
Inflammation, Microcalcification, and Increased Expression of Osteopontin Are Histological Hallmarks of Plaque Vulnerability in Patients with Advanced Carotid Artery Stenosis


Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients and Tissue Fragments
2.2. Patients Data Collection
2.3. Histological Processing
2.4. Investigation of the Osteopontin (OPN) Expression within the Atherosclerotic Plaque by Immunohistochemistry
2.5. Statistical Analysis
3. Results
3.1. Study Group Characteristics
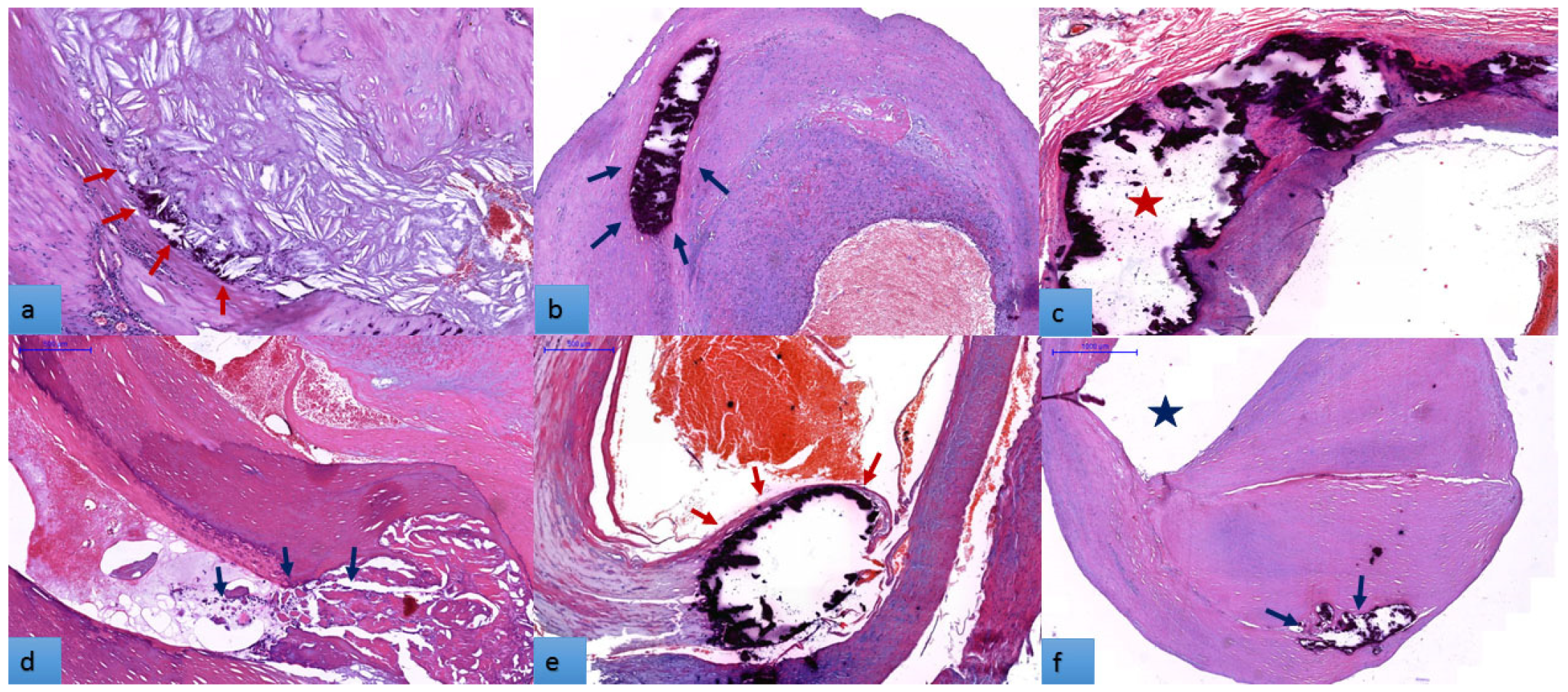
3.2. Histopathogical Features of Carotid Atherosclerotic Plaques
3.3. Osteopontin Expression
3.4. Comparison of the Inflammatory Infiltrate Positive (INF+) and Negative (INF−) Groups
3.5. The Distribution of Atheroma Calcification Patterns
3.6. Treatment Correlations
3.7. Correlations of Calcification Extent, Localization and Patterns with Ulceration, Thrombosis and Hemorrhagic Rupture of the Plaque
3.8. Predictors of Plaque Ulceration
3.9. Predictors of Atherothrombosis
4. Discussion
5. Limitations
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Katan, M.; Luft, A. Global Burden of Stroke. Semin. Neurol. 2018, 38, 208–211. [Google Scholar] [CrossRef] [Green Version]
- Yurdagul, A.; Finney, A.C.; Woolard, M.D.; Orr, A.W. The arterial microenvironment: The where and why of atherosclerosis. Biochem. J. 2016, 473, 1281–1295. [Google Scholar] [CrossRef] [Green Version]
- Jebari-Benslaiman, S.; Galicia-Garcia, U.; Larrea-Sebal, A.; Olaetxea, J.R.; Alloza, I.; Vandenbroeck, K.; Benito-Vicente, A.; Martin, C. Pathophysiology of Atherosclerosis. Int. J. Mol. Sci. 2022, 23, 3346. [Google Scholar] [CrossRef]
- Shi, X.; Gao, J.; Lv, Q.S.; Cai, H.D.; Wang, F.; Ye, R.D.; Liu, X.F. Calcification in Atherosclerotic Plaque Vulnerability: Friend or Foe? Front. Physiol. 2020, 11, 56. [Google Scholar] [CrossRef] [PubMed]
- Akers, E.J.; Nicholls, S.J.; Di Bartolo, B.A. Plaque Calcification Do Lipoproteins Have a Role? Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1902–1910. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Yang, W.J.; Chen, X.Y. Histology-Verified Intracranial Artery Calcification and Its Clinical Relevance With Cerebrovascular Disease. Front. Neurol. 2022, 12, 789035. [Google Scholar] [CrossRef]
- Rambhia, S.H.; Liang, X.; Xenos, M.; Alemu, Y.; Maldonado, N.; Kelly, A.; Chakraborti, S.; Weinbaum, S.; Cardoso, L.; Einav, S.; et al. Microcalcifications Increase Coronary Vulnerable Plaque Rupture Potential: A Patient-Based Micro-CT Fluid-Structure Interaction Study. Ann. Biomed. Eng. 2012, 40, 1443–1454. [Google Scholar] [CrossRef]
- Kwee, R.M. Systematic review on the association between calcification in carotid plaques and clinical ischemic symptoms. J. Vasc. Surg. 2010, 51, 1015–1025. [Google Scholar] [CrossRef] [Green Version]
