
Pathogenesis of Hepatocellular Carcinoma: The Interplay of Apoptosis and Autophagy


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Pathogenesis of HCC
2.1. Endoplasmic Reticulum (ER) and Oxidative Stress
2.2. Abnormalities of Signaling Pathways
- mTOR pathway
- Wnt/β-catenin pathway
- miRNAs
2.3. Additional Factors Are Involved in HCC Pathogenesis
- Exosomes
- Ferroptosis
- Microbiota
- Calcium
- Autophagy and Apoptosis
3. HCC Related to Specific Diseases
3.1. HBV
3.1.1. The Important Role of HBx
3.1.2. The Role of RNAs
3.2. HCV
3.3. NAFLD
3.4. Diabetes
3.5. ALD
3.6. Hemochromatosis
4. Apoptosis
Apoptosis and HCC
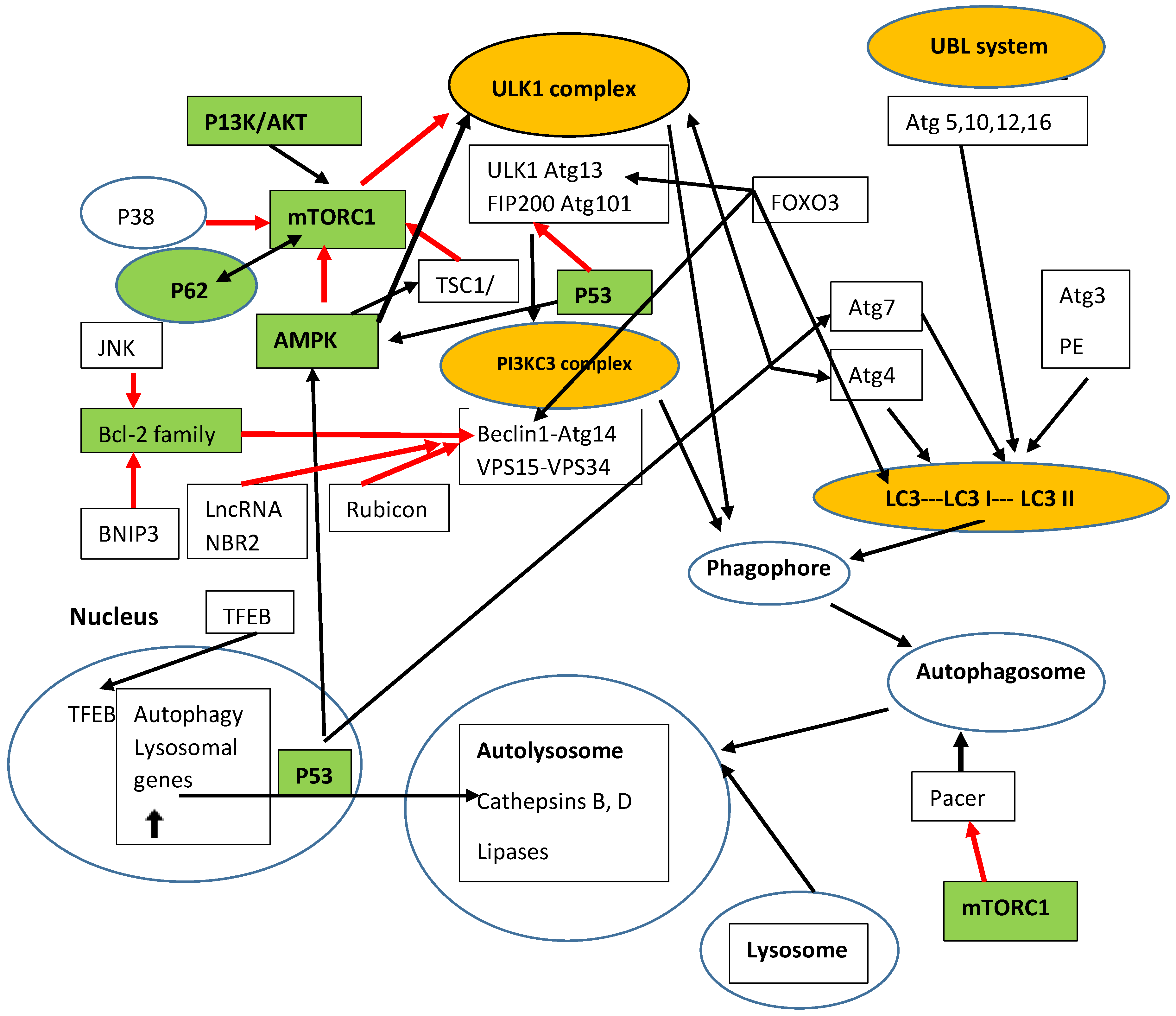
5. Autophagy
Autophagy and HCC
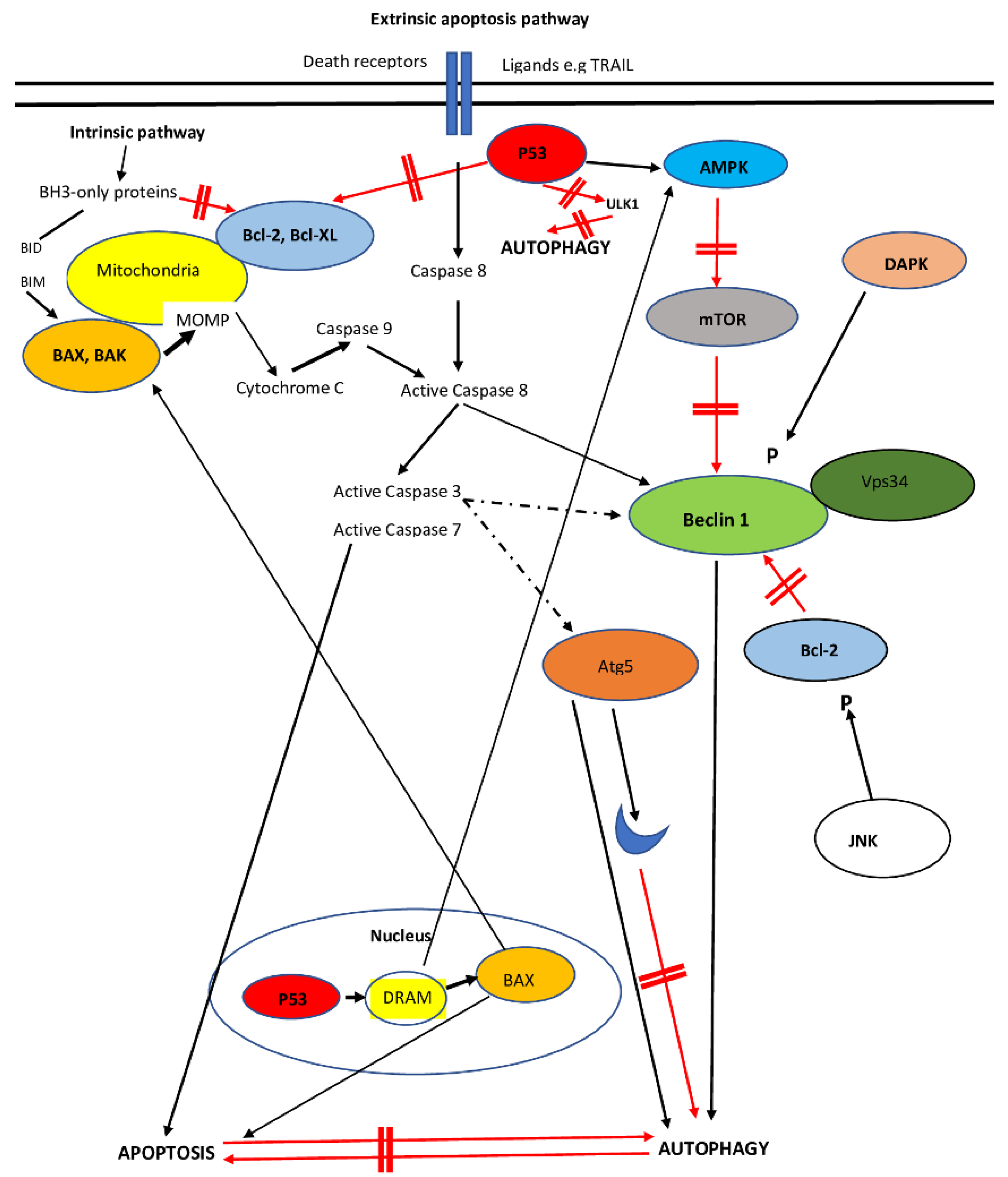
6. Interplay between Apoptosis and Autophagy
6.1. Beclin-1
6.2. Beclin-1 in HCC
6.3. mTOR Interaction with Autophagy–Apoptosis and the Regulation of mTOR in HCC
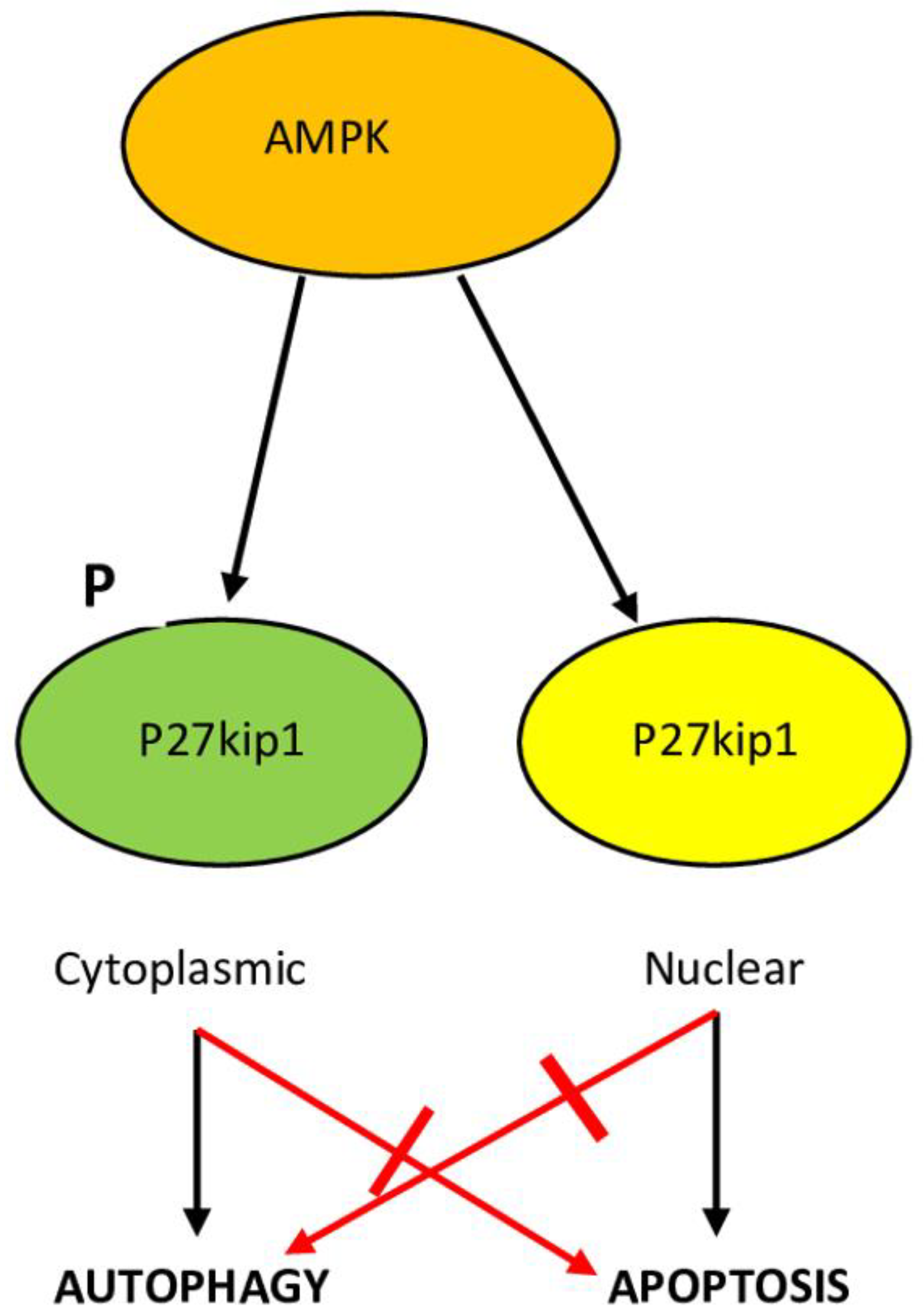
6.4. p27kip1
6.5. The Anti-Apoptotic FLIP
6.6. The Role of the ATG12, ATG5 and ATG3 Proteins in Autophagy
6.7. The Death-Associated Protein Kinase (DAPK) Family in Apoptosis and Autophagy
6.8. p53
6.9. Tumor-Associated Macrophages (TAM) and the Tumor Microenvironment (TEM)
6.10. The Role of Mitochondria
6.11. Other Factors
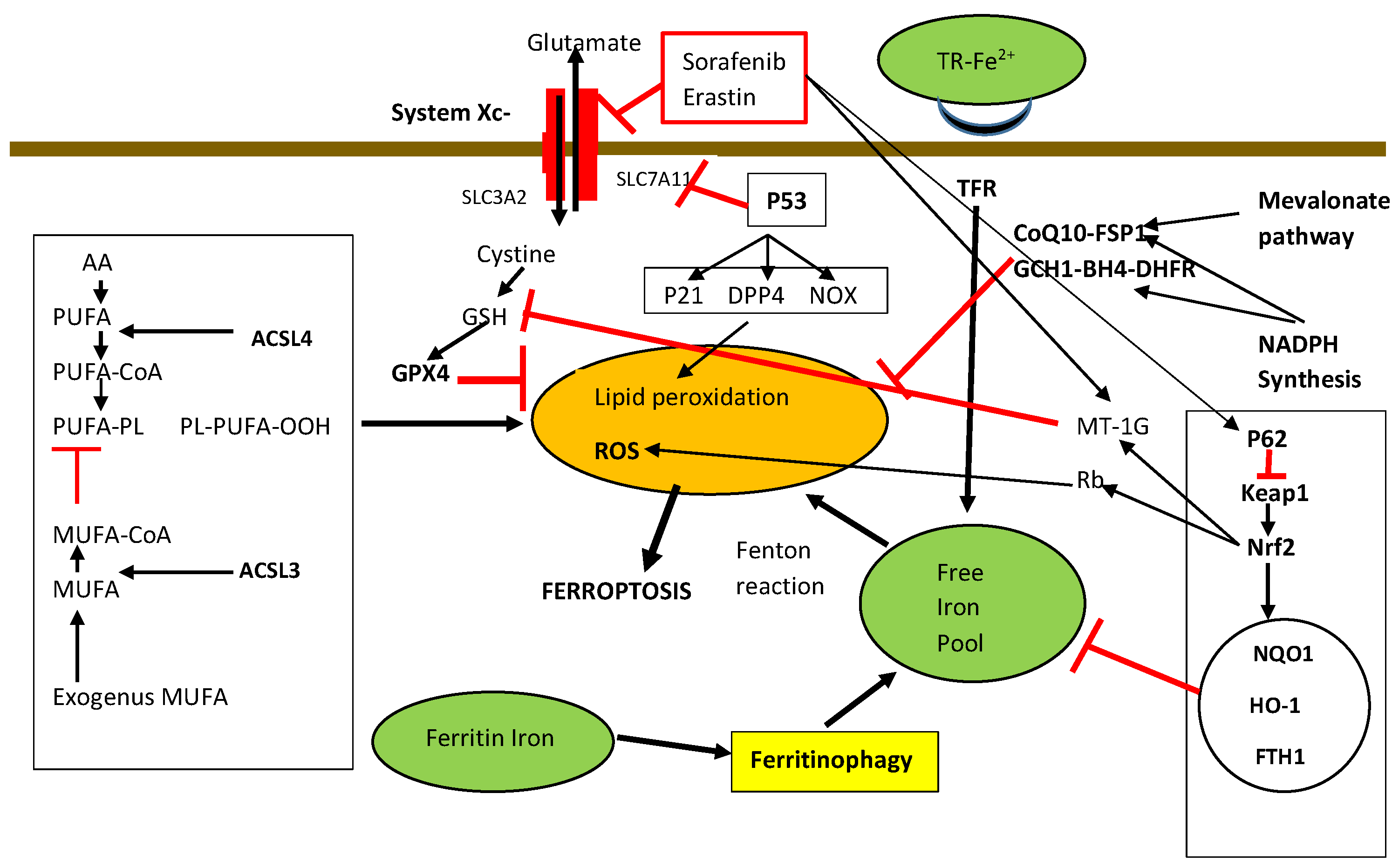
6.12. Ferroptosis
- (1)
- (2)
- Ferritinophagy and other iron metabolism pathways, particularly the p62-Kelch-like ECH-associated protein 1 (Keap1)-Nrf2 regulatory pathways.
- (3)
6.13. Ferroptosis and HCC
7. Implications of Autophagy, Ferroptosis and Apoptosis in the Drug Treatment of HCC
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, A. Hepatocellular carcinoma. N. Engl. J. Med. 2019, 380, 1450–1462. [Google Scholar] [CrossRef] [PubMed]
- Kanda, T.; Goto, T.; Hirotsu, Y.; Moriyama, M.; Omata, M. Molecular mechanisms driving progression of liver cirrhosis towards hepatocellular carcinoma in chronic hepatitis B and C infections: A review. Int. J. Mol. Sci. 2019, 20, 1358. [Google Scholar] [CrossRef] [PubMed]
- Kanda, T.; Goto, T.; Hirotsu, Y.; Masuzaki, R.; Moriyama, M.; Omata, M. Molecular mechanisms: Connections between nonalcoholic fatty liver disease, steatohepatitis and hepatocellular carcinoma. Int. J. Mol. Sci. 2020, 21, 1525. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular carcinoma. Nat. Rev. Dis. Prim. 2021, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Otgonsuren, M.; Henry, L.; Venkatesan, C.; Mishra, A.; Erario, M.; Hunt, S. Association of nonalcoholic fatty liver disease (NAFLD) with hepatocellular carcinoma (HCC) in the United States from 2004 to 2009. Hepatology 2015, 62, 1723–1730. [Google Scholar] [CrossRef] [PubMed]
- Wong, V.W.; Chan, W.K.; Chitturi, S.; Chawla, Y.; Dan, Y.Y.; Duseja, A.; Fan, J.; Goh, K.L.; Hamaguchi, M.; Hashimoto, E.; et al. Asia-Pacific working party on non-alcoholic fatty liver disease guidelines 2017-part 1: Definition, risk factors and assessment. J. Gastroenterol. Hepatol. 2018, 33, 70–85. [Google Scholar] [CrossRef] [PubMed]
- Garuti, F.; Neri, A.; Avanzato, F.; Gramenzi, A.; Rampoldi, D.; Rucci, P.; Farinati, F.; Giannini, E.G.; Piscaglia, F.; Rapaccini, G.L.; et al. The changing scenario of hepatocellular carcinoma in Italy: An update. Liver Int. 2021, 41, 585–597. [Google Scholar] [CrossRef]
- Karageorgos, S.A.; Stratakou, S.; Koulentaki, M.; Voumvouraki, A.; Mantaka, A.; Samonakis, D.; Notas, G.; Kouroumalis, E.A. Long-term change in incidence and risk factors of cirrhosis and hepatocellular carcinoma in Crete, Greece: A 25-year study. Ann. Gastroenterol. 2017, 30, 357–363. [Google Scholar] [CrossRef]
- Obeng, E. Apoptosis (programmed cell death) and its signals-a review. Braz. J. Biol. 2021, 81, 1133–1143. [Google Scholar] [CrossRef]
- Bedoui, S.; Herold, M.J.; Strasser, A. Emerging connectivity of programmed cell death pathways and its physiological implications. Nat. Rev. Mol. Cell Biol. 2020, 21, 678–695. [Google Scholar] [CrossRef]
- Wu, J.; Ye, J.; Kong, W.; Zhang, S.; Zheng, Y. Programmed cell death pathways in hearing loss: A review of apoptosis, autophagy and programmed necrosis. Cell Prolif. 2020, 53, 12915. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Sukumaran, P.; Nascimento Da Conceicao, V.; Sun, Y.; Ahamad, N.; Saraiva, L.R.; Selvaraj, S.; Singh, B.B. Calcium signaling regulates autophagy and apoptosis. Cells 2021, 10, 2125. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Hui, Z.; Wei, S.; Li, D.; Li, W.; Daping, W.; Alahdal, M. IRE1 signaling regulates chondrocyte apoptosis and death fate in the osteoarthritis. J. Cell. Physiol. 2022, 237, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Faruk, M.O.; Ichimura, Y.; Komatsu, M. Selective autophagy. Cancer Sci. 2021, 112, 3972–3978. [Google Scholar] [CrossRef]
- Guo, R.; Wang, H.; Cui, N. Autophagy regulation on pyroptosis: Mechanism and medical implication in sepsis. Mediat. Inflamm. 2021, 2021, 9925059. [Google Scholar] [CrossRef]
- Patra, S.; Praharaj, P.P.; Klionsky, D.J.; Bhutia, S.K. Vorinostat in autophagic cell death: A critical insight into autophagy-mediated, -associated and -dependent cell death for cancer prevention. Drug Discov. Today 2022, 27, 269–279. [Google Scholar] [CrossRef]
- Buratta, S.; Tancini, B.; Sagini, K.; Delo, F.; Chiaradia, E.; Urbanelli, L.; Emiliani, C. Lysosomal exocytosis, exosome release and secretory autophagy: The autophagic-and endo-lysosomal systems go extracellular. Int. J. Mol. Sci. 2020, 21, 2576. [Google Scholar] [CrossRef]
- Schulze, R.J.; Krueger, E.W.; Weller, S.G.; Johnson, K.M.; Casey, C.A.; Schott, M.B.; McNiven, M.A. Direct lysosome-based autophagy of lipid droplets in hepatocytes. Proc. Natl. Acad. Sci. USA 2020, 117, 32443–32452. [Google Scholar] [CrossRef]
- Djulbegovic, M.B.; Uversky, V.N. Ferroptosis-an iron- and disorder-dependent programmed cell death. Int. J. Biol. Macromol. 2019, 135, 1052–1069. [Google Scholar] [CrossRef]
- Zhou, S.Y.; Cui, G.Z.; Yan, X.L.; Wang, X.; Qu, Y.; Guo, Z.N.; Jin, H. Mechanism of ferroptosis and its relationships with other types of programmed cell death: Insights for potential interventions after intracerebral hemorrhage. Front. Neurosci. 2020, 14, 589042. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Kuang, F.; Kroemer, G.; Klionsky, D.J.; Kang, R.; Tang, D. Autophagy-dependent ferroptosis: Machinery and regulation. Cell Chem. Biol. 2020, 27, 420–435. [Google Scholar] [CrossRef] [PubMed]
- Sia, D.; Villanueva, A.; Friedman, S.L.; Llovet, J.M. Liver cancer cell of origin, molecular class, and effects on patient prognosis. Gastroenterology 2017, 152, 745–761. [Google Scholar] [CrossRef] [PubMed]
- Mu, X.; Español-Suñer, R.; Mederacke, I.; Affò, S.; Manco, R.; Sempoux, C.; Lemaigre, F.P.; Adili, A.; Yuan, D.; Weber, A.; et al. Hepatocellular carcinoma originates from hepatocytes and not from the progenitor/biliary compartment. J. Clin. Investig. 2015, 125, 3891–3903. [Google Scholar] [CrossRef]
- Schulze, K.; Nault, J.C.; Villanueva, A. Genetic profiling of hepatocellular carcinoma using next-generation sequencing. J. Hepatol. 2016, 65, 1031–1042. [Google Scholar] [CrossRef]
- Schulze, K.; Imbeaud, S.; Letouzé, E.; Alexandrov, L.B.; Calderaro, J.; Rebouissou, S.; Couchy, G.; Meiller, C.; Shinde, J.; Soysouvanh, F.; et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet. 2015, 47, 505–511. [Google Scholar] [CrossRef]
