
Strategies to Re-Sensitize Castration-Resistant Prostate Cancer to Antiandrogen Therapy
 ,
,
Abstract
1. Introduction
- -
- Finding new drugs with different mechanisms of action from those previously used;
- -
- Using combination therapies to boost synergies, enhancing the action of each drug;
- -
- Research treatments that can resensitize tumor cells to previously used mechanisms of action.
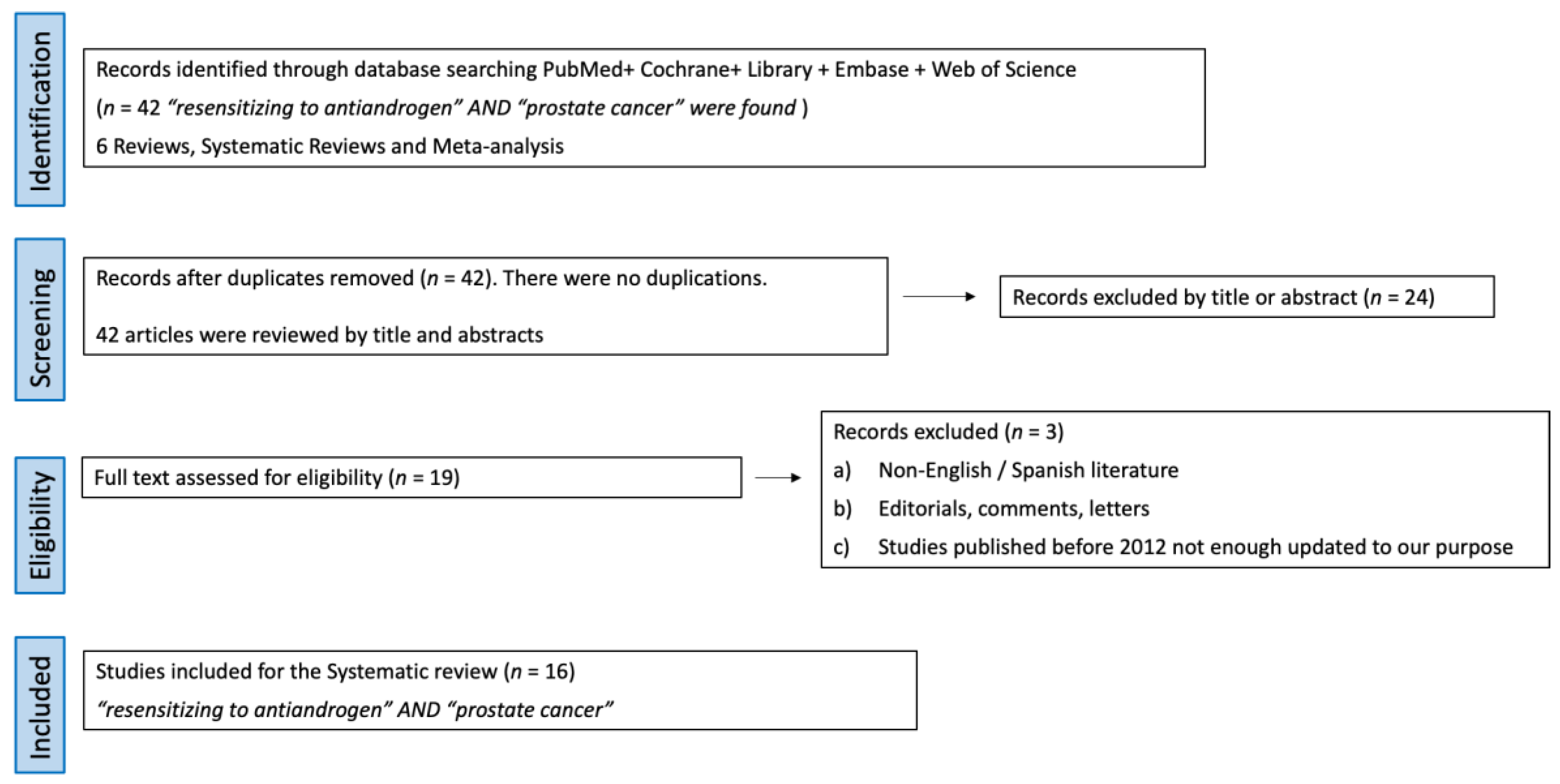
2. Evidence Acquisition
- Studies identification: 42 articles using the terms “resensitizing to antiandrogen” AND “prostate cancer” were found. Out of them, 6 were reviews and systematic reviews, while 36 were original articles. The consistency of this revision is affected by the inherent lack of robust evidence in urology.
- Screening: No articles were duplicated, so 42 articles were screened by title and abstract. Out of them, 24 full-text articles were assessed for eligibility.
- Eligibility: The selection criteria were (a) reviews, original articles, and preclinical studies, (b) studies about castration resistance mechanisms and strategies to resensitize cells to androgen deprivation therapy. Exclusion criteria were (a) non-English/Spanish literature, (b) editorials, comments, and letters, and (c) studies published before 2012, as they were not updated enough for our purpose.
- Study analysis: After applying the above eligibility criteria, 19 studies about castration resistance mechanism and strategies to resensitize cells to androgen deprivation therapy were selected. In total, 16 articles with the highest level of relevance to the discussed topics were selected with the consensus of the authors. A review from the selected studies was conducted.
