

Differential Activation of NRF2 Signaling Pathway in Renal-Cell Carcinoma Caki Cell Lines

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Renal Cancer Cell Lines
2.3. Protein Isolation and Quantification
2.4. Protein Digestion and TMT Labeling of Peptides
2.5. High pH Reversed-Phase Fractionation of TMT Labeled Peptides
2.6. Liquid Chromatography and Tandem Mass Spectrometry
2.7. Data and Bioinformatics Analysis
2.8. Western Blot Analysis
2.9. Immunofluorescence Assay
2.10. Quantitative PCR Analysis
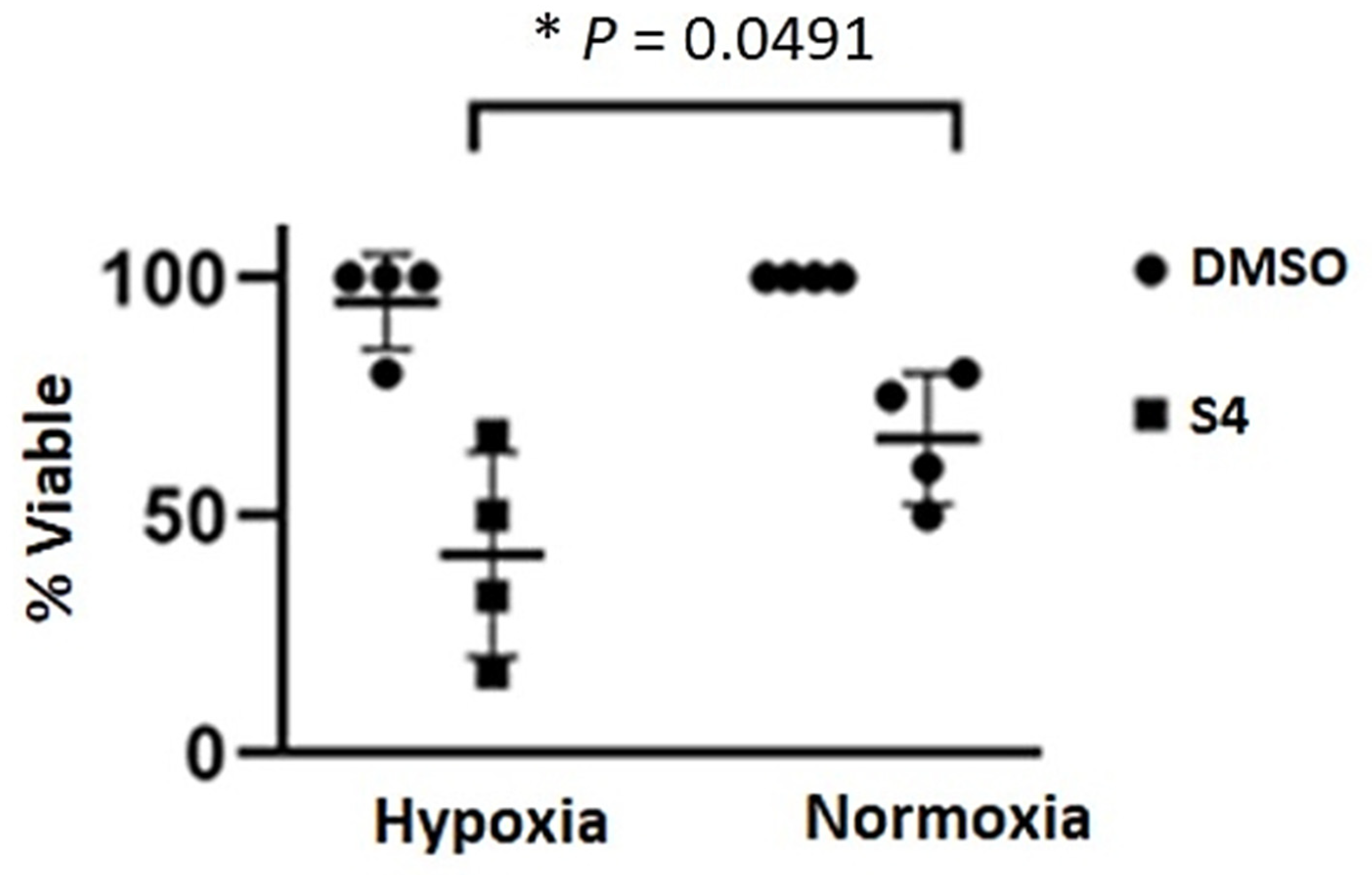
2.11. Effects of Sulfamate S4 Inhibitor on the Viability of Caki-2 Cells
2.12. Statistical Analysis
3. Results
3.1. Qualitative and Quantitative Proteomic Analysis of Caki-1 and Caki-2 Cells
3.2. Western Blot, RT-qPCR, and Immunofluorescence Assay Analysis of Differentially Expressed Proteins
3.3. Sensitivity of Caki-2 Cells to Sulfamate S4 Inhibitor
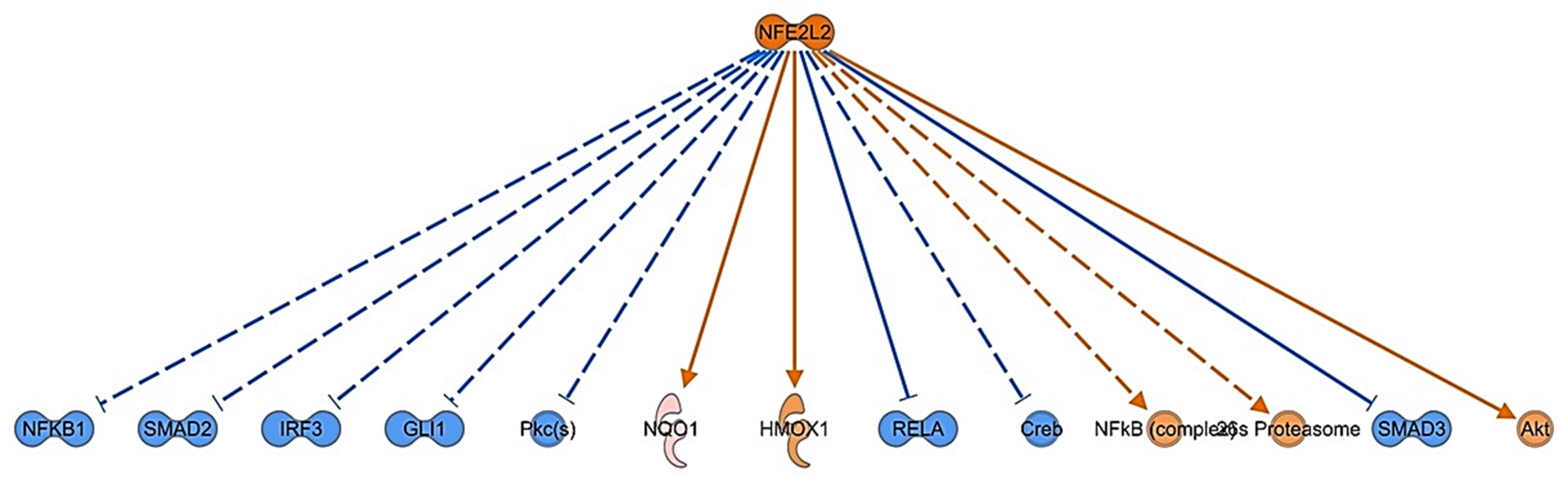
3.4. Bioinformatic Analysis of Differentially Expressed Proteins
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Available online: https://www.wcrf.org/cancer-trends/kidney-cancer-statistics/ (accessed on 20 October 2022).
- Hsieh, J.J.; Purdue, M.P.; Signoretti, S.; Swanton, C.; Albiges, L.; Schmidinger, M.; Heng, D.Y.; Larkin, J.; Ficarra, V. Renal cell carcinoma. Nat. Rev. Dis. Prim. 2017, 3, 17009. [Google Scholar] [CrossRef] [PubMed]
- Clark, D.J.; Dhanasekaran, S.M.; Petralia, F.; Pan, J.; Song, X.; Hu, Y.; da Veiga Leprevost, F.; Reva, B.; Lih, T.-S.M.; Chang, H.-Y.; et al. Integrated Proteogenomic Characterization of Clear Cell Renal Cell Carcinoma. Cell 2019, 179, 964–983.e31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, Y.; Yoshizato, T.; Shiraishi, Y.; Maekawa, S.; Okuno, Y.; Kamura, T.; Shimamura, T.; Sato-Otsubo, A.; Nagae, G.; Suzuki, H.; et al. Integrated molecular analysis of clear-cell renal cell carcinoma. Nat. Genet. 2013, 45, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; Gui, Y.; Gao, S.; Tang, A.; Hu, X.; Huang, Y.; Jia, W.; Li, Z.; He, M.; Sun, L.; et al. Frequent mutations of genes encoding ubiquitin-mediated proteolysis pathway components in clear cell renal cell carcinoma. Nat. Genet. 2011, 44, 17–19. [Google Scholar] [CrossRef] [PubMed]
- Delahunt, B.; Eble, J.N. Papillary renal cell carcinoma: A clinicopathologic and immunohistochemical study of 105 tumors. Mod. Pathol. 1997, 10, 537–544. [Google Scholar]
- Schmidt, L.; Duh, F.-M.; Chen, F.; Kishida, T.; Glenn, G.; Choyke, P.; Scherer, S.W.; Zhuang, Z.; Lubensky, I.; Dean, M.; et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat. Genet. 1997, 16, 68–73. [Google Scholar] [CrossRef]
- Lubensky, I.A.; Schmidt, L.; Zhuang, Z.; Weirich, G.; Pack, S.; Zambrano, N.; Walther, M.M.; Choyke, P.; Linehan, W.M.; Zbar, B. Hereditary and Sporadic Papillary Renal Carcinomas with c-met Mutations Share a Distinct Morphological Phenotype. Am. J. Pathol. 1999, 155, 517–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Research Network; Linehan, W.M.; Spellman, P.T.; Ricketts, C.J.; Creighton, C.J.; Fei, S.S.; Davis, C.; Wheeler, D.A.; Murray, B.A.; Schmidt, L.; et al. Comprehensive Molecular Characterization of Papillary Renal-Cell Carcinoma. N. Engl. J. Med. 2016, 374, 135–145. [Google Scholar] [CrossRef]