- Llovet, J.M.; Montal, R.; Sia, D.; Finn, R.S. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat. Rev. Clin. Oncol. 2018, 15, 599–616. [Google Scholar] [CrossRef]
- Rebouissou, S.; Nault, J.C. Advances in molecular classification and precision oncology in hepatocellular carcinoma. J. Hepatol. 2020, 72, 215–229. [Google Scholar] [CrossRef]
- Yang, Y.M.; Kim, S.Y.; Seki, E. Inflammation and liver cancer: Molecular mechanisms and therapeutic targets. Semin. Liver Dis. 2019, 39, 26–42. [Google Scholar] [CrossRef]
- Zong, W.X.; Thompson, C.B. Necrotic death as a cell fate. Genes Dev. 2006, 20, 1–15. [Google Scholar] [CrossRef]
- Iyer, S.S.; Pulskens, W.P.; Sadler, J.J.; Butter, L.M.; Teske, G.J.; Ulland, T.K.; Eisenbarth, S.C.; Florquin, S.; Flavell, R.A.; Leemans, J.C.; et al. Necrotic cells trigger a sterile inflammatory response through the Nlrp3 inflammasome. Proc. Natl. Acad. Sci. USA 2009, 106, 20388–20393. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Bauernfeind, F.; Halle, A.; Samstad, E.O.; Kono, H.; Rock, K.L.; Fitzgerald, K.A.; Latz, E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 2008, 9, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Hurtado-Navarro, L.; Angosto-Bazarra, D.; Pelegrín, P.; Baroja-Mazo, A.; Cuevas, S. NLRP3 inflammasome and pyroptosis in liver pathophysiology: The emerging relevance of Nrf2 inducers. Antioxidants 2022, 11, 870. [Google Scholar] [CrossRef] [PubMed]
- Papadakos, S.P.; Dedes, N.; Kouroumalis, E.; Theocharis, S. The role of the NLRP3 inflammasome in HCC carcinogenesis and treatment: Harnessing innate immunity. Cancers 2022, 14, 3150. [Google Scholar] [CrossRef]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- García-Pras, E.; Fernández-Iglesias, A.; Gracia-Sancho, J.; Pérez-Del-Pulgar, S. Cell death in hepatocellular carcinoma: Pathogenesis and therapeutic opportunities. Cancers 2021, 14, 48. [Google Scholar] [CrossRef]
- Gufler, S.; Seeboeck, R.; Schatz, C.; Haybaeck, J. The translational bridge between inflammation and hepatocarcinogenesis. Cells 2022, 11, 533. [Google Scholar] [CrossRef]
- Weber, K.; Schilling, J.D. Lysosomes integrate metabolic-inflammatory cross-talk in primary macrophage inflammasome activation. J. Biol. Chem. 2014, 289, 9158–9171. [Google Scholar] [CrossRef]
- Codolo, G.; Plotegher, N.; Pozzobon, T.; Brucale, M.; Tessari, I.; Bubacco, L.; de Bernard, M. Triggering of inflammasome by aggregated α-synuclein, an inflammatory response in synucleinopathies. PLoS ONE 2013, 8, 55375. [Google Scholar] [CrossRef]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 inflammasome: An overview of mechanisms of activation and regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef]
- Dostert, C.; Guarda, G.; Romero, J.F.; Menu, P.; Gross, O.; Tardivel, A.; Suva, M.L.; Stehle, J.C.; Kopf, M.; Stamenkovic, I.; et al. Malarial hemozoin is a Nalp3 inflammasome activating danger signal. PLoS ONE 2009, 4, 6510. [Google Scholar] [CrossRef]
- Orlowski, G.M.; Colbert, J.D.; Sharma, S.; Bogyo, M.; Robertson, S.A.; Rock, K.L. Multiple cathepsins promote pro-IL-1β synthesis and NLRP3-mediated IL-1β activation. J. Immunol. 2015, 195, 1685–1697. [Google Scholar] [CrossRef]
- Barlan, A.U.; Griffin, T.M.; McGuire, K.A.; Wiethoff, C.M. Adenovirus membrane penetration activates the NLRP3 inflammasome. J. Virol. 2011, 85, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Khaled, J.; Kopsida, M.; Lennernäs, H.; Heindryckx, F. Drug resistance and endoplasmic reticulum stress in hepatocellular carcinoma. Cells 2022, 11, 632. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, H.; Umemura, A.; Taniguchi, K.; Font-Burgada, J.; Dhar, D.; Ogata, H.; Zhong, Z.; Valasek, M.A.; Seki, E.; Hidalgo, J.; et al. ER stress cooperates with hypernutrition to trigger TNF-dependent spontaneous HCC development. Cancer Cell 2014, 26, 331–343. [Google Scholar] [CrossRef]
- Pavlović, N.; Heindryckx, F. Exploring the role of endoplasmic reticulum stress in hepatocellular carcinoma through mining of the human protein atlas. Biology 2021, 10, 640. [Google Scholar] [CrossRef]
- Wei, J.; Fang, D. Endoplasmic reticulum stress signaling and the pathogenesis of hepatocarcinoma. Int. J. Mol. Sci. 2021, 22, 1799. [Google Scholar] [CrossRef]
- Wu, J.; Qiao, S.; Xiang, Y.; Cui, M.; Yao, X.; Lin, R.; Zhang, X. Endoplasmic reticulum stress: Multiple regulatory roles in hepatocellular carcinoma. Biomed. Pharmacother 2021, 142, 112005. [Google Scholar] [CrossRef]
- Al-Rawashdeh, F.Y.; Scriven, P.; Cameron, I.C.; Vergani, P.V.; Wyld, L. Unfolded protein response activation contributes to chemoresistance in hepatocellular carcinoma. Eur. J. Gastroenterol. Hepatol. 2010, 22, 1099–1105. [Google Scholar] [CrossRef] [PubMed]
- Rahmani, F.; Ziaeemehr, A.; Shahidsales, S.; Gharib, M.; Khazaei, M.; Ferns, G.A.; Ryzhikov, M.; Avan, A.; Hassanian, S.M. Role of regulatory miRNAs of the PI3K/AKT/mTOR signaling in the pathogenesis of hepatocellular carcinoma. J. Cell. Physiol. 2020, 235, 4146–4152. [Google Scholar] [CrossRef] [PubMed]
- Cassim, S.; Raymond, V.A.; Lacoste, B.; Lapierre, P.; Bilodeau, M. Metabolite profiling identifies a signature of tumorigenicity in hepatocellular carcinoma. Oncotarget 2018, 9, 26868–26883. [Google Scholar] [CrossRef] [PubMed]
- Cassim, S.; Raymond, V.A.; Dehbidi-Assadzadeh, L.; Lapierre, P.; Bilodeau, M. Metabolic reprogramming enables hepatocarcinoma cells to efficiently adapt and survive to a nutrient-restricted microenvironment. Cell Cycle 2018, 17, 903–916. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Hu, Z.Q.; Yu, S.Y.; Mao, L.; Zhou, Z.J.; Wang, P.C.; Gong, Y.; Su, S.; Zhou, J.; Fan, J.; et al. CircRPN2 inhibits aerobic glycolysis and metastasis in hepatocellular carcinoma. Cancer Res. 2022, 82, 1055–1069. [Google Scholar] [CrossRef]
- Feng, J.; Li, J.; Wu, L.; Yu, Q.; Ji, J.; Wu, J.; Dai, W.; Guo, C. Emerging roles and the regulation of aerobic glycolysis in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2020, 39, 126. [Google Scholar] [CrossRef]
- Suzuki, T.; Yano, H.; Nakashima, Y.; Nakashima, O.; Kojiro, M. Beta-catenin expression in hepatocellular carcinoma: A possible participation of beta-catenin in the dedifferentiation process. J. Gastroenterol. Hepatol. 2002, 17, 994–1000. [Google Scholar] [CrossRef]
- Leung, H.W.; Leung, C.O.N.; Lau, E.Y.; Chung, K.P.S.; Mok, E.H.; Lei, M.M.L.; Leung, R.W.H.; Tong, M.; Keng, V.W.; Ma, C.; et al. EPHB2 activates β-catenin to enhance cancer stem cell properties and drive sorafenib resistance in hepatocellular carcinoma. Cancer Res. 2021, 81, 3229–3240. [Google Scholar] [CrossRef]
- Audard, V.; Grimber, G.; Elie, C.; Radenen, B.; Audebourg, A.; Letourneur, F.; Soubrane, O.; Vacher-Lavenu, M.C.; Perret, C.; Cavard, C.; et al. Cholestasis is a marker for hepatocellular carcinomas displaying beta-catenin mutations. J. Pathol. 2007, 212, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Xu, Z.; Zhang, Y.; Evert, M.; Calvisi, D.F.; Chen, X. β-Catenin signaling in hepatocellular carcinoma. J. Clin. Investig. 2022, 132, 154515. [Google Scholar] [CrossRef]