3. The Androgen Receptor
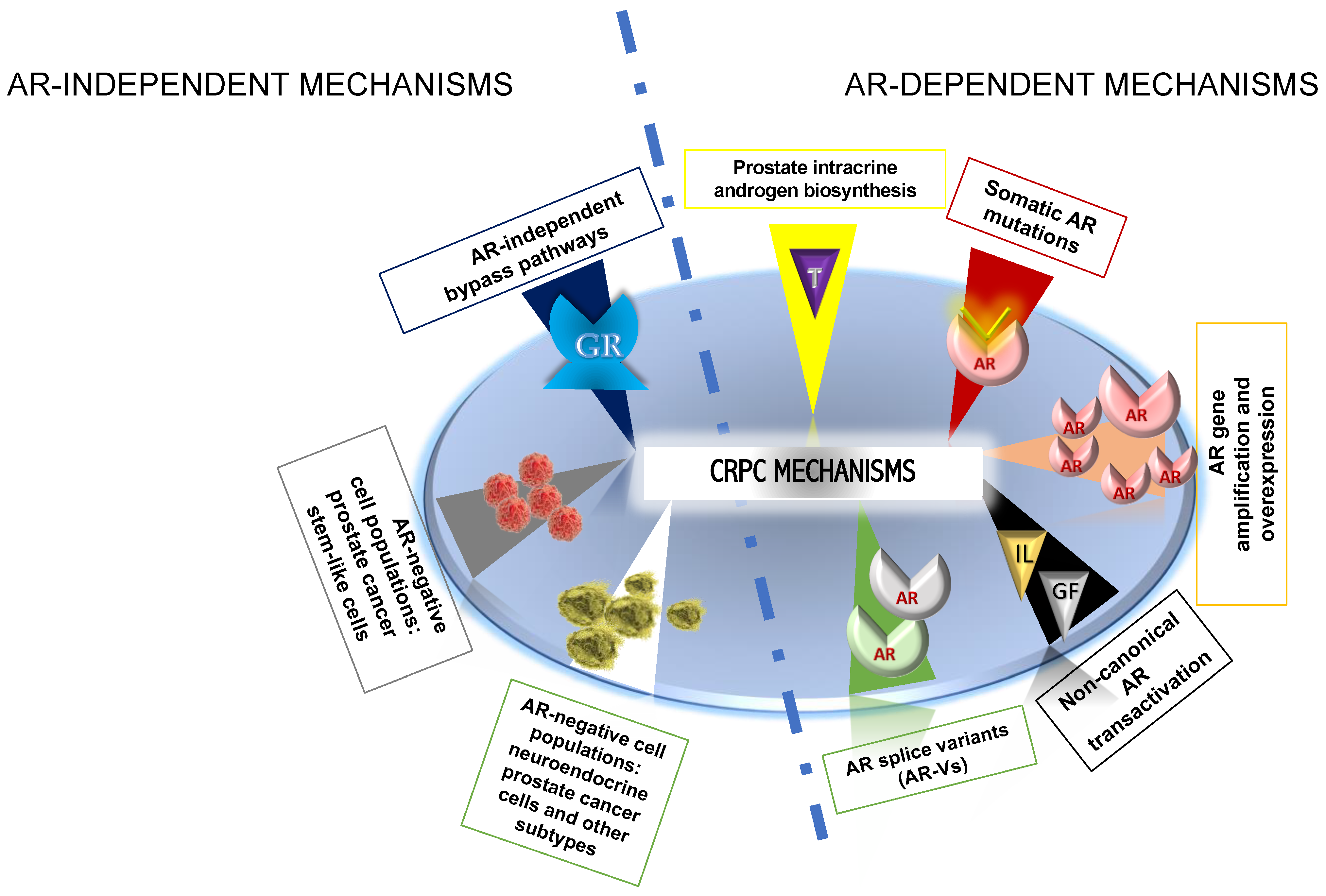
4. Castration Resistance Mechanisms
4.1. AR-Dependent Mechanisms Triggering CRPC
4.1.1. AR gene Amplification and Overexpression
4.1.2. Somatic AR Mutations
4.1.3. Prostate Intracrine Androgen Biosynthesis
4.1.4. AR Splice Variants (AR-Vs)
4.1.5. Non-Canonical AR Transactivation
4.2. AR-Independent Mechanisms Triggering CRPC
4.2.1. AR-Independent Bypass Pathways
4.2.2. AR-Negative Cell Populations: PCa Stem-Like Cells
4.2.3. AR-Negative Cell Populations: Neuroendocrine PCa Cells and Other Subtypes
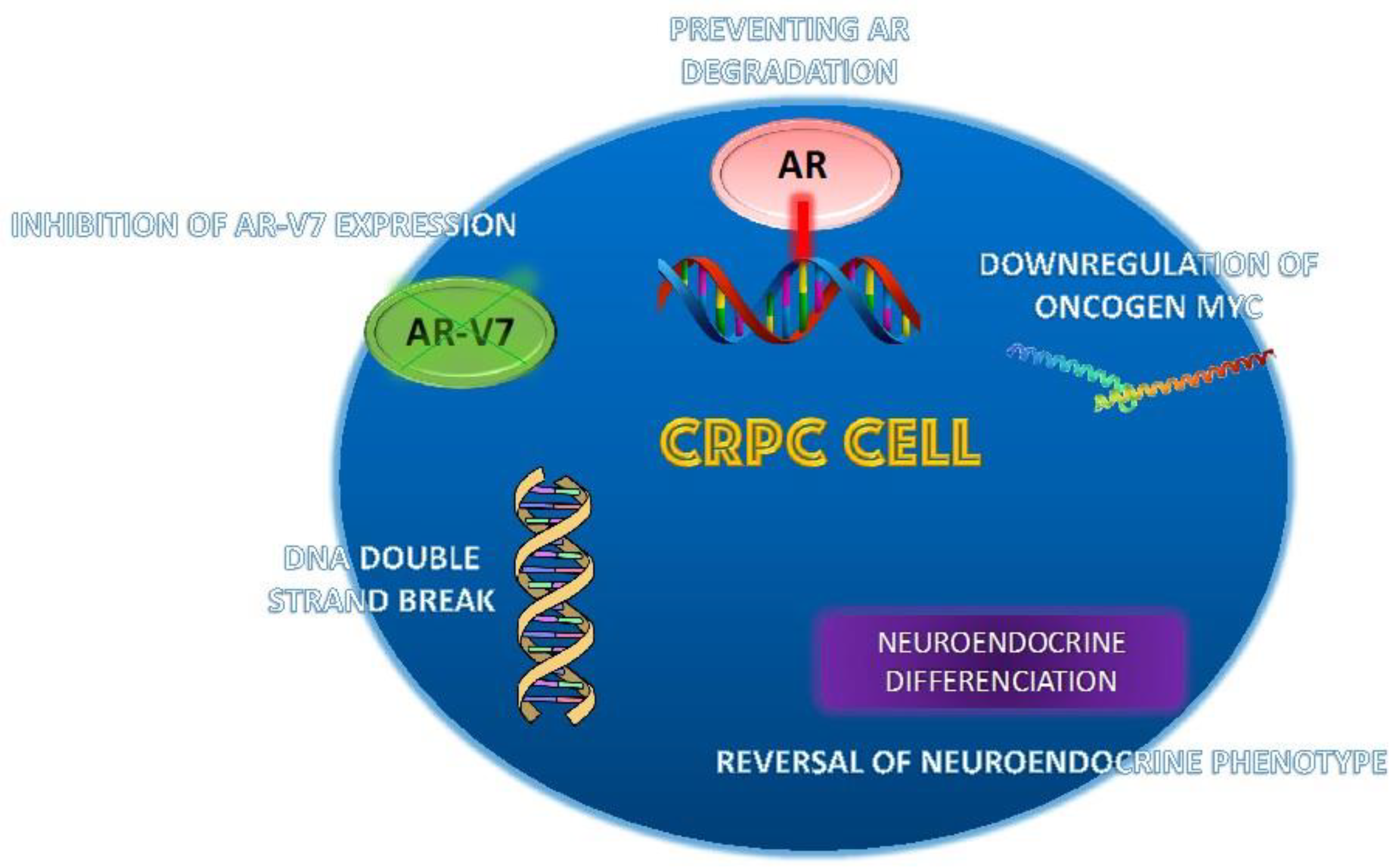
5. Agents and Strategies to Resensitize CRPC to Antiandrogen Therapy
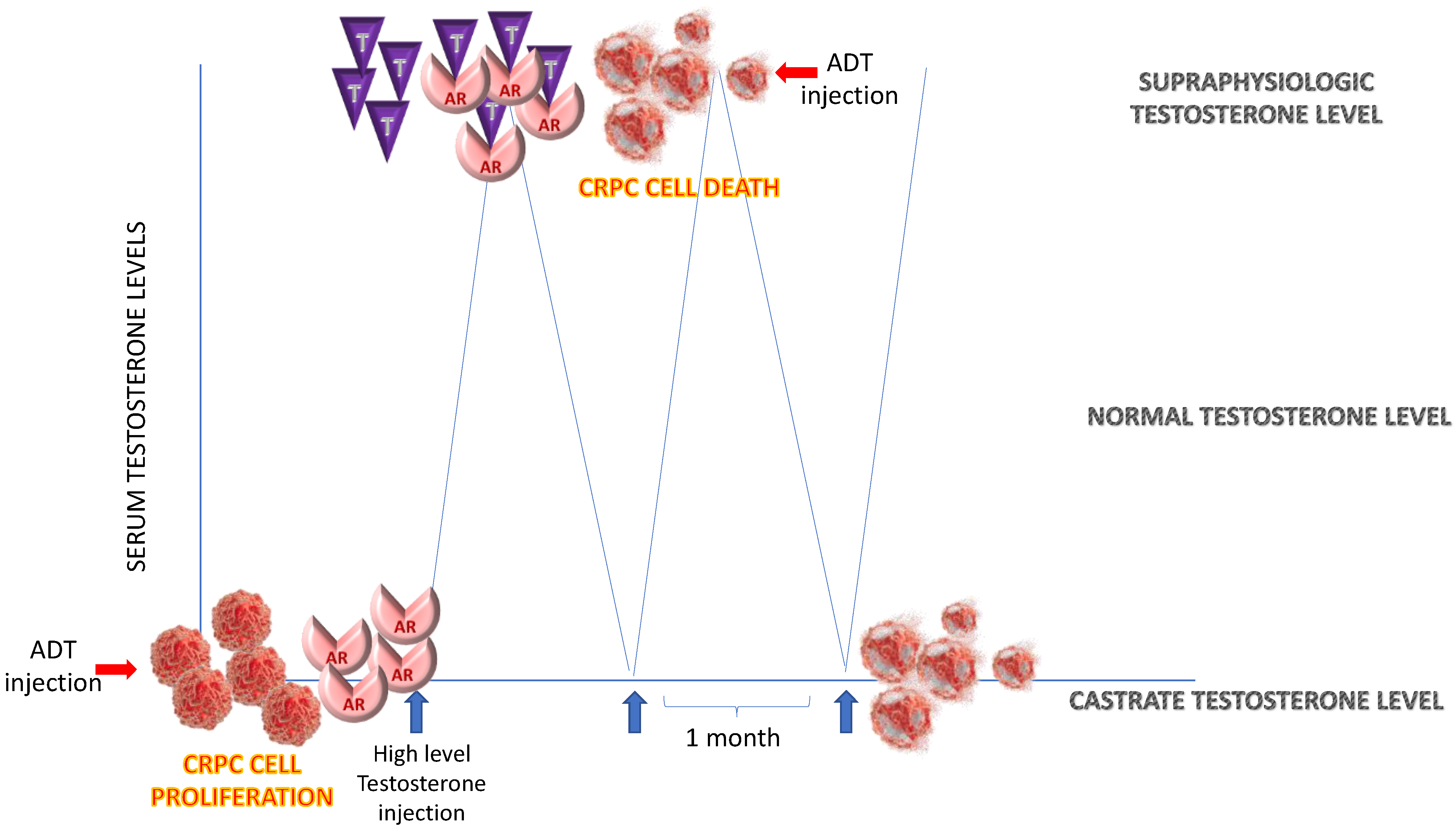
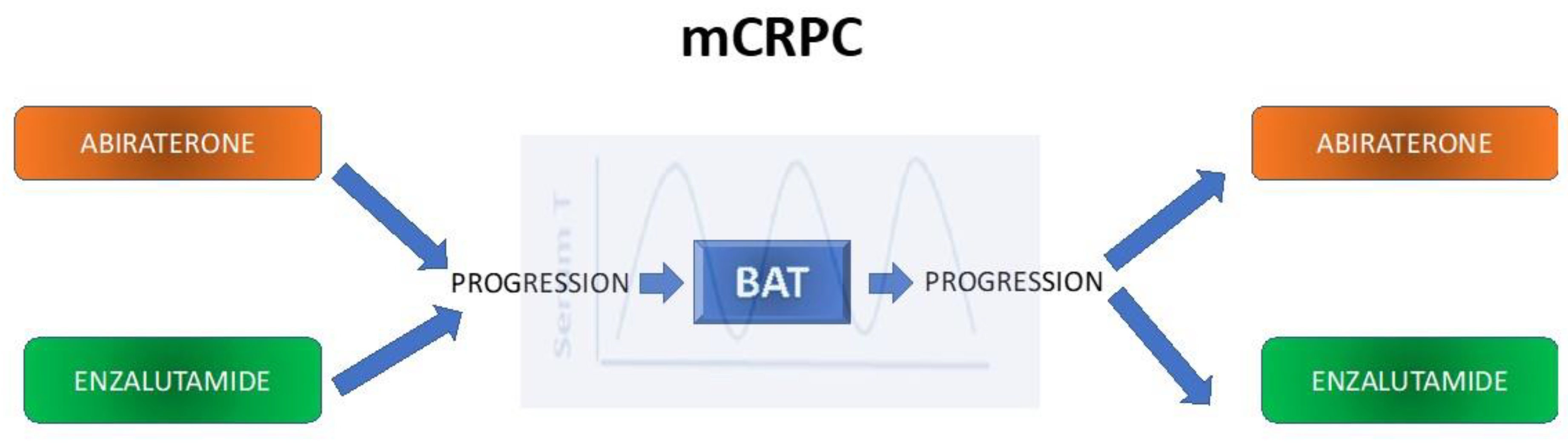
5.1. Bipolar Androgen Therapy
5.2. Other Agents Capable of Resensitizing CRPC to Antiandrogen Therapy
5.2.1. Niclosamide
5.2.2. Monoamine Oxidase A Inhibitors (MAOAIs)
5.2.3. Notch Receptors
5.2.4. Indomethacin
5.2.5. Lapatinib
5.2.6. Panobinostat
5.2.7. EPI-7386
5.2.8. Autophagy Inhibitors
5.2.9. Antisense Oligonucleotides (ASO)
5.2.10. Bet Pathway
5.2.11. Dasatinib
5.2.12. Ipatasertib
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ADT | Androgen deprivation therapy |
| AR | Androgen receptor |
| ARAmw | Androgen receptor activity Mann–Whitney score |
| AREs | androgen response elements |
| ARSis | Androgen receptor signaling inhibitors |
| AR-Vs | Androgen receptor splice variants |
| ASO | Antisense oligonucleotides |
| BAT | Bipolar androgen therapy |
| BET | Bromodomain and extra-terminal |
| CRPC | Castration-resistant prostate cancer |