- Kovacs, G.; Akhtar, M.; Beckwith, B.J.; Bugert, P.; Cooper, C.S.; Delahunt, B.; Eble, J.N.; Fleming, S.; Ljungberg, B.; Medeiros, L.J.; et al. The Heidelberg classification of renal cell tumours. J. Pathol. 1997, 183, 131–133. [Google Scholar] [CrossRef]
- Linehan, W.M.; Walther, M.M.; Zbar, B. The Genetic Basis of Cancer of the Kidney. J. Urol. 2003, 170, 2163–2172. [Google Scholar] [CrossRef] [PubMed]
- Kondo, K.; Yao, M.; Kobayashi, K.; Ota, S.; Yoshida, M.; Kaneko, S.; Baba, M.; Sakai, N.; Kishida, T.; Kawakami, S.; et al. PTEN/MMAC1/TEP1 mutations in human primary renal-cell carcinomas and renal carcinoma cell lines. Int. J. Cancer. 2001, 91, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M.M.; Rathmell, W.K.; Beckermann, K.E. Modeling clear cell renal cell carcinoma and therapeutic implications. Oncogene 2020, 39, 3413–3426. [Google Scholar] [CrossRef] [Green Version]
- Sinha, R.; Winer, A.G.; Chevinsky, M.; Jakubowski, C.; Chen, Y.-B.; Dong, Y.; Tickoo, S.K.; Reuter, V.E.; Russo, P.; Coleman, J.A.; et al. Analysis of renal cancer cell lines from two major resources enables genomics-guided cell line selection. Nat. Commun. 2017, 8, 15165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, S.V.; Haber, D.A.; Settleman, J. Cell line-based platforms to evaluate the therapeutic efficacy of candidate anticancer agents. Nat. Rev. Cancer 2010, 10, 241–253. [Google Scholar] [CrossRef]
- Shoemaker, R.H. The NCI60 human tumour cell line anticancer drug screen. Nat. Rev. Cancer 2006, 6, 813–823. [Google Scholar] [CrossRef]
- Koch, E.; Finne, K.; Eikrem, Ø.; Landolt, L.; Beisland, C.; Leh, S.; Delaleu, N.; Granly, M.; Vikse, B.E.; Osman, T.; et al. Transcriptome-proteome integration of archival human renal cell carcinoma biopsies enables identification of molecular mechanisms. Am. J. Physiol. Physiol. 2019, 316, F1053–F1067. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Liu, X.L.; Fu, Z.Q.; Qiu, P.; Feng, T.; Yan, B.; Wei, J.L. LINC00675 suppresses proliferative, migration and invasion of clear cell renal cell carcinoma via the Wnt/β-catenin pathway. Eur. Rev. Med. Pharmacol. Sci. 2020, 5, 2313–2320. [Google Scholar] [CrossRef]
- Lichner, Z.; Saleh, C.; Subramaniam, V.; Seivwright, A.; Prud’Homme, G.J.; Yousef, G.M. miR-17 inhibition enhances the formation of kidney cancer spheres with stem cell/tumor initiating cell properties. Oncotarget 2014, 6, 5567–5581. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.-H.; Lin, C.-Y.; Lin, W.-C.; Tang, S.-W.; Lai, M.-K.; Lin, J.-Y. Up-Regulation of Vascular Endothelial Growth Factor-D Expression in Clear Cell Renal Cell Carcinoma by CD74: A Critical Role in Cancer Cell Tumorigenesis. J. Immunol. 2008, 181, 6584–6594. [Google Scholar] [CrossRef] [Green Version]