- Fan, Z.; Duan, J.; Wang, L.; Xiao, S.; Li, L.; Yan, X.; Yao, W.; Wu, L.; Zhang, S.; Zhang, Y.; et al. PTK2 promotes cancer stem cell traits in hepatocellular carcinoma by activating Wnt/β-catenin signaling. Cancer Lett. 2019, 450, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Karabicici, M.; Azbazdar, Y.; Ozhan, G.; Senturk, S.; Firtina Karagonlar, Z.; Erdal, E. Changes in Wnt and TGF-β signaling mediate the development of regorafenib resistance in hepatocellular carcinoma cell line HuH7. Front. Cell Dev. Biol. 2021, 9, 639779. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Xu, E.; Zhao, Y.; Singh, S.; Li, X.; Couchy, G.; Chen, X.; Zucman-Rossi, J.; Chikina, M.; Monga, S.P. Modeling a human hepatocellular carcinoma subset in mice through coexpression of met and point-mutant β-catenin. Hepatology 2016, 64, 1587–1605. [Google Scholar] [CrossRef] [PubMed]
- Patil, M.A.; Lee, S.A.; Macias, E.; Lam, E.T.; Xu, C.; Jones, K.D.; Ho, C.; Rodriguez-Puebla, M.; Chen, X. Role of cyclin D1 as a mediator of c-Met- and beta-catenin-induced hepatocarcinogenesis. Cancer Res. 2009, 69, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Y.; Wang, J.; Karagoz, E.; Liang, B.; Song, X.; Shang, R.; Evert, K.; Xu, M.; Che, L.; Evert, M.; et al. Axis inhibition protein 1 (Axin1) deletion-induced hepatocarcinogenesis requires intact β-catenin but not Notch cascade in mice. Hepatology 2019, 70, 2003–2017. [Google Scholar] [CrossRef]
- Tao, J.; Zhang, R.; Singh, S.; Poddar, M.; Xu, E.; Oertel, M.; Chen, X.; Ganesh, S.; Abrams, M.; Monga, S.P. Targeting β-catenin in hepatocellular cancers induced by coexpression of mutant β-catenin and K-Ras in mice. Hepatology 2017, 65, 1581–1599. [Google Scholar] [CrossRef]
- Shang, X.Z.; Zhu, H.; Lin, K.; Tu, Z.; Chen, J.; Nelson, D.R.; Liu, C. Stabilized beta-catenin promotes hepatocyte proliferation and inhibits TNFalpha-induced apoptosis. Lab. Investig. 2004, 84, 332–341. [Google Scholar] [CrossRef]
- Tong, Z.; Li, M.; Wang, W.; Mo, P.; Yu, L.; Liu, K.; Ren, W.; Li, W.; Zhang, H.; Xu, J.; et al. Steroid receptor coactivator 1 promotes human hepatocellular carcinoma progression by enhancing Wnt/β-catenin signaling. J. Biol. Chem. 2015, 290, 18596–18608. [Google Scholar] [CrossRef]
- Vasuri, F.; Visani, M.; Acquaviva, G.; Brand, T.; Fiorentino, M.; Pession, A.; Tallini, G.; D’Errico, A.; de Biase, D. Role of microRNAs in the main molecular pathways of hepatocellular carcinoma. World J. Gastroenterol. 2018, 24, 2647–2660. [Google Scholar] [CrossRef]
- Ruiz-Manriquez, L.M.; Carrasco-Morales, O.; Sanchez, Z.E.A.; Osorio-Perez, S.M.; Estrada-Meza, C.; Pathak, S.; Banerjee, A.; Bandyopadhyay, A.; Duttaroy, A.K.; Paul, S. MicroRNA-mediated regulation of key signaling pathways in hepatocellular carcinoma: A mechanistic insight. Front. Genet. 2022, 13, 910733. [Google Scholar] [CrossRef] [PubMed]
- Ahsani, Z.; Mohammadi-Yeganeh, S.; Kia, V.; Karimkhanloo, H.; Zarghami, N.; Paryan, M. WNT1 Gene from WNT signaling pathway is a direct target of miR-122 in hepatocellular carcinoma. Appl. Biochem. Biotechnol. 2017, 181, 884–897. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Tao, Y.; Shan, L.; Chen, R.; Jiang, H.; Qian, Z.; Cai, F.; Ma, L.; Yu, Y. The role of microRNAs in hepatocellular carcinoma. J. Cancer 2018, 9, 3557–3569. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Xu, Q.; Xiao, S.; Wu, Z.; Gong, J.; Liu, C.; Ren, G.; Wu, H. MicroRNA-424-5p acts as a potential biomarker and inhibits proliferation and invasion in hepatocellular carcinoma by targeting TRIM29. Life Sci. 2019, 224, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Khare, S.; Khare, T.; Ramanathan, R.; Ibdah, J.A. Hepatocellular carcinoma: The role of microRNAs. Biomolecules 2022, 12, 645. [Google Scholar] [CrossRef]
- Sasaki, R.; Kanda, T.; Yokosuka, O.; Kato, N.; Matsuoka, S.; Moriyama, M. Exosomes and hepatocellular carcinoma: From bench to bedside. Int. J. Mol. Sci. 2019, 20, 1406. [Google Scholar] [CrossRef]
- Li, S.; Chen, L. Exosomes in pathogenesis, diagnosis, and treatment of hepatocellular carcinoma. Front. Oncol. 2022, 12, 793432. [Google Scholar] [CrossRef]
- Garrett, W.S. Cancer and the microbiota. Science 2015, 348, 80–86. [Google Scholar] [CrossRef]
- Kamiya, T.; Ohtani, N. The role of immune cells in the liver tumor microenvironment: An involvement of gut microbiota-derived factors. Int. Immunol. 2022, 34, 467–474. [Google Scholar] [CrossRef]
- Liew, W.P.; Mohd-Redzwan, S. Mycotoxin: Its impact on gut health and microbiota. Front. Cell. Infect. Microbiol. 2018, 8, 60. [Google Scholar] [CrossRef]
- Iida, N.; Mizukoshi, E.; Yamashita, T.; Yutani, M.; Seishima, J.; Wang, Z.; Arai, K.; Okada, H.; Yamashita, T.; Sakai, Y.; et al. Chronic liver disease enables gut Enterococcus faecalis colonization to promote liver carcinogenesis. Nat. Cancer 2021, 2, 1039–1054. [Google Scholar] [CrossRef] [PubMed]
- Zheng, R.; Wang, G.; Pang, Z.; Ran, N.; Gu, Y.; Guan, X.; Yuan, Y.; Zuo, X.; Pan, H.; Zheng, J.; et al. Liver cirrhosis contributes to the disorder of gut microbiota in patients with hepatocellular carcinoma. Cancer Med. 2020, 9, 4232–4250. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Guo, S.; Zhou, Y.; Zhao, J.; Wang, M.; Sang, L.; Chang, B.; Wang, B. Hepatocellular carcinoma: How the gut microbiota contributes to pathogenesis, diagnosis, and therapy. Front. Microbiol. 2022, 13, 873160. [Google Scholar] [CrossRef] [PubMed]
- Lai, H.T.; Canoy, R.J.; Campanella, M.; Vassetzky, Y.; Brenner, C. Ca2+ transportome and the interorganelle communication in hepatocellular carcinoma. Cells 2022, 11, 815. [Google Scholar] [CrossRef] [PubMed]
- Tümen, D.; Heumann, P.; Gülow, K.; Demirci, C.N.; Cosma, L.S.; Müller, M.; Kandulski, A. Pathogenesis and current treatment strategies of hepatocellular carcinoma. Biomedicines 2022, 10, 3202. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Markowitz, G.J.; Wang, X.F. The hepatitis B virus-associated tumor microenvironment in hepatocellular carcinoma. Natl. Sci. Rev. 2014, 1, 396–412. [Google Scholar] [CrossRef]
- Xie, Y. Hepatitis B virus-associated hepatocellular carcinoma. Adv. Exp. Med. Biol. 2017, 1018, 11–21. [Google Scholar] [CrossRef]
- Zanetto, A.; Campello, E.; Bulato, C.; Gavasso, S.; Saggiorato, G.; Shalaby, S.; Spiezia, L.; Cillo, U.; Farinati, F.; Russo, F.P.; et al. More pronounced hypercoagulable state and hypofibrinolysis in patients with cirrhosis with versus without HCC. Hepatol. Commun. 2021, 5, 1987–2000. [Google Scholar] [CrossRef]
- Zanetto, A.; Senzolo, M.; Campello, E.; Bulato, C.; Gavasso, S.; Shalaby, S.; Gambato, M.; Vitale, A.; Cillo, U.; Farinati, F.; et al. Influence of hepatocellular carcinoma on platelet aggregation in cirrhosis. Cancers 2021, 13, 1150. [Google Scholar] [CrossRef]
- Tu, T.; Budzinska, M.A.; Shackel, N.A.; Urban, S. HBV DNA integration: Molecular mechanisms and clinical implications. Viruses 2017, 9, 75. [Google Scholar] [CrossRef]
- Ringelhan, M.; O′Connor, T.; Protzer, U.; Heikenwalder, M. The direct and indirect roles of HBV in liver cancer: Prospective markers for HCC screening and potential therapeutic targets. J. Pathol. 2015, 235, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Totoki, Y.; Tatsuno, K.; Covington, K.R.; Ueda, H.; Creighton, C.J.; Kato, M.; Tsuji, S.; Donehower, L.A.; Slagle, B.L.; Nakamura, H.; et al. Trans-ancestry mutational landscape of hepatocellular carcinoma genomes. Nat. Genet. 2014, 46, 1267–1273. [Google Scholar] [CrossRef] [PubMed]
- Sung, W.K.; Zheng, H.; Li, S.; Chen, R.; Liu, X.; Li, Y.; Lee, N.P.; Lee, W.H.; Ariyaratne, P.N.; Tennakoon, C.; et al. Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat. Genet. 2012, 44, 765–769. [Google Scholar] [CrossRef] [PubMed]