| DBD | DNA-binding domain |
| DHT | Dihydrotestosterone |
| DSBs | Double-strand breaks |
| EA | Adverse events |
| EZH2 | Histone lysine-N-methyltransferase |
| GR | Glucocorticoid receptor |
| HDACs | Histone deacetylases |
| HDACIs | HDAC inhibitors |
| IGF-1 | Insulin-like growth factor-1 |
| IL-6 | Interleukin-6 |
| IR | Ionizing radiation |
| LBD | Ligand-binding domain |
| LHRH | Luteinizing hormone-releasing hormone |
| MAOAIs | Monoamine oxidase A inhibitors |
| mCRPC | Metastatic castration-resistant prostate cancer |
| NE | Neuroendocrine |
| NEPC | Neuroendocrine prostate cancer |
| NTD | Terminal transactivation domain |
| NK | Natural killer |
| OR | Objective response |
| ORR | Objective response rate |
| PARP | Poly ADP-ribose polymerase |
| PCa | Prostate cancer |
| PCSCs | Prostate cancer stem-like cells |
| PFS | Progression-free survival |
| PSA50 | ≥50% PSA reduction |
| Retinoblastoma | Rb |
| TICs | Tumor-initiating cells |
References
- Dyba, T.; Randi, G.; Bray, F.; Martos, C.; Giusti, F.; Nicholson, N.; Gavin, A.; Flego, M.; Neamtiu, L.; Dimitrova, N.; et al. The European cancer burden in 2020: Incidence and mortality estimates for 40 countries and 25 major cancers. Eur. J. Cancer 2021, 157, 308–347. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.; Miller, K.; Fuchs, H.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Desai, K.; McManus, J.; Sharifi, N. Hormonal therapy for prostate cancer. Endocr. Rev. 2021, 42, 354–373. [Google Scholar] [CrossRef]
- European Association of Urology. 2023. Available online: https://uroweb.org/guideline/prostate-cancer/ (accessed on 20 March 2023).