- Miyake, M.; Goodison, S.; Lawton, A.; Zhang, G.; Gomes-Giacoia, E.; Rosser, C.J. Erythropoietin is a JAK2 and ERK1/2 effector that can promote renal tumor cell proliferation under hypoxic conditions. J. Hematol. Oncol. 2013, 6, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glube, N.; Giessl, A.; Wolfrum, U.; Langguth, P. Caki-1 Cells Represent an in vitro Model System for Studying the Human Proximal Tubule Epithelium. Nephron Exp. Nephrol. 2007, 107, e47–e56. [Google Scholar] [CrossRef] [PubMed]
- Shinojima, T.; Oya, M.; Takayanagi, A.; Mizuno, R.; Shimizu, N.; Murai, M. Renal cancer cells lacking hypoxia inducible factor (HIF)-1 expression maintain vascular endothelial growth factor expression through HIF-2. Carcinogenesis 2006, 28, 529–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pulkkanen, K.J.; Parkkinen, J.J.; Kettunen, M.; A Kauppinen, R.A.; Lappalainen, M.; Ala-Opas, M.Y.; Ylä-Herttuala, S. Characterization of a new animal model for human renal cell carcinoma. Vivo 2000, 14, 393–400. [Google Scholar]
- Pulkkanen, K.J.; Parkkinen, J.J.; Laukkanen, J.M.; Kettunen, M.I.; Tyynela, K.; Kauppinen, R.A.; Ala-Opas, M.Y.; Yla-Herttuala, S. HSV-tk gene therapy for human renal cell carcinoma in nude mice. Cancer Gene Ther. 2001, 8, 529–536. [Google Scholar] [CrossRef] [Green Version]
- Furge, K.A.; Chen, J.; Koeman, J.; Swiatek, P.; Dykema, K.; Lucin, K.; Kahnoski, R.; Yang, X.J.; Teh, B.T. Detection of DNA Copy Number Changes and Oncogenic Signaling Abnormalities from Gene Expression Data Reveals MYC Activation in High-Grade Papillary Renal Cell Carcinoma. Cancer Res 2007, 67, 3171–3176. [Google Scholar] [CrossRef] [Green Version]
- Looyenga, B.D.; Furge, K.A.; Dykema, K.J.; Koeman, J.; Swiatek, P.J.; Giordano, T.J.; West, A.B.; Resau, J.H.; Teh, B.T.; MacKeigan, J.P. Chromosomal amplification of leucine-rich repeat kinase-2 (LRRK2) is required for oncogenic MET signaling in papillary renal and thyroid carcinomas. Proc. Natl. Acad. Sci. USA 2011, 108, 1439–1444. [Google Scholar] [CrossRef] [Green Version]
- Furge, K.A.; Dykema, K.; Petillo, D.; Westphal, M.; Zhang, Z.; Kort, E.J.; Teh, B.T. Combining differential expression, chromosomal and pathway analyses for the molecular characterization of renal cell carcinoma. Can. Urol. Assoc. J. 2012, 1, S21–S27. [Google Scholar] [CrossRef] [Green Version]
- Kucejova, B.; Peña-Llopis, S.; Yamasaki, T.; Sivanand, S.; Tran, T.A.T.; Alexander, S.; Wolff, N.C.; Lotan, Y.; Xie, X.-J.; Kabbani, W.; et al. Interplay Between pVHL and mTORC1 Pathways in Clear-Cell Renal Cell Carcinoma. Mol. Cancer Res. 2011, 9, 1255–1265. [Google Scholar] [CrossRef] [Green Version]
- Ashida, S.; Nishimori, I.; Tanimura, M.; Onishi, S.; Shuin, T. Effects of von Hippel-Lindau gene mutation and methylation status on expression of transmembrane carbonic anhydrases in renal cell carcinoma. J. Cancer Res. Clin. Oncol. 2002, 128, 561–568. [Google Scholar] [CrossRef]