- Seime, T.; van Wanrooij, M.; Karlof, E.; Kronqvist, M.; Johansson, S.; Matic, L.; Gasser, T.C.; Hedin, U. Biomechanical Assessment of Macro-Calcification in Human Carotid Atherosclerosis and Its Impact on Smooth Muscle Cell Phenotype. Cells 2022, 11, 3279. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Pan, X.J.; Zhang, B.; Yan, Y.H.; Huang, Y.B.; Woolf, A.K.; Gillard, J.H.; Teng, Z.Z.; Hui, P.J. Superficial and multiple calcifications and ulceration associate with intraplaque hemorrhage in the carotid atherosclerotic plaque. Eur. Radiol. 2018, 28, 4968–4977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Böhm, E.W.; Pavlaki, M.; Chalikias, G.; Mikroulis, D.; Georgiadis, G.S.; Tziakas, D.N.; Konstantinides, S.; Schäfer, K. Colocalization of Erythrocytes and Vascular Calcification in Human Atherosclerosis: A Systematic Histomorphometric Analysis. TH Open 2021, 5, e113–e124. [Google Scholar] [CrossRef] [PubMed]
- Shirakawa, K.; Sano, M. Osteopontin in Cardiovascular Diseases. Biomolecules 2021, 11, 1047. [Google Scholar] [CrossRef] [PubMed]
- Steitz, S.A.; Speer, M.Y.; McKee, M.D.; Liaw, L.; Almeida, M.; Yang, H.; Giachelli, C.M. Osteopontin inhibits mineral deposition and promotes regression of ectopic calcification. Am. J. Pathol. 2002, 161, 2035–2046. [Google Scholar] [CrossRef] [Green Version]
- Naylor, R.; Rantner, B.; Ancetti, S.; de Borst, G.J.; De Carlo, M.; Halliday, A.; Kakkos, S.K.; Markus, H.S.; McCabe, D.J.H.; Sillesen, H.; et al. European Society for Vascular Surgery (ESVS) 2023 Clinical Practice Guidelines on the Management of Atherosclerotic Carotid and Vertebral Artery Disease. Eur. J. Vasc. Endovasc. Surg. 2023, 65, 7–111. [Google Scholar] [CrossRef]
- Bonati, L.H.; Kakkos, S.; Berkefeld, J.; de Borst, G.J.; Bulbulia, R.; Halliday, A.; van Herzeele, I.; Koncar, I.; McCabe, D.J.H.; Lal, A.; et al. European Stroke Organisation guideline on endarterectomy and stenting for carotid artery stenosis. Eur. Stroke J. 2021, 6, I–LXII. [Google Scholar] [CrossRef]
- Orrapin, S.; Rerkasem, K. Carotid endarterectomy for symptomatic carotid stenosis. Cochrane Database Syst. Rev. 2017, 6, CD001081. [Google Scholar] [CrossRef]
- Williams, B.; Mancia, G.; Spiering, W.; Rosei, E.A.; Azizi, M.; Burnier, M.; Clement, D.L.; Coca, A.; de Simone, G.; Dominiczak, A.; et al. 2018 ESC/ESH Guidelines for the management of arterial hypertension. Eur. Heart J. 2018, 39, 3021–3104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cosentino, F.; Grant, P.J.; Aboyans, V.; Bailey, C.J.; Ceriello, A.; Delgado, V.; Federici, M.; Filippatos, G.; Grobbee, D.E.; Hansen, T.B.; et al. 2019 ESC Guidelines on diabetes, pre-diabetes, and cardiovascular diseases developed in collaboration with the EASD. Eur. Heart J. 2020, 41, 255–323. [Google Scholar] [CrossRef] [Green Version]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk The Task Force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and European Atherosclerosis Society (EAS). Eur. Heart J. 2020, 41, 111–188. [Google Scholar] [CrossRef] [Green Version]