- Dong, L.; Zieren, R.; Xue, W.; De Reijke, T.; Pienta, K. Metastatic prostate cancer remains incurable, why? Asian J. Urol. 2019, 6, 26–41. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.S. Molecular states underlying androgen receptor activation: A framework for therapeutics targeting androgen signaling in prostate cancer. J. Clin. Oncol. 2012, 30, 644–646. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, K.; Penning, T. Partners in crime: Deregulation of AR activity and androgen synthesis in prostate cancer. Trends Endocrinol. Metab. 2010, 21, 315–324. [Google Scholar] [CrossRef]
- Hoang, D.; Iczkowski, K.; Kilari, D.; See, W.; Nevalainen, M. Androgen receptor-dependent and -independent mechanisms driving prostate cancer progression: Opportunities for therapeutic targeting from multiple angles. Oncotarget 2017, 8, 3724–3745. [Google Scholar] [CrossRef]
- Formaggio, N.; Rubin, M.; Theurillat, J. Loss and revival of androgen receptor signaling in advanced prostate cancer. Oncogene 2021, 40, 1205–1216. [Google Scholar] [CrossRef]
- Crona, D. Androgen receptor targeting drugs in castration-resistant prostate cancer and mechanisms of resistance. Clin. Pharmacol. Ther. 2015, 98, 582–589. [Google Scholar] [CrossRef]
- Congregado, B.; Rivero, I.; Osmán, I.; Sáez, C.; Medina López, R. PARP Inhibitors: A New Horizon for Patients with Prostate Cancer. Biomedicines 2022, 10, 1416. [Google Scholar] [CrossRef]
- Bluemn, E.; Coleman, I.; Lucas, J.; Coleman, R.; Hernandez-Lopez, S.; Tharakan, R.; Bianchi-Frias, D.; Dumpit, R.F.; Kaipainen, A.; Corella, A.N.; et al. Androgen receptor pathway-independent prostate cancer is sustained through FGF signaling. Cancer Cell 2017, 32, 474–489. [Google Scholar] [CrossRef] [PubMed]
- Small, E.; Youngren, J.; Alumkal, J.; Evans, C.; Ryan, C.; Lara, P. Neuroendocrine prostate cancer in patients with metastatic castration resistant prostate cancerresistant to abiraterone or enzalutamide: Preliminary results from the SU2C/PCF/AACR West Coast Prostate Cancer Dream Team. Ann. Oncol. 2014, 25, 255–279. [Google Scholar] [CrossRef]
- Denmeade, S.; Isaacs, J. Bipolar androgen therapy: The rationale for rapid cycling of supraphysiologic androgen/ablation in men with castration resistant prostate cancer. Prostate 2010, 70, 1600–1607. [Google Scholar] [CrossRef]
- Xiong, X.; Qiu, S.; Xianyanling, X.; Hang, H.; Lei, H.; Dazhou Liao, D.; Bai, S.; Ge Peng, G.; Wei, Q.; Ai, J.; et al. Efficacy and safety of bipolar androgen therapy in mCRPC after progression on abiraterone or enzalutamide: A systematic review. Urol. Oncol. Semin. Orig. Investig. 2002, 40, 19–28. [Google Scholar] [CrossRef]
- Wang, B.; Chen, Y.; Kao, W.; Lai, C.; Lin, H.; Hsieh, J. Developing New Treatment Options for Castration-Resistant Prostate Cancer and Recurrent Disease. Biomedicines 2022, 10, 1872. [Google Scholar] [CrossRef] [PubMed]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G.; PRISMA group. Preferred reporting items for systematic reviews and meta-analyses: The PriSMa statement. Int. J. Surg. 2010, 8, 336–341. [Google Scholar] [CrossRef]
- Liberati, A.; Altman, D.G.; Tetzlaff, J.; PRISMA group. Preferred reporting items for systematic review and meta-analysis protocols (PriSMa-P) 2015: Elaboration and explanation. BMJ 2015, 350, 7647. [Google Scholar] [CrossRef]
- Messner, E.; Steele, T.; Tsamouri, M.; Hejazi, N.; Gao, A.; Mudryj, M.; Ghosh, P.M. The Androgen Receptor in Prostate Cancer: Effect of Structure, Ligands and Spliced Variants on Therapy. Biomedicines 2020, 8, 422–441. [Google Scholar] [CrossRef]
- Bennett, N.; Gardiner, R.; Hooper, J.; Johnson, D.; Gobe, G. Molecular cell biology of androgen receptor signalling. Int. J. Biochem. Cell Biol. 2010, 42, 813–827. [Google Scholar] [CrossRef]
- Mostaghel, E.; Page, S.; Lin, D.; Fazli, L.; Coleman, I.; True, L.; Knudsen, B.; Hess, D.; Nelson, C.; Matsumoto, A.; et al. Intraprostatic androgens and androgen-regulated gene expression persist after testosterone suppression: Therapeutic implications for castration-resistant prostate cancer. Cancer Res. 2007, 67, 5033–5041. [Google Scholar] [CrossRef]
- Pinto, F.; Dibitetto, F.; Ragonese, M.; Bassi, P. Mechanisms of Resistance to Second-Generation Antiandrogen Therapy for Prostate Cancer: Actual Knowledge and Perspectives. Med. Sci. 2022, 10, 25–36. [Google Scholar] [CrossRef]
- Schweizer, M.T.; Yu, E. Persistent androgen receptor addiction in castrationresistant prostate cancer. J. Hematol. Oncol. 2015, 8, 128–142. [Google Scholar] [CrossRef]
- Waltering, K.; Helenius, M.; Sahu, B.; Manni, V.; Linja, M.; Janne, O.; Visakorpi, T. Increased expression of androgen receptor sensitizes prostate cancer cells to low levels of androgens. Cancer Res. 2009, 69, 8141–8149. [Google Scholar] [CrossRef] [PubMed]
- Quigley, D. Genomic Hallmarks and Structural Variation in Metastatic Prostate Cancer. Cell 2018, 174, 758–769. [Google Scholar] [CrossRef] [PubMed]
- Yehya, A.; Ghamlouche, F.; Zahwe, A.; Zeid, Y.; Wakimian, K.; Mukherji, D.; Abou-Kheir, W. Drug resistance in metastatic castration-resistant prostate cancer: An update on the status quo. Cancer Drug Resist. 2022, 5, 667–690. [Google Scholar] [CrossRef]
- Koochekpour, S. Androgen receptor signaling and mutations in prostate cancer. Asian J. Androl. 2010, 12, 639–657. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Shi, Y.; Xu, L.L.; Nageswararao, C.; Davis, L.; Segawa, T.; Dobi, A.; McLeod, D.; Srivastava, S. Androgen receptor mutation (T877A) promotes prostate cancer cell growth and cell survival. Oncogene 2006, 25, 3905–3913. [Google Scholar] [CrossRef]
- Steketee, K.; Timmerman, L.; Ziel-van der Made, A.; Doesburg, P.; Brinkmann, A.; Trapman, J. Broadened ligand responsiveness of androgen receptor mutants obtained by random amino acid substitution of H874 and mutation hot spot T877 in prostate cancer. Int. J. Cancer 2002, 100, 309–317. [Google Scholar] [CrossRef]
- Taplin, M.; Bubley, G.; YJ, K.; Small, E.; Upton, M.; Rajeshkumar, B. Selection for androgen receptor mutations in prostate cancers treated with androgen antagonist. Cancer Res. 1999, 59, 2511–2515. [Google Scholar]
- Attard, G.; Reid, A.; Auchus, R.; Hughes, B.; Cassidy, A.; Thompson, E. Clinical and biochemical consequences of CYP17A1 inhibition with abiraterone given with and without exogenous glucocorticoids in castrate men with advanced prostate cancer. J. Clin. Endocrinol. Metab. 2012, 97, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.; Balk, S. Intratumoral androgen biosynthesis in prostate cancer pathogenesis and response to therapy. Endocr. Relat. Cancer 2011, 18, 175–182. [Google Scholar] [CrossRef]
- Sun, S.; Sprenger, C.; Vessella, R.; Haugk, K.; Soriano, K.; Mostaghel, E.; Page, S.; Coleman, I.; Nguyen, H.; Sun, H.; et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J. Clin. Investig. 2010, 120, 2715–2730. [Google Scholar] [CrossRef] [PubMed]