- Blondeau, J.J.; Deng, M.; Syring, I.; Schrödter, S.; Schmidt, D.; Perner, S.; Müller, S.C.; Ellinger, J. Identification of novel long non-coding RNAs in clear cell renal cell carcinoma. Clin. Epigenetics 2015, 7, 10. [Google Scholar] [CrossRef] [Green Version]
- Zaravinos, A.; Pieri, M.; Mourmouras, N.; Anastasiadou, N.; Zouvani, I.; Delakas, D.; Deltas, C. Altered metabolic pathways in clear cell renal cell carcinoma: A meta-analysis and validation study focused on the deregulated genes and their associated networks. Oncoscience 2014, 1, 117–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roos, F.C.; Evans, A.J.; Brenner, W.; Wondergem, B.; Klomp, J.; Heir, P.; Roche, O.; Thomas, C.; Schimmel, H.; Furge, K.A.; et al. Deregulation of E2-EPF Ubiquitin Carrier Protein in Papillary Renal Cell Carcinoma. Am. J. Pathol. 2011, 178, 853–860. [Google Scholar] [CrossRef] [Green Version]
- Brannon, A.R.; Reddy, A.; Seiler, M.; Arreola, A.; Moore, D.T.; Pruthi, R.S.; Wallen, E.M.; Nielsen, M.E.; Liu, H.; Nathanson, K.L.; et al. Molecular Stratification of Clear Cell Renal Cell Carcinoma by Consensus Clustering Reveals Distinct Subtypes and Survival Patterns. Genes Cancer 2010, 1, 152–163. [Google Scholar] [CrossRef] [PubMed]
- Brannon, A.R.; Haake, S.M.; Hacker, K.E.; Pruthi, R.S.; Wallen, E.M.; Nielsen, M.E.; Rathmell, W.K. Meta-analysis of Clear Cell Renal Cell Carcinoma Gene Expression Defines a Variant Subgroup and Identifies Gender Influences on Tumor Biology. Eur. Urol. 2012, 61, 258–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burch, T.C.; Morris, M.A.; Campbell-Thompson, M.; Pugliese, A.; Nadler, J.L.; Nyalwidhe, J.O. Proteomic Analysis of Disease Stratified Human Pancreas Tissue Indicates Unique Signature of Type 1 Diabetes. PLoS ONE 2015, 10, e0135663. [Google Scholar] [CrossRef]
- Burch, T.C.; Rhim, J.S.; Nyalwidhe, J.O. Mitochondria Biogenesis and Bioenergetics Gene Profiles in Isogenic Prostate Cells with Different Malignant Phenotypes. BioMed. Res. Int. 2016, 2016, 1785201. [Google Scholar] [CrossRef] [Green Version]
- Neri, D.; Supuran, C.T. Interfering with pH regulation in tumours as a therapeutic strategy. Nat. Rev. Drug Discov. 2011, 10, 767–777. [Google Scholar] [CrossRef] [Green Version]
- Lou, Y.; McDonald, P.C.; Oloumi, A.; Chia, S.; Ostlund, C.; Ahmadi, A.; Kyle, A.; Keller, U.A.D.; Leung, S.; Huntsman, D.; et al. Targeting Tumor Hypoxia: Suppression of Breast Tumor Growth and Metastasis by Novel Carbonic Anhydrase IX Inhibitors. Cancer Res. 2011, 71, 3364–3376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pacchiano, F.; Carta, F.; McDonald, P.C.; Lou, Y.; Vullo, D.; Scozzafava, A.; Dedhar, S.; Supuran, C.T. Ureido-Substituted Benzenesulfonamides Potently Inhibit Carbonic Anhydrase IX and Show Antimetastatic Activity in a Model of Breast Cancer Metastasis. J. Med. Chem. 2011, 54, 1896–1902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gieling, R.G.; Babur, M.; Mamnani, L.; Burrows, N.; Telfer, B.A.; Carta, F.; Winum, J.-Y.; Scozzafava, A.; Supuran, C.T.; Williams, K.J. Antimetastatic Effect of Sulfamate Carbonic Anhydrase IX Inhibitors in Breast Carcinoma Xenografts. J. Med. Chem. 2012, 55, 5591–5600. [Google Scholar] [CrossRef]