- Coupland, A.P.; Thapar, A.; Qureshi, M.I.; Jenkins, H.; Davies, A.H. The definition of stroke. J. R. Soc. Med. 2017, 110, 9–12. [Google Scholar] [CrossRef] [Green Version]
- Stary, H.C. Natural history and histological classification of atherosclerotic lesions—An update. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1177–1178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lovett, J.K.; Redgrave, J.N.E.; Gallagher, P.J.; Walton, J.; Hands, L.; Rothwell, P.M. Histological correlates of carotid plaque surface morphology on lumen contrast imaging. Stroke 2004, 35, E194. [Google Scholar]
- Kelly-Arnold, A.; Maldonado, N.; Laudier, D.; Aikawa, E.; Cardoso, L.; Weinbaum, S. Revised microcalcification hypothesis for fibrous cap rupture in human coronary arteries. Proc. Natl. Acad. Sci. USA 2013, 110, 10741–10746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, H.E.; Gasbarrino, K.; Veinot, J.P.; Lai, C.; Daskalopoulou, S.S. New Quantitative Digital Image Analysis Method of Histological Features of Carotid Atherosclerotic Plaques. Eur. J. Vasc. Endovasc. Surg. 2019, 58, 654–663. [Google Scholar] [CrossRef]
- Wang, Y.L.; Wang, T.; Luo, Y.M.; Jiao, L.Q. Identification Markers of Carotid Vulnerable Plaques: An Update. Biomolecules 2022, 12, 1192. [Google Scholar] [CrossRef]
- Lee-Rueckert, M.; Lappalainen, J.; Kovanen, P.T.; Escola-Gil, J.C. Lipid-Laden Macrophages and Inflammation in Atherosclerosis and Cancer: An Integrative View. Front. Cardiovasc. Med. 2022, 9, 777822. [Google Scholar] [CrossRef]
- Nakagawa, K.; Tanaka, M.; Hahm, T.H.; Nguyen, H.N.; Matsui, T.; Chen, Y.X.; Nakashima, Y. Accumulation of Plasma-Derived Lipids in the Lipid Core and Necrotic Core of Human Atheroma: Imaging Mass Spectrometry and Histopathological Analyses. Arterioscler. Thromb. Vasc. Biol. 2021, 41, E498–E511. [Google Scholar] [CrossRef]
- Fletcher, E.K.; Wang, Y.L.; Flynn, L.K.; Turner, S.E.; Rade, J.J.; Kimmelstiel, C.D.; Gurbel, P.A.; Bliden, K.P.; Covic, L.; Kuliopulos, A. Deficiency of MMP1a (Matrix Metalloprotease 1a) Collagenase Suppresses Development of Atherosclerosis in Mice Translational Implications for Human Coronary Artery Disease. Arterioscler. Thromb. Vasc. Biol. 2021, 41, E265–E279. [Google Scholar] [CrossRef]
- Gialeli, C.; Shami, A.; Goncalves, I. Extracellular matrix: Paving the way to the newest trends in atherosclerosis. Curr. Opin. Lipidol. 2021, 32, 277–285. [Google Scholar] [CrossRef]
- Tomas, L.; Edsfeldt, A.; Mollet, I.G.; Matic, L.P.; Prehn, C.; Adamski, J.; Paulsson-Berne, G.; Hedin, U.; Nilsson, J.; Bengtsson, E.; et al. Altered metabolism distinguishes high-risk from stable carotid atherosclerotic plaques. Eur. Heart J. 2018, 39, 2301–2310. [Google Scholar] [CrossRef] [Green Version]
