]
- Hung, T.-H.; Shyue, S.-K.; Wu, C.-H.; Chen, C.-C.; Lin, C.-C.; Chang, C.-F.; Chen, S.-F. Deletion or inhibition of soluble epoxide hydrolase protects against brain damage and reduces microglia-mediated neuroinflammation in traumatic brain injury. Oncotarget 2017, 8, 103236–103260. [Google Scholar] [CrossRef]
- Abdallah, C.G.; Sanacora, G.; Duman, R.S.; Krystal, J.H. The neurobiology of depression, ketamine and rapid-acting antidepressants: Is it glutamate inhibition or activation? Pharmacol. Ther. 2018, 190, 148–158. [Google Scholar] [CrossRef]
- Zanos, P.; Gould, T.D. Mechanisms of ketamine action as an antidepressant. Mol. Psychiatry 2018, 23, 801–811. [Google Scholar] [CrossRef]
- Sanacora, G.; Schatzberg, A.F. Ketamine: Promising Path or False Prophecy in the Development of Novel Therapeutics for Mood Disorders? Neuropsychopharmacology 2015, 40, 259–267. [Google Scholar] [CrossRef]
- Lumsden, E.W.; Troppoli, T.A.; Myers, S.J.; Zanos, P.; Aracava, Y.; Kehr, J.; Lovett, J.; Kim, S.; Wang, F.-H.; Schmidt, S.; et al. Antidepressant-relevant concentrations of the ketamine metabolite (2 R,6 R )-hydroxynorketamine do not block NMDA receptor function. Proc. Natl. Acad. Sci. USA 2019, 116, 5160–5169. [Google Scholar] [CrossRef] [PubMed]
- Williams, N.R.; Heifets, B.; Blasey, C.; Sudheimer, K.; Pannu, J.; Pankow, H.; Hawkins, J.; Birnbaum, J.; Lyons, D.M.; Rodriguez, C.I.; et al. Attenuation of Antidepressant Effects of Ketamine by Opioid Receptor Antagonism. Am. J. Psychiatry 2018, 175, 1205–1215. [Google Scholar] [CrossRef]
- Yost, J.G.; Wulf, H.A.; Browne, C.A.; Lucki, I. Antinociceptive and Analgesic Effects of (2R,6R)-Hydroxynorketamine. J. Pharmacol. Exp. Ther. 2022, 382, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Beurel, E.; Song, L.; Jope, R.S. Inhibition of glycogen synthase kinase-3 is necessary for the rapid antidepressant effect of ketamine in mice. Mol. Psychiatry 2011, 16, 1068–1070. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Dong, L.; Wang, N.; Shi, J.-Y.; Yang, J.-J.; Zuo, Z.-Y.; Zhou, Z.-Q. Akt Mediates GSK-3β Phosphorylation in the Rat Prefrontal Cortex during the Process of Ketamine Exerting Rapid Antidepressant Actions. Neuroimmunomodulation 2014, 21, 183–188. [Google Scholar] [CrossRef]
- Choi, M.; Lee, S.H.; Park, M.H.; Kim, Y.-S.; Son, H. Ketamine induces brain-derived neurotrophic factor expression via phosphorylation of histone deacetylase 5 in rats. Biochem. Biophys. Res. Commun. 2017, 489, 420–425. [Google Scholar] [CrossRef]
- Hashimoto, K. Essential Role of Keap1-Nrf2 Signaling in Mood Disorders: Overview and Future Perspective. Front. Pharmacol. 2018, 9, 1182. [Google Scholar] [CrossRef]
- Hoetzel, A.; Schmidt, R. Regulatory role of anesthetics on heme oxygenase-1. Curr. Drug Targets 2010, 11, 1495–1503. [Google Scholar] [CrossRef]
- Wang, N.; Yu, H.-Y.; Shen, X.-F.; Gao, Z.-Q.; Yang, C.; Yang, J.-J.; Zhang, G.-F. The rapid antidepressant effect of ketamine in rats is associated with down-regulation of pro-inflammatory cytokines in the hippocampus. Upsala J. Med. Sci. 2015, 120, 241–248. [Google Scholar] [CrossRef]
- Tan, S.; Wang, Y.; Chen, K.; Long, Z.; Zou, J. Ketamine Alleviates Depressive-Like Behaviors via Down-Regulating Inflammatory Cytokines Induced by Chronic Restraint Stress in Mice. Biol. Pharm. Bull. 2017, 40, 1260–1267. [Google Scholar] [CrossRef]