- Strober, W. Trypan Blue Exclusion Test of Cell Viability. Curr. Protoc. Immunol. 2015, 111, A3.B.1–A3.B.3. [Google Scholar] [CrossRef]
- Lang, M.; Vocke, C.D.; Ricketts, C.J.; Metwalli, A.R.; Ball, M.W.; Schmidt, L.S.; Linehan, W.M. Clinical and Molecular Characterization of Microphthalmia-associated Transcription Factor (MITF)-related Renal Cell Carcinoma. Urology 2020, 149, 89–97. [Google Scholar] [CrossRef]
- Williamson, J.C.; Edwards, A.V.G.; Verano-Braga, T.; Schwämmle, V.; Kjeldsen, F.; Jensen, O.N.; Larsen, M.R. High-performance hybrid Orbitrap mass spectrometers for quantitative proteome analysis: Observations and implications. Proteomics 2016, 16, 907–914. [Google Scholar] [CrossRef] [PubMed]
- Penning, T.M.; Burczynski, M.E.; Jez, J.M.; Hung, C.-F.; Lin, H.-K.; Ma, H.; Moore, M.; Palackal, N.; Ratnam, K. Human 3α-hydroxysteroid dehydrogenase isoforms (AKR1C1–AKR1C4) of the aldo-keto reductase superfamily: Functional plasticity and tissue distribution reveals roles in the inactivation and formation of male and female sex hormones. Biochem. J. 2000, 351, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Duan, L.; Lee, S.H.; Kloosterboer, H.J.; Blair, I.A.; Penning, T.M. Human Cytosolic Hydroxysteroid Dehydrogenases of the Aldo-ketoreductase Superfamily Catalyze Reduction of Conjugated Steroids: Implications for phase I and phase II steroid hormone metabolism. J. Biol. Chem. 2009, 284, 10013–10022. [Google Scholar] [CrossRef] [Green Version]
- Beranič, N.; Gobec, S.; Rižner, T.L. Progestins as inhibitors of the human 20-ketosteroid reductases, AKR1C1 and AKR1C3. Chem. Interact. 2011, 191, 227–233. [Google Scholar] [CrossRef]
- Beranič, N.; Brožič, P.; Brus, B.; Sosič, I.; Gobec, S.; Rižner, T.L. Expression of human aldo-keto reductase 1C2 in cell lines of peritoneal endometriosis: Potential implications in metabolism of progesterone and dydrogesterone and inhibition by progestins. J. Steroid Biochem. Mol. Biol. 2012, 130, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Duan, L.; Chen, M.; Penning, T.M.; Kloosterboer, H.J. Metabolism of the synthetic progestogen norethynodrel by human ketosteroid reductases of the aldo–keto reductase superfamily. J. Steroid Biochem. Mol. Biol. 2012, 129, 139–144. [Google Scholar] [CrossRef] [Green Version]
- Penning, T.M.; Byrns, M.C. Steroid Hormone Transforming Aldo-Keto Reductases and Cancer. Ann. N. Y. Acad. Sci. 2009, 1155, 33–42. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Chang, L.-L.; Yan, F.-J.; Hu, Y.; Zeng, C.-M.; Zhou, T.-Y.; Yuan, T.; Ying, M.-D.; Cao, J.; He, Q.-J.; et al. AKR1C1 Activates STAT3 to Promote the Metastasis of Non-Small Cell Lung Cancer. Theranostics 2018, 8, 676–692. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.-M.; Chang, Y.-C.; Yang, Y.-C.; Lin, S.-K.; Chang, P.M.-H.; Hsiao, M. AKR1C1 controls cisplatin-resistance in head and neck squamous cell carcinoma through cross-talk with the STAT1/3 signaling pathway. J. Exp. Clin. Cancer Res. 2019, 38, 245. [Google Scholar] [CrossRef] [Green Version]