- Saba, L.; Sanfilippo, R.; Sannia, S.; Anzidei, M.; Montisci, R.; Mallarini, G.; Suri, J.S. Association Between Carotid Artery Plaque Volume, Composition, and Ulceration: A Retrospective Assessment With MDCT. Am. J. Roentgenol. 2012, 199, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Poredos, P.; Gregoric, I.D.; Jezovnik, M.K. Inflammation of carotid plaques and risk of cerebrovascular events. Ann. Transl. Med. 2020, 8, 1281. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.K.L.; Thavornpattanapong, P.; Cheung, S.C.P.; Sun, Z.H.; Tu, J.Y. Effect of calcification on the mechanical stability of plaque based on a three-dimensional carotid bifurcation model. BMC Cardiovasc. Disord. 2012, 12, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, M.; McPherson, R.; Abruzzo, A.; Thomas, S.E.; Gorantla, V.R. Carotid Artery Calcification: What We Know So Far. Cureus 2021, 13, e18938. [Google Scholar] [CrossRef] [PubMed]
- Joshi, F.R.; Rajani, N.K.; Abt, M.; Woodward, M.; Bucerius, J.; Mani, V.; Tawakol, A.; Kallend, D.; Fayad, Z.A.; Rudd, J.H.F. Does Vascular Calcification Accelerate Inflammation? A Substudy of the dal-PLAQUE Trial. J. Am. Coll. Cardiol. 2016, 67, 69–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shioi, A.; Ikari, Y. Plaque Calcification During Atherosclerosis Progression and Regression. J. Atheroscler. Thromb. 2018, 25, 294–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- New, S.E.P.; Aikawa, E. Molecular Imaging Insights Into Early Inflammatory Stages of Arterial and Aortic Valve Calcification. Circ. Res. 2011, 108, 1381–1391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, C.F.; Seshadri, V.; Huang, K.; Shao, J.S.; Cai, J.; Vattikuti, R.; Schumacher, A.; Loewy, A.P.; Denhardt, D.T.; Rittling, S.R.; et al. An osteopontin-NADPH oxidase signaling cascade promotes pro-matrix metalloproteinase 9 activation in aortic mesenchymal cells. Circ. Res. 2006, 98, 1479–1489. [Google Scholar] [CrossRef] [Green Version]
- Fehervari, L.; Frigy, A.; Kocsis, L.; Szabo, I.A.; Szabo, T.M.; Urkon, M.; Jako, Z.; Nagy, E.E. Serum Osteoprotegerin and Carotid Intima-Media Thickness Are Related to High Arterial Stiffness in Heart Failure with Reduced Ejection Fraction. Diagnostics 2021, 11, 764. [Google Scholar] [CrossRef]
- Nagy, E.E.; Varga-Fekete, T.; Puskas, A.; Kelemen, P.; Brassai, Z.; Szekeres-Csiki, K.; Gombos, T.; Csanyi, M.C.; Harsfalvi, J. High circulating osteoprotegerin levels are associated with non-zero blood groups. BMC Cardiovasc. Disord. 2016, 16, 106. [Google Scholar] [CrossRef] [Green Version]