- Tamori, A.; Yamanishi, Y.; Kawashima, S.; Kanehisa, M.; Enomoto, M.; Tanaka, H.; Kubo, S.; Shiomi, S.; Nishiguchi, S. Alteration of gene expression in human hepatocellular carcinoma with integrated hepatitis B virus DNA. Clin. Cancer Res. 2005, 11, 5821–5826. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, A.; Totoki, Y.; Abe, T.; Boroevich, K.A.; Hosoda, F.; Nguyen, H.H.; Aoki, M.; Hosono, N.; Kubo, M.; Miya, F.; et al. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat. Genet. 2012, 44, 760–764. [Google Scholar] [CrossRef]
- Wang, M.; Xi, D.; Ning, Q. Virus-induced hepatocellular carcinoma with special emphasis on HBV. Hepatol. Int. 2017, 11, 171–180. [Google Scholar] [CrossRef]
- Nakano, M.; Kawaguchi, T.; Nakamoto, S.; Kawaguchi, A.; Kanda, T.; Imazeki, F.; Kuromatsu, R.; Sumie, S.; Satani, M.; Yamada, S.; et al. Effect of occult hepatitis B virus infection on the early-onset of hepatocellular carcinoma in patients with hepatitis C virus infection. Oncol. Rep. 2013, 30, 2049–2055. [Google Scholar] [CrossRef]
- Wang, H.C.; Huang, W.; Lai, M.D.; Su, I.J. Hepatitis B virus pre-S mutants, endoplasmic reticulum stress and hepatocarcinogenesis. Cancer Sci. 2006, 97, 683–688. [Google Scholar] [CrossRef]
- Wang, H.C.; Wu, H.C.; Chen, C.F.; Fausto, N.; Lei, H.Y.; Su, I.J. Different types of ground glass hepatocytes in chronic hepatitis B virus infection contain specific pre-S mutants that may induce endoplasmic reticulum stress. Am. J. Pathol. 2003, 163, 2441–2449. [Google Scholar] [CrossRef]
- D′souza, S.; Lau, K.C.; Coffin, C.S.; Patel, T.R. Molecular mechanisms of viral hepatitis induced hepatocellular carcinoma. World J. Gastroenterol. 2020, 26, 5759–5783. [Google Scholar] [CrossRef]
- Kanda, T.; Yokosuka, O.; Imazeki, F.; Yamada, Y.; Imamura, T.; Fukai, K.; Nagao, K.; Saisho, H. Hepatitis B virus X protein (HBx)-induced apoptosis in HuH-7 cells: Influence of HBV genotype and basal core promoter mutations. Scand. J. Gastroenterol. 2004, 39, 478–485. [Google Scholar] [CrossRef]
- Koike, K.; Shirakata, Y.; Yaginuma, K.; Arii, M.; Takada, S.; Nakamura, I.; Hayashi, Y.; Kawada, M.; Kobayashi, M. Oncogenic potential of hepatitis B virus. Mol. Biol. Med. 1989, 6, 151–160. [Google Scholar] [PubMed]
- Liu, H.; Shi, W.; Luan, F.; Xu, S.; Yang, F.; Sun, W.; Liu, J.; Ma, C. Hepatitis B virus X protein upregulates transcriptional activation of human telomerase reverse transcriptase. Virus Genes 2010, 40, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Zou, S.Q.; Qu, Z.L.; Li, Z.F.; Wang, X. Hepatitis B virus X gene induces human telomerase reverse transcriptase mRNA expression in cultured normal human cholangiocytes. World J. Gastroenterol. 2004, 10, 2259–2262. [Google Scholar] [CrossRef] [PubMed]
- Kojima, H.; Kaita, K.D.; Xu, Z.; Ou, J.H.; Gong, Y.; Zhang, M.; Minuk, G.Y. The absence of up-regulation of telomerase activity during regeneration after partial hepatectomy in hepatitis B virus X gene transgenic mice. J. Hepatol. 2003, 39, 262–268. [Google Scholar] [CrossRef]
- Miller, R.H.; Robinson, W.S. Common evolutionary origin of hepatitis B virus and retroviruses. Proc. Natl. Acad. Sci. USA 1986, 83, 2531–2535. [Google Scholar] [CrossRef] [PubMed]
- Murakami, S. Hepatitis B virus X protein: A multifunctional viral regulator. J. Gastroenterol. 2001, 36, 651–660. [Google Scholar] [CrossRef]
- Torresi, J.; Tran, B.M.; Christiansen, D.; Earnest-Silveira, L.; Schwab, R.H.M.; Vincan, E. HBV-related hepatocarcinogenesis: The role of signalling pathways and innovative ex vivo research models. BMC Cancer 2019, 19, 707. [Google Scholar] [CrossRef]
- Yen, C.J.; Lin, Y.J.; Yen, C.S.; Tsai, H.W.; Tsai, T.F.; Chang, K.Y.; Huang, W.C.; Lin, P.W.; Chiang, C.W.; Chang, T.T. Hepatitis B virus X protein upregulates mTOR signaling through IKKβ to increase cell proliferation and VEGF production in hepatocellular carcinoma. PLoS ONE 2012, 7, 41931. [Google Scholar] [CrossRef]
- Teng, C.F.; Wu, H.C.; Shyu, W.C.; Jeng, L.B.; Su, I.J. Pre-S2 mutant-induced mammalian target of rapamycin signal pathways as potential therapeutic targets for hepatitis B virus-associated hepatocellular carcinoma. Cell. Transplant. 2017, 26, 429–438. [Google Scholar] [CrossRef]
- Wang, X.; Huo, B.; Liu, J.; Huang, X.; Zhang, S.; Feng, T. Hepatitis B virus X reduces hepatocyte apoptosis and promotes cell cycle progression through the Akt/mTOR pathway in vivo. Gene 2019, 691, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Huo, T.I.; Wang, X.W.; Forgues, M.; Wu, C.G.; Spillare, E.A.; Giannini, C.; Brechot, C.; Harris, C.C. Hepatitis B virus X mutants derived from human hepatocellular carcinoma retain the ability to abrogate p53-induced apoptosis. Oncogene 2001, 20, 3620–3628. [Google Scholar] [CrossRef]
- Wang, X.W.; Gibson, M.K.; Vermeulen, W.; Yeh, H.; Forrester, K.; Stürzbecher, H.W.; Hoeijmakers, J.H.; Harris, C.C. Abrogation of p53-induced apoptosis by the hepatitis B virus X gene. Cancer Res. 1995, 55, 6012–6016. [Google Scholar]
- Kim, H.; Lee, H.; Yun, Y. X-gene product of hepatitis B virus induces apoptosis in liver cells. J. Biol. Chem. 1998, 273, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Ou, J.H. Genetic and epigenetic alterations in hepatitis B virus-associated hepatocellular carcinoma. Virol. Sin. 2015, 30, 85–91. [Google Scholar] [CrossRef]
- Rongrui, L.; Na, H.; Zongfang, L.; Fanpu, J.; Shiwen, J. Epigenetic mechanism involved in the HBV/HCV-related hepatocellular carcinoma tumorigenesis. Curr. Pharm. Des. 2014, 20, 1715–1725. [Google Scholar] [CrossRef]
- Zhang, D.; Guo, S.; Schrodi, S.J. Mechanisms of DNA methylation in virus-host interaction in hepatitis B infection: Pathogenesis and oncogenetic properties. Int. J. Mol. Sci. 2021, 22, 9858. [Google Scholar] [CrossRef]
- Liu, X.Y.; Tang, S.H.; Wu, S.L.; Luo, Y.H.; Cao, M.R.; Zhou, H.K.; Jiang, X.W.; Shu, J.C.; Bie, C.Q.; Huang, S.M.; et al. Epigenetic modulation of insulin-like growth factor-II overexpression by hepatitis B virus X protein in hepatocellular carcinoma. Am. J. Cancer Res. 2015, 5, 956–978. [Google Scholar] [PubMed]
- Sarris, M.E.; Moulos, P.; Haroniti, A.; Giakountis, A.; Talianidis, I. Smyd3 is a transcriptional potentiator of multiple cancer-promoting genes and required for liver and colon cancer development. Cancer Cell 2016, 29, 354–366. [Google Scholar] [CrossRef]
- Wang, Y.; Xie, B.H.; Lin, W.H.; Huang, Y.H.; Ni, J.Y.; Hu, J.; Cui, W.; Zhou, J.; Shen, L.; Xu, L.F.; et al. Amplification of SMYD3 promotes tumorigenicity and intrahepatic metastasis of hepatocellular carcinoma via upregulation of CDK2 and MMP2. Oncogene 2019, 38, 4948–4961. [Google Scholar] [CrossRef]
- Yang, L.; He, J.; Chen, L.; Wang, G. Hepatitis B virus X protein upregulates expression of SMYD3 and C-MYC in HepG2 cells. Med. Oncol. 2009, 26, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.; Wang, T.; Xu, X.; Wu, Y.; Tang, Q.; Chen, K. Long non-coding RNAs in hepatitis B virus-related hepatocellular carcinoma: Regulation, functions, and underlying mechanisms. Int. J. Mol. Sci. 2017, 18, 2505. [Google Scholar] [CrossRef] [PubMed]
- Cui, S.; Qian, Z.; Chen, Y.; Li, L.; Li, P.; Ding, H. Screening of up- and downregulation of circRNAs in HBV-related hepatocellular carcinoma by microarray. Oncol. Lett. 2018, 15, 423–432. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Cui, S.; Zhao, W.; Qian, Z.; Liu, H.; Chen, Y.; Lv, F.; Ding, H.G. Screening and bioinformatics analysis of circular RNA expression profiles in hepatitis B-related hepatocellular carcinoma. Cancer Biomark. 2018, 22, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; An, P.; Winkler, C.A.; Yu, Y. Dysregulated microRNAs in hepatitis B virus-related hepatocellular carcinoma: Potential as biomarkers and therapeutic targets. Front. Oncol. 2020, 10, 1271. [Google Scholar] [CrossRef]