- Mollica, V.; Di Nunno, V.; Cimadamore, A.; Lopez-Beltran, A.; Cheng, L.; Santoni, M.; Scarpelli, M.; Montironi, R.; Massari, F. Molecular Mechanisms Related to Hormone Inhibition Resistance in Prostate Cancer. Cells 2019, 8, 43. [Google Scholar] [CrossRef]
- Verma, S.; Prajapati, K.; Kushwaha, P.; Shuaib, M.; Singh, A.; Kumar, A.; Gupta, S. Resistance to second generation antiandrogens in prostate cancer: Pathways and mechanisms. Cancer Drug Resist. 2020, 3, 742–761. [Google Scholar] [CrossRef]
- Armstrong, C.; Gao, A. Adaptive pathways and emerging strategies overcoming treatment resistance in castration resistant prostate cancer. Asian J. Urol. 2016, 3, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, C.; Gao, A. Current strategies for targeting the activity of androgen receptor variants. Asian J. Urol. 2019, 6, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Antonarakis, E.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef]
- Chen, Y.; Zhou, Q.; Hankey, W.; Fang, X.; Yuan, F. Second generation androgen receptor antagonists and challenges in prostate cancer treatment. Cell Death Dis. 2022, 13, 632–643. [Google Scholar] [CrossRef]
- Zhu, M.L.; Kyprianou, N. Androgen receptor and growth factor signaling cross-talk in prostate cancer cells. Endocr. Relat. Cancer 2008, 15, 841–849. [Google Scholar] [CrossRef]
- Yang, L.; Wang, L.; Lin, H.; Kan, P.; Xie, S.; Tsai, M.; Wang, P.; Chen, Y.; Chang, C. Interleukin-6 differentially regulates androgen receptor transactivation via PI3KAkt, STAT3, and MAPK, three distinct signal pathways in prostate cancer cells. Biochem. Biophys. Res. Commun. 2003, 305, 462–469. [Google Scholar] [CrossRef]
- Comstock, C.; Augello, M.; Benito, R.; Karch, J.; Tran, T.; Utama, F.; Tindall, E.; Wang, Y.; Burd, C.; Groh, E.; et al. Cyclin D1 splice variants: Polymorphism, risk, and isoform-specific regulation in prostate cancer. Clin. Cancer Res. 2009, 15, 5338–5349. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Yeow, W.; Ertel, A.; Coleman, I.; Clegg, N.; Thangavel, C.; Morrissey, C.; Zhang, X.; Comstock, C.; Witkiewicz, A.; et al. The retinoblastoma tumor suppressor controls androgen signaling and human prostate cancer progression. J. Clin. Investig. 2010, 120, 4478–4492. [Google Scholar] [CrossRef] [PubMed]
- Meric-Bernstam, F.; Akcakanat, A.; Chen, H.; Do, K.; Sangai, T. PIK3CA/PTEN mutations and akt activation as markers of sensitivity to allosteric mTOR inhibitors. Clin. Cancer Res. 2012, 18, 1777–1789. [Google Scholar] [CrossRef]
- Carver, B.; Chapinski, C.; Wongvipat, J.; Hieronymus, H.; Chen, Y. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell 2011, 19, 575–586. [Google Scholar] [CrossRef]
- Tucci, M.; Zichi, C.; Buttigliero, C.; Vignani, F.; Scagliotti, G. Enzalutamide-resistant castration-resistant prostate cancer: Challenges and solutions. OncoTargets Ther. 2018, 11, 7353–7368. [Google Scholar] [CrossRef]
- Toren, P.; Zoubeidi, A. Targeting the PI3K/Akt pathway in prostate cancer: Challenges and opportunities. Int. J. Oncol. 2014, 45, 1793–1801. [Google Scholar] [CrossRef] [PubMed]
- Leung, J.; Sadar, M. Non-genomic actions of the androgen receptor in prostate cancer. Front. Endocrinol. 2017, 8, 2–8. [Google Scholar] [CrossRef]
- Thomas, C.; Lamoureux, F.; Crafter, C.; Davies, B.; Beraldi, E. Synergistic targeting of PI3K/AKT pathway and androgen receptor axis significantly delays castrationresistant prostate cancer progression In Vivo. Mol. Cancer Ther. 2013, 12, 2342–2355. [Google Scholar] [CrossRef]
- Hernando Polo, S.; Moreno Muñoz, D.; Rosero Rodríguez, A.; Silva Ruiz, J.; Rosero Rodríguez, D.; Couñago, F. Changing the History of Prostate Cancer with New Targeted Therapies. Biomedicines 2021, 9, 392–412. [Google Scholar] [CrossRef]
- Arora, V.; Schenkein, E.; Murali, R.; Subudhi, S.; Wongvipat, J.; Balbas, M.; Shah, N.; Cai, L.; Efstathiou, E.; Logothetis, C.; et al. Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell 2013, 155, 1309–1322. [Google Scholar] [CrossRef]
- Isikbay, M.; Otto, K.; Kregel, S.; Kach, J.; Cai, Y.; Vander Griend, D.; Conzen, S.; Szmulewitz, R. Glucocorticoid receptor activity contributes to resistance to androgen-targeted therapy in prostate cancer. Horm. Cancer 2014, 5, 72–89. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; Morrison, S.; Clarke, M.; Weissman, I. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef]
- Chen, X.; Rycaj, K.; Liu, X.; Tang, D. New insights into prostate cancer stem cells. Cell Cycle 2013, 12, 579–586. [Google Scholar] [CrossRef]
- Dean, M.; Fojo, T.; Bates, S. Tumour stem cells and drug resistance. Nat. Rev. Cancer 2005, 5, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Patrawala, L.; Calhoun, T.; Schneider-Broussard, R.; Li, H.; Bhatia, B.; Tang, S.; Reilly, J.; Chandra, D.; Zhou, J.; Claypool, K.; et al. Highly purified CD44+ prostate cancer cells from xenograft human tumors are enriched in tumorigenic and metastatic progenitor cells. Oncogene 2006, 25, 1696–1708. [Google Scholar] [CrossRef]
- Kushwaha, P.; Verma, S.; Kumar, S.; Gupta, S. Role of prostate cancer stem-like cells in the development of antiandrogen resistance. Cancer Drug Resist. 2022, 5, 459–471. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Park, D.; Zhong, Y.; Lu, Y.; Rycaj, K.; Gong, S.; Chen, X.; Liu, X.; Chao, H.; Whitney, P.; et al. Stem cell and neurogenic geneexpression profiles link prostate basal cells to aggressive prostate cancer. Nat. Commun. 2016, 7, 10798. [Google Scholar] [CrossRef]
- Sun, Y.; Niu, J.; Huang, J. Neuroendocrine differentiation in prostate cancer. Am. J. Transl. Res. 2009, 1, 148–162. [Google Scholar]
- Beltran, H.; Tomlins, S.; Aparicio, A.; Arora, V.; Rickman, D.; Ayala, G. Aggressive variants of castration-resistant prostate cancer. Clin. Cancer Res. 2014, 20, 2846–2850. [Google Scholar] [CrossRef]
- Epstein, J.; Amin, M.; Beltran, H.; Lotan, T.; Mosquera, J.; Reuter, V. Proposed morphologic classification of prostate cancer with neuroendocrine differentiation. Am. J. Surg. Pathol. 2014, 38, 756–767. [Google Scholar] [CrossRef]
- Lipianskaya, J.; Cohen, A.; Chen, C.; Hsia, E.; Squires, J.; Li, Z.; Zhang, Y.; Li, W.; Chen, X.; Xu, H.; et al. Androgen-deprivation therapy-induced aggressive prostate cancer with neuroendocrine differentiation. Asian J. Androl. 2014, 16, 541–544. [Google Scholar] [CrossRef] [PubMed]
- Zou, M.; Toivanen, R.; Mitrofanova, A.; Floch, N.; Hayati, S.; Sun, Y. Transdifferentiation as a mechanism of treatment resistance in a mouse model of castration-resistant prostate cancer. Cancer Discov. 2017, 7, 736–749. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Kondrikov, D.; Yuan, T.; Lin, F.; Hansen, J.; Lin, M. Receptor protein tyrosine phosphatase alpha signaling is involved in androgen depletion-induced neuroendocrine differentiation of androgen-sensitive LNCaP human prostate cáncer cells. Oncogene 2003, 22, 6704–6716. [Google Scholar] [CrossRef]