- Morony, S.; Tintut, Y.; Zhang, Z.; Cattley, R.C.; Van, G.; Dwyer, D.; Stolina, M.; Kostenuik, P.J.; Demer, L.L. Osteoprotegerin inhibits vascular calcification without affecting atherosclerosis in ldlr((-/-)) mice. Circulation 2008, 117, 411–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lok, Z.S.Y.; Lyle, A.N. Osteopontin in Vascular Disease Friend or Foe? Arterioscler. Thromb. Vasc. Biol. 2019, 39, 613–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolak, T.; Sion-Vardi, N.; Novack, V.; Greenberg, G.; Szendro, G.; Tarnovscki, T.; Nov, O.; Shelef, I.; Paran, E.; Rudich, A. N-Terminal Rather Than Full-Length Osteopontin or Its C-Terminal Fragment Is Associated With Carotid-Plaque Inflammation in Hypertensive Patients. Am. J. Hypertens. 2013, 26, 326–333. [Google Scholar] [CrossRef] [Green Version]
- Strobescu-Ciobanu, C.; Giusca, S.E.; Caruntu, I.D.; Amalinei, C.; Rusu, A.; Cojocaru, E.; Popa, R.F.; Lupascu, C.D. Osteopontin and osteoprotegerin in atherosclerotic plaque—Are they significant markers of plaque vulnerability? Rom. J. Morphol. Embryol. 2020, 61, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Cosarca, M.C.; Horvath, E.; Molnar, C.; Molnar, G.B.; Russu, E.; Muresan, V.A. Calcification patterns in femoral and carotid atheromatous plaques: A comparative morphometric study. Exp. Ther. Med. 2021, 22, 865. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Median (Quartile Range)/Mean ± SE | INF+ Group (n = 75) | INF− Group (n = 44) | p Values |
|---|---|---|---|---|
| Demographic and lifestyle variables | ||||
| Age (years) | 67 (61–72) | 67 (60–73) | 67.5 (63–71) | 0.412 |
| Gender (f/m) | 37 (31.1)/82 (68.9) | 23 (30.7)/52 (69.3) | 14 (31.8)/30 (68.2) | 1.000 |
| Smoking (yes/no) | 58 (48.7)/61 (51.3) | 40 (53.3)/35 (46.7) | 18 (40.9)/26 (59.1) | 0.254 |
| Disease characteristics and comorbidities | ||||
| Grade of stenosis * (%) | 81.3 ± 0.7 | 80.3 ± 0.9 | 83.1 ± 1.2 | 0.062 |
| Carotid atherosclerosis, uni- vs. bilateral (u/b) | 84 (70.6)/35 (29.4) | 50 (66.7)/25 (33.3) | 34 (77.3)/10 (22.7) | 0.297 |
| Stroke history (y/n) | 76 (63.9)/43 (36.1) | 51 (68)/24 (32) | 25 (56.8)/19 (43.2) | 0.240 |
| Occurrence of aphasia (yes/no) | 17 (14.3)/102 (85.7) | 15 (20)/60 (80) | 2 (4.5)/42 (95.5) | 0.027 |
| Occurrence of paresis/plegia (yes/no) | 13 (10.9)/106 (89.1) | 11 (14.7)/64 (85.3) | 2 (4.5)/42 (95.5) | 0.128 |
| Hypertension (y/n) | 110 (92.4)/9 (7.6) | 67 (89.3)/8 (10.7) | 43 (97.7)/1 (2.3) | 0.151 |
| Diabetes (y/n) | 33 (27.7)/86 (72.3) | 18 (24)/57 (76) | 15 (34.1)/29 (65.9) | 0.471 |
| Polyvascular disease (1/2/3 arterial beds affected) | 80 (67.2)/28 (23.5)/11 (9.3) | 58 (77.3)/15 (20)/2 (2.7) | 22 (50)/13 (29.5)/9(20.5) | 0.001 |
| Plaque calcification | ||||
| Calcification extent (grade 3–4/grade 0–2) | 54 (45.4)/65 (54.6) | 30 (40)/45 (60) | 24 (54.5)/19 (45.5) | 0.142 |
| Superficial/deep calcification | 65 (54.6)/54 (45.4) | 48 (64)/27 (36) | 17(38.6)/27 (61.4) | 0.008 |