- Zhu, H.T.; Liu, R.B.; Liang, Y.Y.; Hasan, A.M.E.; Wang, H.Y.; Shao, Q.; Zhang, Z.C.; Wang, J.; He, C.Y.; Wang, F.; et al. Serum microRNA profiles as diagnostic biomarkers for HBV-positive hepatocellular carcinoma. Liver Int. 2017, 37, 888–896. [Google Scholar] [CrossRef]
- Rana, M.A.; Ijaz, B.; Daud, M.; Tariq, S.; Nadeem, T.; Husnain, T. Interplay of Wnt β-catenin pathway and miRNAs in HBV pathogenesis leading to HCC. Clin. Res. Hepatol. Gastroenterol. 2019, 43, 373–386. [Google Scholar] [CrossRef]
- Baskiran, A.; Atay, A.; Baskiran, D.Y.; Akbulut, S. Hepatitis B/D-related hepatocellular carcinoma. A clinical literature review. J. Gastrointest. Cancer 2021, 52, 1192–1197. [Google Scholar] [CrossRef]
- Diaz, G.; Engle, R.E.; Tice, A.; Melis, M.; Montenegro, S.; Rodriguez-Canales, J.; Hanson, J.; Emmert-Buck, M.R.; Bock, K.W.; Moore, I.N.; et al. Molecular signature and mechanisms of hepatitis D virus-associated hepatocellular carcinoma. Mol. Cancer Res. 2018, 16, 1406–1419. [Google Scholar] [CrossRef]
- Rizzo, G.E.M.; Cabibbo, G.; Craxì, A. Hepatitis B virus-associated hepatocellular carcinoma. Viruses 2022, 14, 986. [Google Scholar] [CrossRef]
- Goossens, N.; Hoshida, Y. Hepatitis C virus-induced hepatocellular carcinoma. Clin. Mol. Hepatol. 2015, 21, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.; Dolganiuc, A.; Szabo, G. Toll-like receptors 1 and 6 are involved in TLR2-mediated macrophage activation by hepatitis C virus core and NS3 proteins. J. Leukoc. Biol. 2007, 82, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Hosomura, N.; Kono, H.; Tsuchiya, M.; Ishii, K.; Ogiku, M.; Matsuda, M.; Fujii, H. HCV-related proteins activate Kupffer cells isolated from human liver tissues. Dig. Dis. Sci. 2011, 56, 1057–1064. [Google Scholar] [CrossRef] [PubMed]
- Goto, K.; Roca Suarez, A.A.; Wrensch, F.; Baumert, T.F.; Lupberger, J. Hepatitis C virus and hepatocellular carcinoma: When the host loses its grip. Int. J. Mol. Sci. 2020, 21, 3057. [Google Scholar] [CrossRef]
- Tian, Z.; Xu, C.; Yang, P.; Lin, Z.; Wu, W.; Zhang, W.; Ding, J.; Ding, R.; Zhang, X.; Dou, K. Molecular pathogenesis: Connections between viral hepatitis-induced and non-alcoholic steatohepatitis-induced hepatocellular carcinoma. Front. Immunol. 2022, 13, 984728. [Google Scholar] [CrossRef]
- Sur, S.; Sasaki, R.; Devhare, P.; Steele, R.; Ray, R.; Ray, R.B. Association between microRNA-373 and long noncoding RNA NORAD in hepatitis C virus-infected hepatocytes impairs Wee1 expression for growth promotion. J. Virol. 2018, 92, 01215–01218. [Google Scholar] [CrossRef]
- Kanda, T.; Tada, M.; Imazeki, F.; Yokosuka, O.; Nagao, K.; Saisho, H. 5-aza-2′-deoxycytidine sensitizes hepatoma and pancreatic cancer cell lines. Oncol. Rep. 2005, 14, 975–979. [Google Scholar] [CrossRef] [PubMed]
- Kanda, T.; Yokosuka, O.; Omata, M. Hepatitis C virus and hepatocellular carcinoma. Biology 2013, 2, 304–316. [Google Scholar] [CrossRef]
- Wirth, T.C.; Manns, M.P. The impact of the revolution in hepatitis C treatment on hepatocellular carcinoma. Ann. Oncol. 2016, 27, 1467–1474. [Google Scholar] [CrossRef]
- Moriya, K.; Fujie, H.; Shintani, Y.; Yotsuyanagi, H.; Tsutsumi, T.; Ishibashi, K.; Matsuura, Y.; Kimura, S.; Miyamura, T.; Koike, K. The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nat. Med. 1998, 4, 1065–1067. [Google Scholar] [CrossRef]
- Kanda, T.; Steele, R.; Ray, R.; Ray, R.B. Hepatitis C virus core protein augments androgen receptor-mediated signaling. J. Virol. 2008, 82, 11066–11072. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Majumder, M.; Steele, R.; Meyer, K.; Ray, R.; Ray, R.B. Hepatitis C virus NS5A protein protects against TNF-alpha mediated apoptotic cell death. Virus Res. 2000, 67, 173–178. [Google Scholar] [CrossRef]
- Majumder, M.; Ghosh, A.K.; Steele, R.; Ray, R.; Ray, R.B. Hepatitis C virus NS5A physically associates with p53 and regulates p21/waf1 gene expression in a p53-dependent manner. J. Virol. 2001, 75, 1401–1407. [Google Scholar] [CrossRef]
- Zhu, Z.; Tran, H.; Mathahs, M.M.; Moninger, T.O.; Schmidt, W.N. HCV induces telomerase reverse transcriptase, increases its catalytic activity, and promotes caspase degradation in infected human hepatocytes. PLoS ONE 2017, 12, 0166853. [Google Scholar] [CrossRef]
- Wang, W.; Pan, Q.; Fuhler, G.M.; Smits, R.; Peppelenbosch, M.P. Action and function of Wnt/β-catenin signaling in the progression from chronic hepatitis C to hepatocellular carcinoma. J. Gastroenterol. 2017, 52, 419–431. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Ding, X.; Tang, J.; Cao, Y.; Hu, P.; Zhou, F.; Shan, X.; Cai, X.; Chen, Q.; Ling, N.; et al. Enhancement of canonical Wnt/β-catenin signaling activity by HCV core protein promotes cell growth of hepatocellular carcinoma cells. PLoS ONE 2011, 6, 27496. [Google Scholar] [CrossRef]
- Park, C.Y.; Choi, S.H.; Kang, S.M.; Kang, J.I.; Ahn, B.Y.; Kim, H.; Jung, G.; Choi, K.Y.; Hwang, S.B. Nonstructural 5A protein activates beta-catenin signaling cascades: Implication of hepatitis C virus-induced liver pathogenesis. J. Hepatol. 2009, 51, 853–864. [Google Scholar] [CrossRef]
- Street, A.; Macdonald, A.; Crowder, K.; Harris, M. The hepatitis C virus NS5A protein activates a phosphoinositide 3-kinase-dependent survival signaling cascade. J. Biol. Chem. 2004, 279, 12232–12241. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Nakao, H.; Tan, S.L.; Polyak, S.J.; Neddermann, P.; Vijaysri, S.; Jacobs, B.L.; Katze, M.G. Subversion of cell signaling pathways by hepatitis C virus nonstructural 5A protein via interaction with Grb2 and P85 phosphatidylinositol 3-kinase. J. Virol. 2002, 76, 9207–9217. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Tong, W.; Zhang, X.; Chen, L.; Yi, Z.; Pan, T.; Hu, Y.; Xiang, L.; Yuan, Z. Hepatitis C virus non-structural protein NS5A interacts with FKBP38 and inhibits apoptosis in Huh7 hepatoma cells. FEBS Lett. 2006, 580, 4392–4400. [Google Scholar] [CrossRef]
- Higgs, M.R.; Lerat, H.; Pawlotsky, J.M. Hepatitis C virus-induced activation of β-catenin promotes c-Myc expression and a cascade of pro-carcinogenetic events. Oncogene 2013, 32, 4683–4693. [Google Scholar] [CrossRef]
- Cotler, S.J.; Hay, N.; Xie, H.; Chen, M.L.; Xu, P.Z.; Layden, T.J.; Guzman, G. Immunohistochemical expression of components of the Akt-mTORC1 pathway is associated with hepatocellular carcinoma in patients with chronic liver disease. Dig. Dis. Sci. 2008, 53, 844–849. [Google Scholar] [CrossRef]
- Zhou, L.; Huang, Y.; Li, J.; Wang, Z. The mTOR pathway is associated with the poor prognosis of human hepatocellular carcinoma. Med. Oncol. 2010, 27, 255–261. [Google Scholar] [CrossRef]
- Aydin, Y.; Chatterjee, A.; Chandra, P.K.; Chava, S.; Chen, W.; Tandon, A.; Dash, A.; Chedid, M.; Moehlen, M.W.; Regenstein, F.; et al. Interferon-alpha-induced hepatitis C virus clearance restores p53 tumor suppressor more than direct-acting antivirals. Hepatol. Commun. 2017, 1, 256–269. [Google Scholar] [CrossRef] [PubMed]