- Labrecque, M.; Coleman, I.; Brown, L.; True, L.; Kollath, L.; Lakely, B. Molecular profiling stratifies diverse phenotypes of treatment-refractory metastatic castration-resistant prostate cancer. J. Clin. Investig. 2019, 129, 4492–4505. [Google Scholar] [CrossRef]
- Teply, B.; Kachhap, S.; Eisenberger, M.; Denmeade, S. Extreme response to highdose testosterone in BRCA2- and ATM-mutated prostate cancer. Eur. Urol. 2017, 71, 499. [Google Scholar] [CrossRef]
- Hedayati, M.; Haffner, M.; Coulter, J.; Raval, R.; Zhang, Y.; Zhou, H. Androgen deprivation followed by acute androgen stimulation selectively sensitizes ARpositive prostate cancer cells to ionizing radiation. Clin. Cancer Res. 2016, 22, 3310–3319. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, J.; D’Antonio, J.; Chen, S. Adaptive auto-regulation of androgen receptor provides a paradigm shifting rationale for bipolar androgen therapy (BAT) for castrate resistant human prostate cancer. Prostate 2012, 72, 1491–1505. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Pan, Y.; Regan, K.; Wu, C.; Zhang, X.; Tindall, D. Androgens repress expression of the F-box protein Skp2 via p107 dependent and independent mechanisms in LNCaP prostate cancer cells. Prostate 2012, 72, 225–232. [Google Scholar] [CrossRef]
- Chen, L.; Sun, Y.; Tang, M. High-dose-androgen-induced autophagic cell death to suppress the Enzalutamide-resistant prostate cancer growth via altering the circRNABCL2/ miRNA-198/AMBRA1 signaling. Cell Death Discov. 2022, 8, 128. [Google Scholar] [CrossRef] [PubMed]
- Wolf, D. Synthetic androgens suppress the transformed phenotype in the human prostate carcinoma cell line LNCaP. Br. J. Cancer 1991, 64, 47–53. [Google Scholar] [CrossRef]
- Kokontis, J. Increased androgen receptor activity and altered c-myc expression in prostate cancer cells after long-term androgen deprivation. Cancer Res. 1994, 54, 1566–1573. [Google Scholar]
- Umekita, Y. Human prostate tumor growth in athymic mice: Inhibition by androgens and stimulation by finasteride. Proc. Natl. Acad. Sci. USA 1996, 93, 11802–11807. [Google Scholar] [CrossRef] [PubMed]
- Feltquate, D.; Nordquist, L.; Eicher, C.; Morris, M.; Smaletz, O.; Slovin, S. Rapid androgen cycling as treatment for patients with prostate cancer. Clin. Cancer Res. 2006, 12, 7414–7421. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.; Huang, D.; Kelly, W.; Slovin, S.; Stephenson, R. Phase 1 trial of high-dose exogenous testosterone in patients with castration-resistant metastatic prostate cancer. Eur. Urol. 2009, 56, 237–244. [Google Scholar] [CrossRef]
- Denmeade, S. Bipolar androgen therapy (BAT): A patient’s guide. Prostate 2022, 82, 753–762. [Google Scholar] [CrossRef] [PubMed]
- Schweizer, M.; Antonarakis, E.S.; Wang, H.; Ajiboye, A.S.; Spitz, A.; Cao, H.; Luo, J.; Haffner, M.C.; Yegnasubramanian, S.; Carducci, M.A.; et al. Effect of bipolar androgen therapy for asymptomatic men with castration-resistant prostate cancer: Results from a pilot clinical study. Sci. Transl. Med. 2015, 7, 269. [Google Scholar] [CrossRef]
- Benjamin, A.T.; Wang, H.; Luber, B.; Sullivan, R.; Rifkind, I. Bipolar androgen therapy in men with metastatic castration-resistant prostate cancer after progression on enzalutamide: An open-label, phase 2, multicohort study. Lancet Oncol. 2018, 19, 76–86. [Google Scholar] [CrossRef]
- Markowski, M.; Wang, H.; Sullivan, R. A multicohort open-label phase II trial of bipolar androgen therapy in men with metastatic castration-resistant prostate cancer (RESTORE): A comparison of post-abiraterone versus post-enzalutamide cohorts. Eur. Urol. 2021, 79, 692–699. [Google Scholar] [CrossRef]
- Sena, L. Bipolar androgen therapy sensitizes castration-resistant prostate cancer to subsequent androgen receptor ablative therapy. Eur. J. Cancer 2021, 144, 302–309. [Google Scholar] [CrossRef]
- Denmeade, S.; Wang, H.; Agarwal, N. TRANSFORMER: A randomized phase II study comparing bipolar androgen therapy versus enzalutamide in asymptomatic men with castrationresistant metastatic prostate cancer. J. Clin. Oncol. 2021, 39, 1371–1382. [Google Scholar] [CrossRef]
- Zarbá, M.; Angel, M.; Losco, F.; Zarbá, J.J.; Pupilli, J.C.; Rodrigo Chacon, M.; Sade, J.P. Experience of bipolar androgen therapy (BAT) in Argentinian oncology centres. eCancer 2022, 16, 1480–1489. [Google Scholar] [CrossRef] [PubMed]
- Kiang Chua, M.L.; Bristow, R.G. Testosterone in Androgen Receptor Signaling and DNA Repair: Enemy or Frenemy? Clin. Cancer Res. 2016, 22, 3124–3126. [Google Scholar] [CrossRef] [PubMed]
- Gongora, A.B.; Marshall, C.H.; Isaacsson, P.; Velh, C.D.; Lopes, J.F.; Marin, A.A. Extreme Responses to a Combination of DNA-Damaging Therapy and Immunotherapy in CDK12-Altered Metastatic Castration-Resistant Prostate Cancer: A Potential Therapeutic Vulnerability. Clin. Genitourin. Cancer 2022, 20, 183–188. [Google Scholar] [CrossRef]
- Chen, W.; Beck, I.; Schober, W.; Brockow, K.; Effner, R.; Buters, J. Human mast cells express androgen receptors but treatment with testosterone exerts no influence on IgE-independent mast cell degranulation elicited by neuromuscular blocking agents. Exp. Dermatol. 2010, 19, 302–304. [Google Scholar] [CrossRef] [PubMed]
- Pu, Y.; Xu, M.; Liang, Y.; Yang, K.; Guo, Y.; Yang, X. Androgen receptor antagonists compromise T cell response against prostate cancer leading to early tumor relapse. Sci. Transl. Med. 2016, 8, 333. [Google Scholar] [CrossRef] [PubMed]
- Seki, T.; Shimizu, Y.; Ishii, K.; Takahama, Y.; Kato, K.; Yano, T. NK cells can preferentially target prostate cancer stem-like cells via the TRAIL/DR5 signalling pathway. Biomolecules 2021, 11, 1702. [Google Scholar] [CrossRef]
- Tang, M.; Sun, Y.; Huang, C. High dose androgen suppresses natural killer cytotoxicity of castration-resistant prostate cancer cells via altering AR/circFKBP5/miRNA-513a-5p/PD-L1 signals. Cell Death Dis. 2022, 13, 746–757. [Google Scholar] [CrossRef]
- Markowski, M.C.; Taplin, M.E.; Aggarwal, R.R.; Wang, H.; Lalji, A.; Judith Paller, C. COMBAT-CRPC: Concurrent administration of bipolar androgen therapy (BAT) and nivolumab in men with metastatic castration-resistant prostate cancer (mCRPC). J. Clin. Oncol. 2021, 39, 5014. [Google Scholar] [CrossRef]
- Markowski, M. Molecular and clinical characterization of patients with metastatic castration resistant prostate cancer achieving deep responses to bipolar androgen therapy. Clin. Genitourin. Cancer 2022, 20, 97–101. [Google Scholar] [CrossRef]
- Qiu, X. Response to supraphysiological testosterone is predicted by a distinct androgen receptor cistrome. JCI Insight 2022, 7, e157164. [Google Scholar] [CrossRef]