| Microcalcification (yes/no) | 54 (45.4)/65 (54.6) | 42 (56)/33 (44) | 12 (27.3)/32 (72.7) | 0.004 |
| Nodular calcification (yes/no) | 71 (59.7)/48 (40.3) | 49 (65.3)/26 (34.7) | 22 (50)/22 (50) | 0.122 |
| Extended/confluent calcification (yes/no) | 41 (34.5)/78 (65.5) | 20 (26.7)/55 (73.3) | 21 (47.4)/23 (52.3) | 0.027 |
| Metaplasia (yes/no) | 33 (27.7)/86 (72.3) | 17 (22.6)/58 (77.3) | 16 (40.9)/28 (59.1) | 0.138 |
| OPN expression (grade 1/2/3) | 57(47.9)/36 (30.3)/26(21.8) | 21(28)/29(38.7)/25(33.3) | 36(81.8)/7(15.9)/1(2.3) | <0.001 |
| Biological variables and medication | ||||
| Hypercholesterolemia (yes/no) | 115 (96.6)/4 (3.4) | 74 (98.7)/1 (1.3) | 41 (93.2)/3 (6.8) | 0.142 |
| Abs. neutrophil count (109/L) | 5.29 (4.05–6.66) | 5.51 (4.04–6.8) | 5.07 (4.05–6.38) | 0.293 |
| Abs. lymphocyte count (109/L) | 1.95 (1.56–2.53) | 1.95 (1.64–2.47) | 1.93 (1.45–2.64) | 0.686 |
| Neutrophil/Lymphocyte ratio | 2.70 (1.91–3.62) | 2.8 (1.95–3.62) | 2.55 (1.65–3.75) | 0.338 |
| Anti-hypertensive drugs (y/n) | 110 (92.4)/9 (7.6) | 67 (89.3)/8 (10.7) | 43 (97.7)/1 (2.3) | 0.151 |
| Anticoagulant pre.op.(y/n) | 7 (5.8)/112 (94.2) | 0 (0)/ 75 (17.1) | 37 (89.2)/7 (10.8) | 0.077 |
| Anti-aggregants pre.op.(y/n) | 111 (93.2)/8 (6.8) | 71 (94.6)/4 (5.4) | 40 (90.9)/4 (9.1) | 0.465 |
| Anticoagulant post.op. (y/n) | 95 (79.8)/24 (20.2) | 62 (82.6)/13 (17.4) | 33 (75)/11 (25) | 0.349 |
| Anti-aggregants post.op. (y/n) | 119 (100)/0 | 75 (100)/0 | 44 (100)/0 | - |
| Hypolipidemics (y/n) | 116 (97.4)/3 (2.6) | 73 (97.3)/2 (2.7) | 43 (97.7)/1 (2.3) | 1.000 |
| Ulceration | Thrombosis | Hemorrhage | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No | Yes | p | No | Yes | p | No | Yes | p | ||||
| Localization | superficial | 32 | 33 | 0.064 | superficial | 57 | 8 | 0.789 | superficial | 38 | 27 | 0.031 |
| deep | 36 | 18 | deep | 46 | 8 | deep | 42 | 12 | ||||
| Microcalcification | No | 47 | 18 | <0.001 | No | 58 | 7 | 0.422 | No | 48 | 17 | 0.117 |
| Yes | 20 | 34 | Yes | 45 | 9 | Yes | 32 | 22 | ||||
| Nodular pattern | No | 28 | 20 | 0.85 | No | 40 | 8 | 0.422 | No | 36 | 12 | 0.165 |
| Yes | 39 | 32 | Yes | 63 | 8 | Yes | 44 | 27 | ||||
| Extended/confluent pattern | No | 40 | 38 | 0.173 | No | 67 | 11 | 1 | No | 47 | 31 | 0.019 |
| Yes | 27 | 14 | Yes | 36 | 5 | Yes | 33 | 8 | ||||
| Osteoid metaplasia | No | 47 | 39 | 0.68 | No | 77 | 9 | 0.14 | No | 55 | 31 | 0.277 |
| Yes | 20 | 13 | Yes | 26 | 7 | Yes | 25 | 8 | ||||
| OPN expression | 1+ | 40 | 17 | 0.022 | 1+ | 50 | 7 | 0.924 | 1+ | 42 | 15 | 0.299 |
| 2+ | 16 | 20 | 2+ | 31 | 5 | 2+ | 21 | 15 | ||||
| 3+ | 12 | 14 | 3+ | 22 | 4 | 3+ | 17 | 9 | ||||
| Cumulative patterns: Nodular AND/OR extended/confluent AND/OR Osteoid metaplasia | No | 47 | 39 | No | 76 | 10 | No | 55 | 31 | |||