- Dash, S.; Aydin, Y.; Wu, T. Integrated stress response in hepatitis C promotes Nrf2-related chaperone-mediated autophagy: A novel mechanism for host-microbe survival and HCC development in liver cirrhosis. Semin. Cell. Dev. Biol. 2020, 101, 20–35. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.L. Metabolic alterations and hepatitis C: From bench to bedside. World J. Gastroenterol. 2016, 22, 1461–1476. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, S.; Krajewski, M.; Scherer, C.; Scholz, V.; Mordhorst, V.; Truschow, P.; Schöbel, A.; Reimer, R.; Schwudke, D.; Herker, E. Complex lipid metabolic remodeling is required for efficient hepatitis C virus replication. Biochim. Biophys. Acta Mol. Cell. Biol. Lipids 2018, 1863, 1041–1056. [Google Scholar] [CrossRef]
- Luedde, T.; Schwabe, R.F. NF-κB in the liver--linking injury, fibrosis and hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 108–118. [Google Scholar] [CrossRef]
- Hassan, M.; Selimovic, D.; Ghozlan, H.; Abdel-kader, O. Hepatitis C virus core protein triggers hepatic angiogenesis by a mechanism including multiple pathways. Hepatology 2009, 49, 1469–1482. [Google Scholar] [CrossRef]
- Munakata, T.; Liang, Y.; Kim, S.; McGivern, D.R.; Huibregtse, J.; Nomoto, A.; Lemon, S.M. Hepatitis C virus induces E6AP-dependent degradation of the retinoblastoma protein. PLoS Pathog. 2007, 3, 1335–1347. [Google Scholar] [CrossRef]
- Luna-Cuadros, M.A.; Chen, H.W.; Hanif, H.; Ali, M.J.; Khan, M.M.; Lau, D.T. Risk of hepatocellular carcinoma after hepatitis C virus cure. World J. Gastroenterol. 2022, 28, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Dash, S.; Aydin, Y.; Widmer, K.E.; Nayak, L. Hepatocellular carcinoma mechanisms associated with chronic HCV infection and the impact of direct-acting antiviral treatment. J. Hepatocell. Carcinoma 2020, 7, 45–76. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef] [PubMed]
- Margini, C.; Dufour, J.F. The story of HCC in NAFLD: From epidemiology, across pathogenesis, to prevention and treatment. Liver Int. 2016, 36, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Kutlu, O.; Kaleli, H.N.; Ozer, E. Molecular pathogenesis of nonalcoholic steatohepatitis-(NASH-)related hepatocellular carcinoma. Can. J. Gastroenterol. Hepatol. 2018, 2018, 8543763. [Google Scholar] [CrossRef]
- Pinyol, R.; Torrecilla, S.; Wang, H.; Montironi, C.; Piqué-Gili, M.; Torres-Martin, M.; Wei-Qiang, L.; Willoughby, C.E.; Ramadori, P.; Andreu-Oller, C.; et al. Molecular characterisation of hepatocellular carcinoma in patients with non-alcoholic steatohepatitis. J. Hepatol. 2021, 75, 865–878. [Google Scholar] [CrossRef]
- Singal, A.G.; Manjunath, H.; Yopp, A.C.; Beg, M.S.; Marrero, J.A.; Gopal, P.; Waljee, A.K. The effect of PNPLA3 on fibrosis progression and development of hepatocellular carcinoma: A meta-analysis. Am. J. Gastroenterol. 2014, 109, 325–334. [Google Scholar] [CrossRef]
- Sun, H.; Yang, W.; Tian, Y.; Zeng, X.; Zhou, J.; Mok, M.T.S.; Tang, W.; Feng, Y.; Xu, L.; Chan, A.W.H.; et al. An inflammatory-CCRK circuitry drives mTORC1-dependent metabolic and immunosuppressive reprogramming in obesity-associated hepatocellular carcinoma. Nat. Commun. 2018, 9, 5214. [Google Scholar] [CrossRef]
- Pei, Y.; Zhang, T.; Renault, V.; Zhang, X. An overview of hepatocellular carcinoma study by omics-based methods. Acta Biochim. Biophys. Sin. 2009, 41, 1–15. [Google Scholar] [CrossRef]
- Tian, Y.; Arai, E.; Makiuchi, S.; Tsuda, N.; Kuramoto, J.; Ohara, K.; Takahashi, Y.; Ito, N.; Ojima, H.; Hiraoka, N.; et al. Aberrant DNA methylation results in altered gene expression in non-alcoholic steatohepatitis-related hepatocellular carcinomas. J. Cancer Res. Clin. Oncol. 2020, 146, 2461–2477. [Google Scholar] [CrossRef]
- de Conti, A.; Dreval, K.; Tryndyak, V.; Orisakwe, O.E.; Ross, S.A.; Beland, F.A.; Pogribny, I.P. Inhibition of the cell death pathway in nonalcoholic steatohepatitis (NASH)-related hepatocarcinogenesis is associated with histone H4 lysine 16 deacetylation. Mol. Cancer Res. 2017, 15, 1163–1172. [Google Scholar] [CrossRef] [PubMed]
- Takaki, Y.; Saito, Y.; Takasugi, A.; Toshimitsu, K.; Yamada, S.; Muramatsu, T.; Kimura, M.; Sugiyama, K.; Suzuki, H.; Arai, E.; et al. Silencing of microRNA-122 is an early event during hepatocarcinogenesis from non-alcoholic steatohepatitis. Cancer Sci. 2014, 105, 1254–1260. [Google Scholar] [CrossRef]
- Kuramoto, J.; Arai, E.; Tian, Y.; Funahashi, N.; Hiramoto, M.; Nammo, T.; Nozaki, Y.; Takahashi, Y.; Ito, N.; Shibuya, A.; et al. Genome-wide DNA methylation analysis during non-alcoholic steatohepatitis-related multistage hepatocarcinogenesis: Comparison with hepatitis virus-related carcinogenesis. Carcinogenesis 2017, 38, 261–270. [Google Scholar] [CrossRef]
- Liu, F.; Li, H.; Chang, H.; Wang, J.; Lu, J. Identification of hepatocellular carcinoma-associated hub genes and pathways by integrated microarray analysis. Tumori J. 2015, 101, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Xiong, Y.; Sheng, Q.; Zhao, S.; Wattacheril, J.; Flynn, C.R. A micro-RNA expression signature for human NAFLD progression. J. Gastroenterol. 2016, 51, 1022–1030. [Google Scholar] [CrossRef]
- Ringelhan, M.; Pfister, D.; O′Connor, T.; Pikarsky, E.; Heikenwalder, M. The immunology of hepatocellular carcinoma. Nat. Immunol. 2018, 19, 222–232. [Google Scholar] [CrossRef]
- Taniguchi, K.; Karin, M. NF-κB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef] [PubMed]
- Das, M.; Garlick, D.S.; Greiner, D.L.; Davis, R.J. The role of JNK in the development of hepatocellular carcinoma. Genes Dev. 2011, 25, 634–645. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Wong, V.W.; Wong, G.L.; Choi, P.C.; Chan, A.W.; Li, M.K.; Chan, H.Y.; Chim, A.M.; Yu, J.; Sung, J.J.; Chan, H.L. Disease progression of non-alcoholic fatty liver disease: A prospective study with paired liver biopsies at 3 years. Gut 2010, 59, 969–974. [Google Scholar] [CrossRef]
- Pais, R.; Charlotte, F.; Fedchuk, L.; Bedossa, P.; Lebray, P.; Poynard, T.; Ratziu, V. LIDO Study Group. A systematic review of follow-up biopsies reveals disease progression in patients with non-alcoholic fatty liver. J. Hepatol. 2013, 59, 550–556. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Allen, A.M.; Wang, Z.; Prokop, L.J.; Murad, M.H.; Loomba, R. Fibrosis progression in nonalcoholic fatty liver vs nonalcoholic steatohepatitis: A systematic review and meta-analysis of paired-biopsy studies. Clin. Gastroenterol. Hepatol. 2015, 13, 643–654. [Google Scholar] [CrossRef] [PubMed]
- Chettouh, H.; Lequoy, M.; Fartoux, L.; Vigouroux, C.; Desbois-Mouthon, C. Hyperinsulinaemia and insulin signalling in the pathogenesis and the clinical course of hepatocellular carcinoma. Liver Int. 2015, 35, 2203–2217. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, H.; Xiao, H. Metformin actions on the liver: Protection mechanisms emerging in hepatocytes and immune cells against NASH-related HCC. Int. J. Mol. Sci. 2021, 22, 5016. [Google Scholar] [CrossRef]
- Anstee, Q.M.; Reeves, H.L.; Kotsiliti, E.; Govaere, O.; Heikenwalder, M. From NASH to HCC: Current concepts and future challenges. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 411–428. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N.; Yada, N.; Hagiwara, S.; Sakurai, T.; Kitano, M.; Kudo, M. Unique features associated with hepatic oxidative DNA damage and DNA methylation in non-alcoholic fatty liver disease. J. Gastroenterol. Hepatol. 2016, 31, 1646–1653. [Google Scholar] [CrossRef]