- Guo, H. Androgen receptor and MYC equilibration centralizes on developmental superenhancer. Nat. Commun. 2021, 12, 7308. [Google Scholar] [CrossRef]
- Stine, Z. MYC, metabolism, and cancer. Cancer Discov. 2015, 5, 1024–1039. [Google Scholar] [CrossRef] [PubMed]
- Sena, L.A.; Kumar, R.; Sanin, D.E.; Thompson, E.A.; Rosen, D.M.; Dalrymple, S.L. Androgen receptor activity in prostate cancer dictates efficacy of bipolar androgen therapy through MYC. J. Clin. Investig. 2022, 132, e162396. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X. MYC drives aggressive prostate cancer by disrupting transcriptional pause release at androgen receptor targets. Nat. Commun. 2022, 13, 2559. [Google Scholar] [CrossRef]
- Peinetti, N.; Bilusic, M.; Burnstein, K.L. Is androgen receptor activity in metastatic prostate cancer a good biomarker for bipolar androgen therapy? J. Clin. Investig. 2022, 132, e165357. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, P.; Schweizer, M.T.; Lucas, J.M.; Coleman, I.; Nyquist, M.D.; Frank, S.B.; Tharakan, R.; Mostaghel, E.; Luo, J.; Pritchard, C.C.; et al. Supraphysiological androgens suppress prostate cancer growth through androgen receptor-mediated DNA damage. J. Clin. Investig. 2019, 129, 4245–4260. [Google Scholar] [CrossRef] [PubMed]
- Rice, M.; Malhotra, S.; Stoyanova, T. Second-Generation Antiandrogens: From Discovery to Standard of Care in Castration Resistant Prostate Cancer. Front. Oncol. 2019, 9, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Lou, W.; Zhu, Y.; Nadiminty, N.; Schwartz, C.; Evans, C. Niclosamide inhibits androgen receptor variants expression and overcomes enzalutamide resistance in castrationresistant prostate cancer. Clin. Cancer Res. 2014, 20, 3198–3210. [Google Scholar] [CrossRef]
- Liu, C.; Armstrong, C.; Zhu, Y.; Lou, W.; Gao, A. Niclosamide enhances abiraterone treatment via inhibition of androgen receptor variants in castration resistant prostate cancer. Oncotarget 2016, 7, 32210–32220. [Google Scholar] [CrossRef]
- Liu, C.; Armstrong, C.; Lou, W.; Lombard, A.; Cucchiara, V.; Gu, X.; Yang, J. Niclosamide and Bicalutamide Combination Treatment Overcomes Enzalutamide- and Bicalutamide-Resistant Prostate Cancer. Mol. Cancer Ther. 2017, 16, 1521–1530. [Google Scholar] [CrossRef]
- Mamta, P.; Chengfei, L.; Chun-Yi, W.; Christopher, E.P.; Marc, D. Phase Ib trial of reformulated niclosamide with abiraterone/prednisone in men with castration-resistant prostate cancer. Sci. Rep. 2021, 11, 6377–6390. [Google Scholar] [CrossRef]
- Schweizer, M. A phase I study of niclosamide in combination with enzalutamide in men with castration-resistant prostate cancer. PLoS ONE 2018, 13, e0198389. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Lou, W.; Armstrong, C.; Zhu, Y.; Evans, C.; Gao, A. Niclosamide suppresses cell migration and invasion in enzalutamide resistant prostate cancer cells via Stat3-AR axis inhibition. Prostate 2015, 75, 1341–1353. [Google Scholar] [CrossRef] [PubMed]
- Gaur, S.; Gross, M.; Liao, C.; Qian, B.; Shih, J. Effect of Monoamine oxidase A (MAOA) inhibitors on androgen-sensitive and castration-resistant prostate cancer cells. Prostate 2019, 79, 667–677. [Google Scholar] [CrossRef] [PubMed]
- Rice, M.; Hsu, E.; Aslan, M.; Ghoochani, A.; Su, A.; Stoyanova, T. Loss of Notch1 activity inhibits prostate cancer growth and metastasis and sensitizes prostate cancer cells to antiandrogen therapies. Mol. Cancer Ther. 2019, 18, 1230–1242. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Armstrong, C.; Lou, W.; Lombard, A.; Evans, C.; Gao, A. Inhibition of AKR1C3 activation overcomes resistance to abiraterone in advanced prostate cancer. Mol. Cancer Ther. 2017, 16, 35–44. [Google Scholar] [CrossRef]
- Liu, C.; Lou, W.; Zhu, Y.; Yang, J.; Nadiminty, N.; Gaikwad, N. Intracrine Androgens and AKR1C3 activation confer resistance to enzalutamide in prostate cancer. Cancer Res. 2015, 75, 1413–1422. [Google Scholar] [CrossRef]
- Armstrong, C.M.; Gao, A.C. Dysregulated androgen synthesis and anti-androgen resistance in advanced prostate cancer. Am. J. Clin. Exp. Urol. 2021, 9, 292–300. [Google Scholar]
- Liu, C.; Lou, W.; Pan, C.-X.; Lara, P.; Evans, C.; Parikh, M.; White, R.d.V.; Dall’Era, M.; Gao, A. Combination of indomethacin and enzalutamide to treat castration-resistant prostate cancer. J. Urol. 2018, 199, 3694. [Google Scholar] [CrossRef]
- Gao, S.; Ye, H.; Gerrin, S.; Wang, H.; Sharma, A.; Chen, S. ErbB2 signaling increases androgen receptor expression in abiraterone-resistant prostate cancer. Clin. Cancer Res. 2016, 22, 3672–3682. [Google Scholar] [CrossRef]
- Shiota, M.; Bishop, J.; Takeuchi, A.; Nip, K.; Cordonnier, T.; Beraldi, E. Inhibition of the HER2-YB1-AR axis with Lapatinib synergistically enhances Enzalutamide anti-tumor efficacy in castration resistant prostate cancer. Oncotarget 2015, 6, 9086–9098. [Google Scholar] [CrossRef] [PubMed]
- Biersack, B.; Nitzsche, B.; Höpfner, M. HDAC inhibitors with potential to overcome drug resistance in castration-resistant prostate cancer. Cancer Drug Resist. 2022, 5, 64–79. [Google Scholar] [CrossRef] [PubMed]
- Welsbie, D.; Xu, J.; Chen, Y.; Borsu, L.; Scher, H.; Rosen, N.; Sawyers, C. Histone deacetylases are required for androgen receptor function in hormone-sensitive and castrate-resistant prostate cancer. Cancer Res. 2009, 69, 958–966. [Google Scholar] [CrossRef]
- Ferrari, A.C.; Alumkal, J.J.; Stein, M.N.; Taplin, M.E.; Babb, J.; Barnett, E.S. Epigenetic Therapy with Panobinostat Combined with Bicalutamide Rechallenge in Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2019, 25, 52–63. [Google Scholar] [CrossRef]
- Oronsky, B.; Oronsky, N.; Scicinski, J.; Fanger, G.; Lybeck, M.; Reid, T. Rewriting the Epigenetic Code for Tumor Resensitization: A Review. Transl. Oncol. 2014, 7, 626–631. [Google Scholar] [CrossRef]
- Ferrari, A.C. Reversing resistance to antiandrogens with a histone deacetylase inhibitor. Oncotarget 2018, 9, 37284–37285. [Google Scholar] [CrossRef]
- Yang, Y.; Banuelos, C.; Mawji, N.; Wang, J.; Kato, M.; Haile, S. Targeting Androgen Receptor Activation Function-1 with EPI to Overcome Resistance Mechanisms in Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2016, 22, 4466–4477. [Google Scholar] [CrossRef]
- Le Moigne, R.; Banuelos, C.A.; Mawji, N.R.; Tam, T.; Wang, J.; Jian, K.; Andersen, R.J.; Cesano, A.; Sadar, M.D.; Zhou, H.J.; et al. Treatment of castrated resistant prostate cancer with EPI-7386, a second generation N-terminal domain androgen receptor inhibitor. Mol. Cancer Ther. 2019, 18, 117–118. [Google Scholar] [CrossRef]
- Hong, N.; Le Moigne, R.; Banuelos, C. Pre-clinical development of the second generation N-terminal domain androgen receptor inhibitor, EPI-7386, for the treatment of prostate cancer. Cancer Res. 2020, 80, 1953. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT05075577 (accessed on 21 January 2023).