| Yes | 21 | 12 | 0.493 | Yes | 27 | 6 | 0.47 | Yes | 25 | 8 | 0.46 | |
| Variables | Coefficient | SD | OR (95%CI) | p-Level |
|---|---|---|---|---|
| Age (Q4/Q1) | 0.089 | 0.251 | 1.19 (0.45–3.18) | 0.721 |
| Gender (M/F) | 0.182 | 0.408 | 0.83 (0.38–1.82) | 0.656 |
| Diabetes (Yes/No) | −0.044 | 0.227 | 0.82 (0.36–1.86) | 0.846 |
| Smoking (Yes/No) | −0.019 | 0.387 | 1.02 (0.49–2.11) | 0.959 |
| Hypertension (Yes/No) | −1.060 | 0.733 | 0.34 (0.08–1.45) | 0.150 |
| Carotid atherosclerosis, unilateral/bilateral (u/b) | −0.335 | 0.412 | 0.71 (0.32–1.60) | 0.418 |
| Stroke history (Yes/No) | −0.084 | 0.385 | 0.92 (0.43–1.90) | 0.826 |
| Polyvascular disease (≥2 a. beds/single bed) | −0.647 | 0.315 | 26.68 (1.52–468.19) | 0.042 |
| Stenosis grade (Q4/Q1) | −0.416 | 0.244 | 0.43 (0.16–1.14) | 0.091 |
| Hypercholesterolemia (Yes/No) | 0.836 | 1.170 | 2.30 (0.23–22.85) | 0.476 |
| Revascularization (Yes/No) | −0.284 | 0.391 | 0.75 (0.35–1.62) | 0.468 |
| Calcification extent (High/Low) | −0.297 | 0.373 | 0.43 (0.15–1.21) | 0.427 |
| Nodular (Yes/No) | 0.225 | 0.379 | 1.15 (0.55–2.41) | 0.554 |
| Extended/confluent (Yes/No) | −0.716 | 0.405 | 0.54 (0.25–1.19) | 0.079 |
| Osteoid metaplasia (Yes/No) | −0.197 | 0.417 | 0.78 (0.34–1.77) | 0.637 |
| Mixed calcification pattern (Yes/No) | −0.451 | 0.336 | 0.68 (0.30–1.57) | 0.182 |
| Microcalcification (Yes/No) | 1.568 | 0.398 | 4.44 (2.04–9.63) | <0.001 |
| Superficial/deep | −0.721 | 0.380 | 2.06 (0.98–4.35) | 0.059 |
| Lipid core (Yes/No) | 1.190 | 0.402 | 3.28 (1.49–7.24) | 0.004 |
| Inflammatory infiltrate (INF+/INF−) | 2.015 | 0.474 | 7.50 (2.96–19.00) | <0.001 |
| Model 1. Variables | Estimate | SD | Odds Ratio (95% CI) | p Value |
|---|---|---|---|---|
| Microcalcification | 1.470 | 0.491 | 4.44 (2.04–9.63) | 0.003 |
| Lipid core (Yes/No) | 0.104 | 0.526 | 3.28 (1.49–7.24) | 0.843 |
| Superficial/deep | 0.376 | 0.509 | 2.06 (0.98–4.35) | 0.460 |
| Extended/confluent calcification | −0.048 | 0.496 | 0.54 (0.25–1.19) | 0.922 |
| Inflammatory infiltrate (INF+/INF−) | 1.575 | 0.574 | 7.50 (2.96–19.00) | 0.007 |
| Stenosis grade (Q4:Q1) | −0.376 | 0.300 | 0.43 (0.16–1.14) | 0.212 |
| Polyvascular disease (≥2 a. beds/single bed) | −0.390 | 0.409 | 26.68 (1.52–468.19) | 0.342 |
| Hypertension (Yes/No) | −0.042 | 0.880 | 0.34 (0.08–1.45) | 0.961 |
| Variables | Estimate | SD | OR (95%CI) | p-Level |
|---|---|---|---|---|
| Age (Q4/Q1) | 0.150 | 0.364 | 1.60 (0.25–10.29) | 0.681 |
| Gender (M/F) | −1.284 | 0.408 | 1.44 (0.29–7.06) | 0.656 |
| Diabetes (Yes/No) | 0.313 | 0.301 | 1.69 (0.56–5.09) | 0.300 |
| Smoking (Yes/No) | 0.230 | 0.343 | 0.79 (0.27–2.29) | 0.675 |
| Hypertension (Yes/No) | 0.233 | 1.095 | 1.26 (0.14–10.83) | 0.821 |
| Carotid atherosclerosis, unilateral/bilateral | −0.669 | 0.675 | 1.95 (0.52–7.33) | 0.323 |