- Gentric, G.; Maillet, V.; Paradis, V.; Couton, D.; L’Hermitte, A.; Panasyuk, G.; Fromenty, B.; Celton-Morizur, S.; Desdouets, C. Oxidative stress promotes pathologic polyploidization in nonalcoholic fatty liver disease. J. Clin. Investig. 2015, 125, 981–992. [Google Scholar] [CrossRef]
- Nelson, J.E.; Wilson, L.; Brunt, E.M.; Yeh, M.M.; Kleiner, D.E.; Unalp-Arida, A.; Kowdley, K.V. Nonalcoholic Steatohepatitis Clinical Research Network. Relationship between the pattern of hepatic iron deposition and histological severity in nonalcoholic fatty liver disease. Hepatology 2011, 53, 448–457. [Google Scholar] [CrossRef]
- Hamaguchi, K.; Miyanishi, K.; Osuga, T.; Tanaka, S.; Ito, R.; Sakamoto, H.; Kubo, T.; Ohnuma, H.; Murase, K.; Takada, K.; et al. Association between hepatic oxidative stress related factors and activation of Wnt/β-catenin signaling in NAFLD-induced hepatocellular carcinoma. Cancers 2022, 14, 2066. [Google Scholar] [CrossRef]
- Koike, K.; Moriya, K. Metabolic aspects of hepatitis C viral infection: Steatohepatitis resembling but distinct from NASH. J. Gastroenterol. 2005, 40, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Simon, T.G.; King, L.Y.; Chong, D.Q.; Nguyen, L.H.; Ma, Y.; VoPham, T.; Giovannucci, E.L.; Fuchs, C.S.; Meyerhardt, J.A.; Corey, K.E.; et al. Diabetes, metabolic comorbidities, and risk of hepatocellular carcinoma: Results from two prospective cohort studies. Hepatology 2018, 67, 1797–1806. [Google Scholar] [CrossRef] [PubMed]
- Kramer, J.R.; Natarajan, Y.; Dai, J.; Yu, X.; Li, L.; El-Serag, H.B.; Kanwal, F. Effect of diabetes medications and glycemic control on risk of hepatocellular cancer in patients with nonalcoholic fatty liver disease. Hepatology 2022, 75, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Song, S.; Li, X.; Bian, D.; Wu, X. Association of metabolic traits with occurrence of nonalcoholic fatty liver disease-related hepatocellular carcinoma: A systematic review and meta-analysis of longitudinal cohort studies. Saudi J. Gastroenterol. 2022, 28, 92–100. [Google Scholar] [CrossRef]
- Davila, J.A.; Morgan, R.O.; Shaib, Y.; McGlynn, K.A.; El-Serag, H.B. Diabetes increases the risk of hepatocellular carcinoma in the United States: A population based case control study. Gut 2005, 54, 533–539. [Google Scholar] [CrossRef]
- El-Serag, H.B.; Hampel, H.; Javadi, F. The association between diabetes and hepatocellular carcinoma: A systematic review of epidemiologic evidence. Clin. Gastroenterol. Hepatol. 2006, 4, 369–380. [Google Scholar] [CrossRef]
- Doycheva, I.; Zhang, T.; Amjad, W.; Thuluvath, P.J. Diabetes and hepatocellular carcinoma: Incidence trends and impact of liver disease etiology. J. Clin. Exp. Hepatol. 2020, 10, 296–303. [Google Scholar] [CrossRef]
- Tateishi, R.; Matsumura, T.; Okanoue, T.; Shima, T.; Uchino, K.; Fujiwara, N.; Senokuchi, T.; Kon, K.; Sasako, T.; Taniai, M.; et al. Hepatocellular carcinoma development in diabetic patients: A nationwide survey in Japan. J. Gastroenterol. 2021, 56, 261–273. [Google Scholar] [CrossRef]
- Vetrano, E.; Rinaldi, L.; Mormone, A.; Giorgione, C.; Galiero, R.; Caturano, A.; Nevola, R.; Marfella, R.; Sasso, F.C. Non-alcoholic fatty liver disease (NAFLD), type 2 diabetes, and non-viral hepatocarcinoma: Pathophysiological mechanisms and new therapeutic strategies. Biomedicines 2023, 11, 468. [Google Scholar] [CrossRef]
- Ngo, M.T.; Jeng, H.Y.; Kuo, Y.C.; Diony Nanda, J.; Brahmadhi, A.; Ling, T.Y.; Chang, T.S.; Huang, Y.H. The role of IGF/IGF-1R signaling in hepatocellular carcinomas: Stemness-related properties and drug resistance. Int. J. Mol. Sci. 2021, 22, 1931. [Google Scholar] [CrossRef]
- Lai, S.; Quan, Z.; Hao, Y.; Liu, J.; Wang, Z.; Dai, L.; Dai, H.; He, S.; Tang, B. Long non-coding RNA LINC01572 promotes hepatocellular carcinoma progression via sponging miR-195-5p to enhance PFKFB4-mediated glycolysis and PI3K/AKT activation. Front. Cell Dev. Biol. 2021, 9, 783088. [Google Scholar] [CrossRef] [PubMed]
- Global Burden of Disease Liver Cancer Collaboration; Akinyemiju, T.; Abera, S.; Ahmed, M.; Alam, N.; Alemayohu, M.A.; Allen, C.; Al-Raddadi, R.; Alvis-Guzman, N.; Amoako, Y.; et al. The burden of primary liver cancer and underlying etiologies from 1990 to 2015 at the global, regional, and national level: Results from the Global Burden of Disease Study 2015. JAMA Oncol. 2017, 3, 1683–1691. [Google Scholar] [CrossRef]
- Goutté, N.; Sogni, P.; Bendersky, N.; Barbare, J.C.; Falissard, B.; Farges, O. Geographical variations in incidence, management and survival of hepatocellular carcinoma in a Western country. J. Hepatol. 2017, 66, 537–544. [Google Scholar] [CrossRef]
- Sifaki-Pistolla, D.; Karageorgos, S.A.; Koulentaki, M.; Samonakis, D.; Stratakou, S.; Digenakis, E.; Kouroumalis, E. Geoepidemiology of hepatocellular carcinoma in the island of Crete, Greece. A possible role of pesticides. Liver Int. 2016, 36, 588–594. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H.K.; Bataller, R.; Cortez-Pinto, H.; Gao, B.; Gual, A.; Lackner, C.; Mathurin, P.; Mueller, S.; Szabo, G.; Tsukamoto, H. Alcoholic liver disease. Nat. Rev. Dis. Prim. 2018, 4, 16. [Google Scholar] [CrossRef]
- Sasaki-Tanaka, R.; Ray, R.; Moriyama, M.; Ray, R.B.; Kanda, T. Molecular changes in relation to alcohol consumption and hepatocellular carcinoma. Int. J. Mol. Sci. 2022, 23, 9679. [Google Scholar] [CrossRef] [PubMed]
- Sarsour, E.H.; Kumar, M.G.; Chaudhuri, L.; Kalen, A.L.; Goswami, P.C. Redox control of the cell cycle in health and disease. Antioxid. Redox Signal. 2009, 11, 2985–3011. [Google Scholar] [CrossRef]
- Parlesak, A.; Schäfer, C.; Schütz, T.; Bode, J.C.; Bode, C. Increased intestinal permeability to macromolecules and endotoxemia in patients with chronic alcohol abuse in different stages of alcohol-induced liver disease. J. Hepatol. 2000, 32, 742–747. [Google Scholar] [CrossRef]
- Wheeler, M.D. Endotoxin and Kupffer cell activation in alcoholic liver disease. Alcohol. Res. Health 2003, 27, 300–306. [Google Scholar]
- Fukui, H. Relation of endotoxin, endotoxin binding proteins and macrophages to severe alcoholic liver injury and multiple organ failure. Alcohol. Clin. Exp. Res. 2005, 29, 172–179. [Google Scholar] [CrossRef]
- Méndez-Sánchez, N.; Valencia-Rodriguez, A.; Vera-Barajas, A.; Abenavoli, L.; Scarpellini, E.; Ponciano-Rodriguez, G.; Wang, D.Q. The mechanism of dysbiosis in alcoholic liver disease leading to liver cancer. Hepatoma Res. 2020, 6, 5. [Google Scholar] [CrossRef]
- Mandrekar, P.; Szabo, G. Signalling pathways in alcohol-induced liver inflammation. J. Hepatol. 2009, 50, 1258–1266. [Google Scholar] [CrossRef]
- Park, E.J.; Lee, J.H.; Yu, G.Y.; He, G.; Ali, S.R.; Holzer, R.G.; Osterreicher, C.H.; Takahashi, H.; Karin, M. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell 2010, 140, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Petrasek, J.; Bala, S.; Csak, T.; Lippai, D.; Kodys, K.; Menashy, V.; Barrieau, M.; Min, S.Y.; Kurt-Jones, E.A.; Szabo, G. IL-1 receptor antagonist ameliorates inflammasome-dependent alcoholic steatohepatitis in mice. J. Clin. Investig. 2012, 122, 3476–3489. [Google Scholar] [CrossRef]
- Homann, N.; Stickel, F.; König, I.R.; Jacobs, A.; Junghanns, K.; Benesova, M.; Schuppan, D.; Himsel, S.; Zuber-Jerger, I.; Hellerbrand, C.; et al. Alcohol dehydrogenase 1C*1 allele is a genetic marker for alcohol-associated cancer in heavy drinkers. Int. J. Cancer 2006, 118, 1998–2002. [Google Scholar] [CrossRef] [PubMed]