- Qu, Y.; Shan, J.; Wang, S.; Chang, L.; Pu, Y.; Wang, X.; Tan, Y.; Yamamoto, M.; Hashimoto, K. Rapid-acting and long-lasting antidepressant-like action of (R)-ketamine in Nrf2 knock-out mice: A role of TrkB signaling. Eur. Arch. Psychiatry Clin. Neurosci. 2021, 271, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.K.; Budac, D.P.; Bisulco, S.; Lee, A.W.; Smith, R.A.; Beenders, B.; Kelley, K.W.; Dantzer, R. NMDA Receptor Blockade by Ketamine Abrogates Lipopolysaccharide-Induced Depressive-Like Behavior in C57BL/6J Mice. Neuropsychopharmacology 2013, 38, 1609–1616. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.-J.; Chen, T.-L.; Ueng, Y.-F.; Chen, R.-M. Ketamine inhibits tumor necrosis factor-α and interleukin-6 gene expressions in lipopolysaccharide-stimulated macrophages through suppression of toll-like receptor 4-mediated c-Jun N-terminal kinase phosphorylation and activator protein-1 activation. Toxicol. Appl. Pharmacol. 2008, 228, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Welters, I.D.; Hafer, G.; Menzebach, A.; Mühling, J.; Neuhäuser, C.; Browning, P.; Goumon, Y. Ketamine Inhibits Transcription Factors Activator Protein 1 and Nuclear Factor-κB, Interleukin-8 Production, as well as CD11b and CD16 Expression: Studies in Human Leukocytes and Leukocytic Cell Lines. Anesth. Analg. 2010, 110, 934–941. [Google Scholar] [CrossRef]
- Beilin, B.; Rusabrov, Y.; Shapira, Y.; Roytblat, L.; Greemberg, L.; Yardeni, I.Z.; Bessler, H. Low-dose ketamine affects immune responses in humans during the early postoperative period. Br. J. Anaesth. 2007, 99, 522–527. [Google Scholar] [CrossRef]
- Zunszain, P.A.; Horowitz, M.A.; Cattaneo, A.; Lupi, M.M.; Pariante, C.M. Ketamine: Synaptogenesis, immunomodulation and glycogen synthase kinase-3 as underlying mechanisms of its antidepressant properties. Mol. Psychiatry 2013, 18, 1236–1241. [Google Scholar] [CrossRef]
- Yang, J.-J.; Wang, N.; Yang, C.; Shi, J.-Y.; Yu, H.-Y.; Hashimoto, K. Serum Interleukin-6 Is a Predictive Biomarker for Ketamine’s Antidepressant Effect in Treatment-Resistant Patients With Major Depression. Biol. Psychiatry 2015, 77, e19–e20. [Google Scholar] [CrossRef]
- Zhan, Y.; Zhou, Y.; Zheng, W.; Liu, W.; Wang, C.; Lan, X.; Deng, X.; Xu, Y.; Zhang, B.; Ning, Y. Alterations of multiple peripheral inflammatory cytokine levels after repeated ketamine infusions in major depressive disorder. Transl. Psychiatry 2020, 10, 246. [Google Scholar] [CrossRef]
- Yuskaitis, C.J.; Jope, R.S. Glycogen synthase kinase-3 regulates microglial migration, inflammation, and inflammation-induced neurotoxicity. Cell. Signal. 2009, 21, 264–273. [Google Scholar] [CrossRef]
- VanderZwaag, J.; Halvorson, T.; Dolhan, K.; Šimončičová, E.; Ben-Azu, B.; Tremblay, M. The Missing Piece? A Case for Microglia’s Prominent Role in the Therapeutic Action of Anesthetics, Ketamine, and Psychedelics. Neurochem. Res. 2022, 1–38. [Google Scholar] [CrossRef]
- Urs, N.M.; Snyder, J.C.; Jacobsen, J.P.R.; Peterson, S.M.; Caron, M.G. Deletion of GSK3β in D2R-expressing neurons reveals distinct roles for β-arrestin signaling in antipsychotic and lithium action. Proc. Natl. Acad. Sci. USA 2012, 109, 20732–20737. [Google Scholar] [CrossRef]