| Stroke history (Yes/No) | 0.603 | 0.612 | 1.82 (0.55–6.06) | 0.326 |
| Polyvascular disease (≥2 a. beds/single bed) | 0.545 | 0.547 | 1.67 (0.57–4.88) | 0.320 |
| Stenosis grade (Q4/Q1) | −0.189 | 0.375 | 0.68 (0.15–2.98) | 0.615 |
| Revascularization (Yes/No) | 1.409 | 0.783 | 4.09 (0.88–18.98) | 0.074 |
| Nodular (Yes/No) | −0.454 | 0.539 | 0.63 (0.22–1.82) | 0.401 |
| Extended/confluent (Yes/No) | −0.167 | 0.577 | 0.84 (0.27–2.62) | 0.772 |
| Osteoid metaplasia (Yes/No) | 0.834 | 0.552 | 2.30 (0.78–6.80) | 0.133 |
| Mixed calcification pattern (Yes/No) | 0.524 | 0.562 | 1.69 (0.56–5.09) | 0.353 |
| Microcalcification (Yes/No) | 0.505 | 0.541 | 1.65 (0.57–4.79) | 0.353 |
| Superficial/deep | 0.214 | 0.537 | 0.80 (0.28–2.31) | 0.690 |
| Lipid core (Yes/No) | 1.252 | 0.670 | 3.49 (0.93–13.01) | 0.064 |
| Inflammatory infiltrate (INF+/INF−) | 1.572 | 0.782 | 4.81 (1.04–22.33) | 0.046 |
| Ulceration (Yes/No) | 23.78 | - | 61.02 (3.55–1046) | 0.005 |
| Variables | Estimate | SD | Odds Ratio (95% CI) | p Value |
|---|---|---|---|---|
| Lipid core (Yes/No) | 0.609 | 0.823 | 3.28 (1.49–7.24) | 0.843 |
| Inflammatory infiltrate (INF+/INF−) | 0.623 | 0.971 | 7.50 (2.96–19.00) | 0.007 |
| Revascularization (Yes/No) | 2.021 | 0.832 | 0.43 (0.16–1.14) | 0.212 |
| Ulceration (Yes/No) | 3.335 | 1.082 | 61.02 (0.08–1.45) | 0.002 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balmos, I.A.; Horváth, E.; Brinzaniuc, K.; Muresan, A.V.; Olah, P.; Molnár, G.B.; Nagy, E.E. Inflammation, Microcalcification, and Increased Expression of Osteopontin Are Histological Hallmarks of Plaque Vulnerability in Patients with Advanced Carotid Artery Stenosis. Biomedicines 2023, 11, 881. https://doi.org/10.3390/biomedicines11030881
Balmos IA, Horváth E, Brinzaniuc K, Muresan AV, Olah P, Molnár GB, Nagy EE. Inflammation, Microcalcification, and Increased Expression of Osteopontin Are Histological Hallmarks of Plaque Vulnerability in Patients with Advanced Carotid Artery Stenosis. Biomedicines. 2023; 11(3):881. https://doi.org/10.3390/biomedicines11030881
Chicago/Turabian StyleBalmos, Ioan Alexandru, Emőke Horváth, Klara Brinzaniuc, Adrian Vasile Muresan, Peter Olah, Gyopár Beáta Molnár, and Előd Ernő Nagy. 2023. "Inflammation, Microcalcification, and Increased Expression of Osteopontin Are Histological Hallmarks of Plaque Vulnerability in Patients with Advanced Carotid Artery Stenosis" Biomedicines 11, no. 3: 881. https://doi.org/10.3390/biomedicines11030881
APA StyleBalmos, I. A., Horváth, E., Brinzaniuc, K., Muresan, A. V., Olah, P., Molnár, G. B., & Nagy, E. E. (2023). Inflammation, Microcalcification, and Increased Expression of Osteopontin Are Histological Hallmarks of Plaque Vulnerability in Patients with Advanced Carotid Artery Stenosis. Biomedicines, 11(3), 881. https://doi.org/10.3390/biomedicines11030881