- Huebbers, C.U.; Verhees, F.; Poluschkin, L.; Olthof, N.C.; Kolligs, J.; Siefer, O.G.; Henfling, M.; Ramaekers, F.C.; Preuss, S.F.; Beutner, D.; et al. Upregulation of AKR1C1 and AKR1C3 expression in OPSCC with integrated HPV16 and HPV-negative tumors is an indicator of poor prognosis. Int. J. Cancer 2019, 144, 2465–2477. [Google Scholar] [CrossRef] [PubMed]
- Ji, Q.; Aoyama, C.; Nien, Y.-D.; Liu, P.I.; Chen, P.K.; Chang, L.; Stanczyk, F.Z.; Stolz, A. Selective Loss of AKR1C1 and AKR1C2 in Breast Cancer and Their Potential Effect on Progesterone Signaling. Cancer Res 2004, 64, 7610–7617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, Y.; Lin, M.; Ou, D.; Huang, Z.; Shen, P. A novel ferroptosis-related 12-gene signature predicts clinical prognosis and reveals immune relevancy in clear cell renal cell carcinoma. BMC Cancer 2021, 21, 831. [Google Scholar] [CrossRef]
- Endo, S.; Matsunaga, T.; Nishinaka, T. The Role of AKR1B10 in Physiology and Pathophysiology. Metabolites 2021, 11, 332. [Google Scholar] [CrossRef]
- Cao, D.; Fan, S.T.; Chung, S.S.M. Identification and Characterization of a Novel Human Aldose Reductase-like Gene. J. Biol. Chem. 1998, 273, 11429–11435. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, D.; Ding, L.; Sivaprasad, U.; Geh, E.; Myers, J.B.; Bernstein, J.A.; Hershey, G.K.K.; Mersha, T.B. Multiple Transcriptome Data Analysis Reveals Biologically Relevant Atopic Dermatitis Signature Genes and Pathways. PLoS ONE 2015, 10, e0144316. [Google Scholar] [CrossRef] [Green Version]
- Penning, T.M. The aldo-keto reductases (AKRs): Overview. Chem. Interact. 2014, 234, 236–246. [Google Scholar] [CrossRef] [Green Version]
- Soares, C.T.; Fachin, L.R.V.; Trombone, A.P.F.; Rosa, P.S.; Ghidella, C.C.; Belone, A.F.F. Potential of AKR1B10 as a Biomarker and Therapeutic Target in Type 2 Leprosy Reaction. Front. Med. 2018, 5, 263. [Google Scholar] [CrossRef]
- Ohashi, T.; Idogawa, M.; Sasaki, Y.; Suzuki, H.; Tokino, T. AKR1B10, a Transcriptional Target of p53, Is Downregulated in Colorectal Cancers Associated with Poor Prognosis. Mol. Cancer Res. 2013, 11, 1554–1563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, Y.T.; Matkowskyj, K.A.; Li, H.; Bai, H.; Zhang, W.; Tsao, M.-S.; Liao, J.; Yang, G.-Y. Overexpression and oncogenic function of aldo-keto reductase family 1B10 (AKR1B10) in pancreatic carcinoma. Mod. Pathol. 2012, 25, 758–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, S.M.U.; Jiang, Z.N.; Zheng, Z.H.; Li, Y.; Wang, X.J.; Tang, X. AKR1B10 expression predicts response of gastric cancer to neoadjuvant chemotherapy. Oncol. Lett. 2019, 17, 773–780. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Yan, R.; Al-Salman, A.; Shen, Y.; Bu, Y.; Ma, J.; Luo, D.-X.; Huang, C.; Jiang, Y.; Wilber, A.; et al. Epidermal growth factor induces tumour marker AKR1B10 expression through activator protein-1 signalling in hepatocellular carcinoma cells. Biochem. J. 2012, 442, 273–282. [Google Scholar] [CrossRef]