- Beaulieu, J.-M. A role for Akt and glycogen synthase kinase-3 as integrators of dopamine and serotonin neurotransmission in mental health. J. Psychiatry Neurosci. 2012, 37, 7–16. [Google Scholar] [CrossRef]
- Meltzer, H.Y.; Matsubara, S.; Lee, J.C. The ratios of serotonin2 and dopamine2 affinities differentiate atypical and typical antipsychotic drugs. Psychopharmacol. Bull. 1989, 25, 390–392. [Google Scholar] [PubMed]
- Kapur, S.; Zipursky, R.B.; Remington, G. Clinical and Theoretical Implications of 5-HT2 and D2 Receptor Occupancy of Clozapine, Risperidone, and Olanzapine in Schizophrenia. Am. J. Psychiatry 1999, 156, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Alimohamad, H.; Rajakumar, N.; Seah, Y.-H.; Rushlow, W. Antipsychotics alter the protein expression levels of β-catenin and GSK-3 in the rat medial prefrontal cortex and striatum. Biol. Psychiatry 2005, 57, 533–542. [Google Scholar] [CrossRef]
- Li, X.; Rosborough, K.M.; Friedman, A.B.; Zhu, W.; Roth, K. Regulation of mouse brain glycogen synthase kinase-3 by atypical antipsychotics. Int. J. Neuropsychopharmacol. 2007, 10, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Schmid, C.L.; Bohn, L.M. Serotonin, But Not N-Methyltryptamines, Activates the Serotonin 2A Receptor Via a β-Arrestin2/Src/Akt Signaling Complex In Vivo. J. Neurosci. 2010, 30, 13513–13524. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhu, W.; Roh, M.-S.; Friedman, A.B.; Rosborough, K.; Jope, R.S. In Vivo Regulation of Glycogen Synthase Kinase-3β (GSK3β) by Serotonergic Activity in Mouse Brain. Neuropsychopharmacology 2004, 29, 1426–1431. [Google Scholar] [CrossRef]
- Polter, A.M.; Li, X. Glycogen Synthase Kinase-3 is an Intermediate Modulator of Serotonin Neurotransmission. Front. Mol. Neurosci. 2011, 4, 31. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Receptor | Citations for Expression by Microglia | Citations for Gq-PLC-PKC Coupling | Citations for Inflammatory Cytokines Decrease | Citation for Antidepressant Activity |
|---|---|---|---|---|
| Cannabinoid CBR2 | [146,153] | [147,161] | [148,149,161] | [144,162] |
| Opioid μ-receptor | [103,163] | [164] | [165] | [166,167,168] |
| Serotonin 5HT2B | [101,102] | [169,170,171] | [101,102] | [172,173,174] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalkman, H.O. Inhibition of Microglial GSK3β Activity Is Common to Different Kinds of Antidepressants: A Proposal for an In Vitro Screen to Detect Novel Antidepressant Principles. Biomedicines 2023, 11, 806. https://doi.org/10.3390/biomedicines11030806
Kalkman HO. Inhibition of Microglial GSK3β Activity Is Common to Different Kinds of Antidepressants: A Proposal for an In Vitro Screen to Detect Novel Antidepressant Principles. Biomedicines. 2023; 11(3):806. https://doi.org/10.3390/biomedicines11030806
Chicago/Turabian StyleKalkman, Hans O. 2023. "Inhibition of Microglial GSK3β Activity Is Common to Different Kinds of Antidepressants: A Proposal for an In Vitro Screen to Detect Novel Antidepressant Principles" Biomedicines 11, no. 3: 806. https://doi.org/10.3390/biomedicines11030806
APA StyleKalkman, H. O. (2023). Inhibition of Microglial GSK3β Activity Is Common to Different Kinds of Antidepressants: A Proposal for an In Vitro Screen to Detect Novel Antidepressant Principles. Biomedicines, 11(3), 806. https://doi.org/10.3390/biomedicines11030806