- Nguyen, H.; Yang, J.; Kung, H.; Shi, X.; Tilki, D. Targeting autophagy overcomes Enzalutamide resistance in castration-resistant prostate cancer cells and improves therapeutic response in a xenograft model. Oncogene 2014, 33, 4521–4530. [Google Scholar] [CrossRef]
- Xie, Y.; Wang, L.; Khan, M.A.; Hamburger, A.W.; Guang, W.; Passaniti, A.; Munir, K.; Ross, D.D.; Dean, M.; Hussain, A. Metformin and Androgen Receptor-Axis-Targeted (ARAT) Agents Induce Two PARP-1-Dependent Cell Death Pathways in Androgen-Sensitive Human Prostate Cancer Cells. Cancers 2021, 13, 633. [Google Scholar] [CrossRef] [PubMed]
- Varambally, S.; Dhanasekaran, S.; Zhou, M.; Barrette, T.; Kumar-Sinha, C.; Sanda, M. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 2022, 419, 624–629. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Tien, J.C.; Vo, J.; Tan, M.; Parolia, A.; Zhang, Y.; Wang, L.; Qiao, Y.; Shukla, S.; Wang, X.; et al. Epigenetic Reprogramming with Antisense Oligonucleotides Enhances the Effectiveness of Androgen Receptor Inhibition in Castration-Resistant Prostate Cancer. Cancer Res. 2018, 78, 5731–5740. [Google Scholar] [CrossRef] [PubMed]
- Dardenne, E.; Beltran, H.; Benelli, M.; Gayvert, K.; Berger, A.; Puca, L. N-Myc induces an EZH2-mediated transcriptional program driving neuroendocrine prostate cancer. Cancer Cell 2016, 30, 563–577. [Google Scholar] [CrossRef] [PubMed]
- Ku, S.; Rosario, S.; Wang, Y.; Mu, P.; Seshadri, M.; Goodrich, Z. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 2017, 355, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Doroshow, D.; Eder, J.; LoRusso, P.M. BET inhibitors: A novel epigenetic approach. Ann. Oncol. 2017, 28, 1776–1787. [Google Scholar] [CrossRef] [PubMed]
- Asangani, I.; Dommeti, V.; Wang, X.; Malik, R.; Cieslik, M.; Yang, R. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature 2014, 510, 278–282. [Google Scholar] [CrossRef]
- Szafran, A.; Stephan, C.; Bolt, M.; Mancini, M.; Marcelli, M.; Mancini, M. High-Content Screening Identifies Src Family Kinases as Potential Regulators of AR-V7 Expression and Androgen-Independent Cell Growth. Prostate 2017, 77, 82–93. [Google Scholar] [CrossRef]
- Chattopadhyay, I.; Wang, J.; Qin, M.; Gao, L.; Holtz, R.; Vessella, R.; Leach, R.; Gelman, I. Src promotes castration-recurrent prostate cancer through androgen receptor-dependent canonical and non-canonical transcriptional signatures. Oncotarget 2017, 8, 10324–10347. [Google Scholar] [CrossRef]
- Moreira-Silva, F.; Henrique, R.; Jeronimo, C. From Therapy Resistance to Targeted Therapies in Prostate Cancer. Front. Oncol. 2022, 12, 877379. [Google Scholar] [CrossRef]
- Adelaiye-Ogala, R.; Gryder, B.; Nguyen, Y.; Alilin, A.; Grayson, A.; Bajwa, W. Targeting the PI3K/AKT Pathway Overcomes Enzalutamide Resistance by Inhibiting Induction of the Glucocorticoid Receptor. Mol. Cancer Ther. 2020, 19, 1436–1447. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Target | Mechanism of action | Reference |
|---|---|---|---|
| BAT (high level of testosterone followed to castrate range) | Androgen Receptor (AR) |
| Schweizer, MT. Sci Transl Med, 2015 [77] Benjamin AT. Lancet Oncol, 2018 [78] Markowski, MC. Eur Urol, 2021 [79] Denmeade, SR. J Clin Oncol, 2021 [81] |
| Niclosamide | Androgen receptor (AR) |
| Liu, C. Mol Cancer Ther, 2017 [101] Liu, C. Prostate 2015 [104] |
| Monoamine oxidase A inhibitors (MAOAIs) | Monoamine oxidase A in prostate | Sensitizes cells to enzalutamide | Gaur, S. Prostate, 2019 [105] |
| Inhibitors of Notch receptors | Notch receptors | Inhibition of gamma secretase | Rice, MA. Mol Cancer Ther, 2019 [106] |
| Indomethacin | Intracrine androgens | Inhibition of AKR1C3 enzymatic activity | Liu, C. Cancer Res, 2015 [108] |
| Lapatinib | HER2 signaling axis |
| Shiota, M. Oncotarget, 2015 [112] |
| Panobinostat | Histone deacetylases (HDAC) pathway | HDAC inhibition | Ferrari, AC. Clin Cancer Res, 2019 [115] |
| EPI-7386 | AR N-terminal domain | Inhibition of the N-terminal domain (NTD) of the AR | Hong, NH. Cancer Res, 2020 [120] |
| Clomipramine and metformine | Autophagy inhibition | Autophagy inhibition | Nguyen, HG. Oncogene, 2014 [122] |
| Antisense oligonucleotides (ASO) | Epigenetic modifications | Inhibition of the histone lysine-N-methyltransferase EZH2 | Xiao, L. Cancer Res, 2018 [125] |
| ZEN003694/JQ1 | Bromodomain and extra-terminal (BET) pathway | Inhibition of BET and BRD4 | Asangani, I.A. Nature, 2014 [129] |
| Dasatinib | SRC gene | inhibition of proto-oncogene tyrosine protein kinase Src | Chattopadhyay, I. Oncotarget. 2017 [131] |
| Ipatasertib | PI3K-Akt-mTOR pathway | Akt inhibitor | Adelaiye-Ogala, R. Mol Cancer Ther, 2020 [133] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Congregado Ruiz, B.; Rivero Belenchón, I.; Lendínez Cano, G.; Medina López, R.A. Strategies to Re-Sensitize Castration-Resistant Prostate Cancer to Antiandrogen Therapy. Biomedicines 2023, 11, 1105. https://doi.org/10.3390/biomedicines11041105
Congregado Ruiz B, Rivero Belenchón I, Lendínez Cano G, Medina López RA. Strategies to Re-Sensitize Castration-Resistant Prostate Cancer to Antiandrogen Therapy. Biomedicines. 2023; 11(4):1105. https://doi.org/10.3390/biomedicines11041105
Chicago/Turabian StyleCongregado Ruiz, Belén, Inés Rivero Belenchón, Guillermo Lendínez Cano, and Rafael Antonio Medina López. 2023. "Strategies to Re-Sensitize Castration-Resistant Prostate Cancer to Antiandrogen Therapy" Biomedicines 11, no. 4: 1105. https://doi.org/10.3390/biomedicines11041105
APA StyleCongregado Ruiz, B., Rivero Belenchón, I., Lendínez Cano, G., & Medina López, R. A. (2023). Strategies to Re-Sensitize Castration-Resistant Prostate Cancer to Antiandrogen Therapy. Biomedicines, 11(4), 1105. https://doi.org/10.3390/biomedicines11041105