- He, Y.-C.; Shen, Y.; Cao, Y.; Tang, F.-Q.; Tian, D.-F.; Huang, C.-F.; Tao, H.; Zhou, F.-L.; Zhang, B.; Song, L.; et al. Overexpression of AKR1B10 in nasopharyngeal carcinoma as a potential biomarker. Cancer Biomark. 2016, 16, 127–135. [Google Scholar] [CrossRef]
- de la Vega, M.R.; Cholanians, A.B.; Schmidlin, C.J.; Chapman, E.; Zhang, D.D. Modulating NRF2 in Disease: Timing Is Everything. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 555–575. [Google Scholar] [CrossRef]
- De La Vega, M.R.; Chapman, E.; Zhang, D.D. NRF2 and the Hallmarks of Cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar] [CrossRef]
- Schmidlin, C.J.; Dodson, M.B.; Zhang, D.D. Filtering through the role of NRF2 in kidney disease. Arch. Pharmacal Res. 2019, 43, 361–369. [Google Scholar] [CrossRef]
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.-L.; Kensler, T.W.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317. [Google Scholar] [CrossRef] [Green Version]
- Zimta, A.-A.; Cenariu, D.; Irimie, A.; Magdo, L.; Nabavi, S.M.; Atanasov, A.G.; Berindan-Neagoe, I. The Role of Nrf2 Activity in Cancer Development and Progression. Cancers 2019, 11, 1755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menegon, S.; Columbano, A.; Giordano, S. The Dual Roles of NRF2 in Cancer. Trends Mol. Med. 2016, 22, 578–593. [Google Scholar] [CrossRef]
- Taguchi, K.; Yamamoto, M. The KEAP1–NRF2 System in Cancer. Front. Oncol. 2017, 7, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kansanen, E.; Kuosmanen, S.M.; Leinonen, H.; Levonen, A.-L. The Keap1-Nrf2 pathway: Mechanisms of activation and dysregulation in cancer. Redox Biol. 2013, 1, 45–49. [Google Scholar] [CrossRef] [Green Version]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjöstedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A pathology atlas of the human cancer transcriptome. Science 2017, 357, eaan2507. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Shen, J.; Chen, Y.; Gao, J.; Yao, J. Fatty acid metabolism-related signature predicts survival in patients with clear cell renal carcinoma. Aging 2022, 14, 9969–9979. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Huang, M.; Kong, L.; Li, Y. miR-372 suppresses tumour proliferation and invasion by targeting IGF2BP1 in renal cell carcinoma. Cell Prolif. 2015, 48, 593–599. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Protein Accession | Abundance Ratio (Caki-2/Caki-1). | Gene Symbol | Protein Accession | Abundance Ratio (Caki-2/Caki-1) |
|---|---|---|---|---|---|
| AKR1C2 | P52895 | 37.660 | TCEAL3 | Q969E4 | −50.000 |
| CA12 | O43570 | 28.951 | CA3 | P07451 | −37.037 |
| TPPP3 | Q9BW30 | 27.533 | TPM2 | P07951 | −37.037 |
| AKR1B10 | O60218 | 26.012 | ADAMTSL1 | Q8N6G6 | −33.333 |
| PDZK1 | Q5T2W1 | 25.480 | YLPM1 | P49750 | −33.333 |
| FABP6 | P51161 | 24.132 | IGF2BP1 | Q9NZI8 | −32.258 |
| CYP2S1 | Q96SQ9 | 23.136 | VAMP5 | O95183 | −30.303 |
| SNCG | O76070 | 22.877 | CXCL6 | P80162 | −30.303 |
| SFN | P31947 | 21.316 | PRDX2 | P32119 | −27.778 |
| SPINT2 | O43291 | 21.130 | FBN2 | P35556 | −25.000 |
| SERPINB5 | P36952 | 19.572 | BASP1 | P80723 | −25.000 |
| NEFL | P07196 | 17.122 | BCAT1 | P54687 | −22.222 |
| GLDC | P23378 | 16.902 | TRIM22 | Q8IYM9 | −21.277 |
| NAPRT | Q6XQN6 | 16.742 | SERPINE1 | P05121 | −19.608 |
| TYMP | P19971 | 16.403 | GSTP1 | P09211 | −19.231 |
| Top Canonical Pathways | ||
|---|---|---|
| Name | p-Value | Overlap |
| NRF2-mediated Oxidative Stress Response | 5.76 × 10−11 | 7.6% 18/237 |
| LPS/IL-1-mediated Inhibition of RXR Function | 1.40 × 10−9 | 6.7% 17/254 |
| Xenobiotic Metabolism AHR Signaling Pathway | 2.29 × 10−8 | 11.5% 10/87 |
| Xenobiotic Metabolism CAR Signaling Pathway | 1.04 × 10−7 | 6.8% 13/191 |
| Xenobiotic Metabolism PXR Signaling Pathway | 1.10 × 10−7 | 6.8% 13/192 |
| Molecular and Cellular Functions | ||
| Name | p-value range | # Molecules |
| Cellular Movement | 1.46 × 10−6–9.26 × 10−25 | 117 |
| Cell Death and Survival | 6.02 × 10−7–3.90 × 10−21 | 128 |
| Cell Morphology | 8.27 × 10−7–1.04 × 10−17 | 71 |
| Cell-To-Cell Signaling and Interaction | 1.53 × 10−6–1.46 × 10−15 | 84 |
| Cellular Development | 1.59 × 10−6–4.45 × 10−14 | 123 |
| Top Networks | ||
| Associated Network Functions | Score | |
| Cellular Assembly and Organization, Cellular Comprise, Cellular Function, and Maintenance | 60 | |
| Cell-To-Cell Signaling and Interaction, Cancer, Endocrine System Disorders | 46 | |
| Cellular Assembly and Organization, Cell Morphology, Cellular Movement | 41 | |
| Drug Metabolism, Liver Hyperbilirubinemia, Metabolomics Disease | 32 | |
| Vitamin and Mineral Metabolism, Carbohydrate Metabolism, Drug Metabolism | 27 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hitefield, N.L.; Mackay, S.; Hays, L.E.; Chen, S.; Oduor, I.O.; Troyer, D.A.; Nyalwidhe, J.O. Differential Activation of NRF2 Signaling Pathway in Renal-Cell Carcinoma Caki Cell Lines. Biomedicines 2023, 11, 1010. https://doi.org/10.3390/biomedicines11041010
Hitefield NL, Mackay S, Hays LE, Chen S, Oduor IO, Troyer DA, Nyalwidhe JO. Differential Activation of NRF2 Signaling Pathway in Renal-Cell Carcinoma Caki Cell Lines. Biomedicines. 2023; 11(4):1010. https://doi.org/10.3390/biomedicines11041010
Chicago/Turabian StyleHitefield, Naomi L., Stephen Mackay, Lauren E. Hays, Shimin Chen, Ian O. Oduor, Dean A. Troyer, and Julius O. Nyalwidhe. 2023. "Differential Activation of NRF2 Signaling Pathway in Renal-Cell Carcinoma Caki Cell Lines" Biomedicines 11, no. 4: 1010. https://doi.org/10.3390/biomedicines11041010
APA StyleHitefield, N. L., Mackay, S., Hays, L. E., Chen, S., Oduor, I. O., Troyer, D. A., & Nyalwidhe, J. O. (2023). Differential Activation of NRF2 Signaling Pathway in Renal-Cell Carcinoma Caki Cell Lines. Biomedicines, 11(4), 1010. https://doi.org/10.3390/biomedicines11041010