- Munaka, M.; Kohshi, K.; Kawamoto, T.; Takasawa, S.; Nagata, N.; Itoh, H.; Oda, S.; Katoh, T. Genetic polymorphisms of tobacco- and alcohol-related metabolizing enzymes and the risk of hepatocellular carcinoma. J. Cancer Res. Clin. Oncol. 2003, 129, 355–360. [Google Scholar] [CrossRef]
- Sakamoto, T.; Hara, M.; Higaki, Y.; Ichiba, M.; Horita, M.; Mizuta, T.; Eguchi, Y.; Yasutake, T.; Ozaki, I.; Yamamoto, K.; et al. Influence of alcohol consumption and gene polymorphisms of ADH2 and ALDH2 on hepatocellular carcinoma in a Japanese population. Int. J. Cancer 2006, 118, 1501–1507. [Google Scholar] [CrossRef] [PubMed]
- Salameh, H.; Raff, E.; Erwin, A.; Seth, D.; Nischalke, H.D.; Falleti, E.; Burza, M.A.; Leathert, J.; Romeo, S.; Molinaro, A.; et al. PNPLA3 gene polymorphism is associated with predisposition to and severity of alcoholic liver disease. Am. J. Gastroenterol. 2015, 110, 846–856. [Google Scholar] [CrossRef] [PubMed]
- Stickel, F.; Buch, S.; Lau, K.; Meyer zu Schwabedissen, H.; Berg, T.; Ridinger, M.; Rietschel, M.; Schafmayer, C.; Braun, F.; Hinrichsen, H.; et al. Genetic variation in the PNPLA3 gene is associated with alcoholic liver injury in caucasians. Hepatology 2011, 53, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Buch, S.; Stickel, F.; Trépo, E.; Way, M.; Herrmann, A.; Nischalke, H.D.; Brosch, M.; Rosendahl, J.; Berg, T.; Ridinger, M.; et al. A genome-wide association study confirms PNPLA3 and identifies TM6SF2 and MBOAT7 as risk loci for alcohol-related cirrhosis. Nat. Genet. 2015, 47, 1443–1448. [Google Scholar] [CrossRef] [PubMed]
- Ganne-Carrié, N.; Nahon, P. Hepatocellular carcinoma in the setting of alcohol-related liver disease. J. Hepatol. 2019, 70, 284–293. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H.K.; Stickel, F. Molecular mechanisms of alcohol-mediated carcinogenesis. Nat. Rev. Cancer 2007, 7, 599–612. [Google Scholar] [CrossRef]
- Wu, J.; Wang, Y.; Jiang, R.; Xue, R.; Yin, X.; Wu, M.; Meng, Q. Ferroptosis in liver disease: New insights into disease mechanisms. Cell Death Discov. 2021, 7, 276. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; An, P.; Xie, E.; Wu, Q.; Fang, X.; Gao, H.; Zhang, Z.; Li, Y.; Wang, X.; Zhang, J.; et al. Characterization of ferroptosis in murine models of hemochromatosis. Hepatology 2017, 66, 449–465. [Google Scholar] [CrossRef]
- Kowdley, K.V. Iron, hemochromatosis, and hepatocellular carcinoma. Gastroenterology 2004, 127, S79–S86. [Google Scholar] [CrossRef]
- Elmberg, M.; Hultcrantz, R.; Ekbom, A.; Brandt, L.; Olsson, S.; Olsson, R.; Lindgren, S.; Lööf, L.; Stål, P.; Wallerstedt, S.; et al. Cancer risk in patients with hereditary hemochromatosis and in their first-degree relatives. Gastroenterology 2003, 125, 1733–1741. [Google Scholar] [CrossRef]
- Haider, M.B.; Al Sbihi, A.; Chaudhary, A.J.; Haider, S.M.; Edhi, A.I. Hereditary hemochromatosis: Temporal trends, sociodemographic characteristics, and independent risk factor of hepatocellular cancer-nationwide population-based study. World J. Hepatol. 2022, 14, 1804–1816. [Google Scholar] [CrossRef] [PubMed]
- D′Arcy, M.S. Cell death: A review of the major forms of apoptosis, necrosis and autophagy. Cell. Biol. Int. 2019, 43, 582–592. [Google Scholar] [CrossRef]
- Adams, J.M. Ways of dying: Multiple pathways to apoptosis. Genes Dev. 2003, 17, 2481–2495. [Google Scholar] [CrossRef]
- Kroemer, G.; Galluzzi, L.; Brenner, C. Mitochondrial membrane permeabilization in cell death. Physiol. Rev. 2007, 87, 99–163. [Google Scholar] [CrossRef]
- Marquardt, J.U.; Edlich, F. Predisposition to apoptosis in hepatocellular carcinoma: From mechanistic insights to therapeutic strategies. Front. Oncol. 2019, 9, 1421. [Google Scholar] [CrossRef] [PubMed]
- Czabotar, P.E.; Lessene, G.; Strasser, A.; Adams, J.M. Control of apoptosis by the BCL-2 protein family: Implications for physiology and therapy. Nat. Rev. Mol. Cell. Biol. 2014, 15, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of apoptosis in health and disease: The balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell. Biol. 2019, 20, 175–193. [Google Scholar] [CrossRef]
- Cosentino, K.; García-Sáez, A.J. Bax and Bak pores: Are we closing the circle? Trends Cell. Biol. 2017, 27, 266–275. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, K.L.; Huang, K.; Zhang, J.; Chen, Y.; Luo, X. Inactivation of prosurvival Bcl-2 proteins activates Bax/Bak through the outer mitochondrial membrane. Genes Dev. 2016, 30, 973–988. [Google Scholar] [CrossRef]
- Bertheloot, D.; Latz, E.; Franklin, B.S. Necroptosis, pyroptosis and apoptosis: An intricate game of cell death. Cell. Mol. Immunol. 2021, 18, 1106–1121. [Google Scholar] [CrossRef]
- Wang, Y.; Kanneganti, T.D. From pyroptosis, apoptosis and necroptosis to PANoptosis: A mechanistic compendium of programmed cell death pathways. Comput. Struct. Biotechnol. J. 2021, 19, 4641–4657. [Google Scholar] [CrossRef]
- Ketelut-Carneiro, N.; Fitzgerald, K.A. Apoptosis, pyroptosis, and necroptosis-oh my! The many ways a cell can die. J. Mol. Biol. 2022, 434, 167378. [Google Scholar] [CrossRef]
- Farazi, P.A.; DePinho, R.A. Hepatocellular carcinoma pathogenesis: From genes to environment. Nat. Rev. Cancer 2006, 6, 674–687. [Google Scholar] [CrossRef]
- Moreno-Càceres, J.; Fabregat, I. Apoptosis in liver carcinogenesis and chemotherapy. Hepat. Oncol. 2015, 2, 381–397. [Google Scholar] [CrossRef]
- Locatelli, I.; Sutti, S.; Vacchiano, M.; Bozzola, C.; Albano, E. NF-κB1 deficiency stimulates the progression of non-alcoholic steatohepatitis (NASH) in mice by promoting NKT-cell-mediated responses. Clin. Sci. 2013, 124, 279–287. [Google Scholar] [CrossRef]
- Grohmann, M.; Wiede, F.; Dodd, G.T.; Gurzov, E.N.; Ooi, G.J.; Butt, T.; Rasmiena, A.A.; Kaur, S.; Gulati, T.; Goh, P.K.; et al. Obesity drives STAT-1-dependent NASH and STAT-3-dependent HCC. Cell 2018, 175, 1289–1306. [Google Scholar] [CrossRef] [PubMed]
- Tang, G.; Minemoto, Y.; Dibling, B.; Purcell, N.H.; Li, Z.; Karin, M.; Lin, A. Inhibition of JNK activation through NF-kappaB target genes. Nature 2001, 414, 313–317. [Google Scholar] [CrossRef]
- Maeda, S.; Kamata, H.; Luo, J.L.; Leffert, H.; Karin, M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell 2005, 121, 977–990. [Google Scholar] [CrossRef] [PubMed]
- Vucur, M.; Reisinger, F.; Gautheron, J.; Janssen, J.; Roderburg, C.; Cardenas, D.V.; Kreggenwinkel, K.; Koppe, C.; Hammerich, L.; Hakem, R.; et al. RIP3 inhibits inflammatory hepatocarcinogenesis but promotes cholestasis by controlling caspase-8- and JNK-dependent compensatory cell proliferation. Cell. Rep. 2013, 4, 776–790. [Google Scholar] [CrossRef] [PubMed]
- Schneider, A.T.; Gautheron, J.; Feoktistova, M.; Roderburg, C.; Loosen, S.H.; Roy, S.; Benz, F.; Schemmer, P.; Büchler, M.W.; Nachbur, U.; et al. RIPK1 suppresses a TRAF2-dependent pathway to liver cancer. Cancer Cell 2017, 31, 94–109. [Google Scholar] [CrossRef] [PubMed]
- Cubero, F.J.; Zhao, G.; Nevzorova, Y.A.; Hatting, M.; Al Masaoudi, M.; Verdier, J.; Peng, J.; Schaefer, F.M.; Hermanns, N.; Boekschoten, M.V.; et al. Haematopoietic cell-derived Jnk1 is crucial for chronic inflammation and carcinogenesis in an experimental model of liver injury. J. Hepatol. 2015, 62, 140–149. [Google Scholar] [CrossRef]
- Kanda, T.; Matsuoka, S.; Yamazaki, M.; Shibata, T.; Nirei, K.; Takahashi, H.; Kaneko, T.; Fujisawa, M.; Higuchi, T.; Nakamura, H.; et al. Apoptosis and non-alcoholic fatty liver diseases. World J. Gastroenterol. 2018, 24, 2661–2672. [Google Scholar] [CrossRef]
- Lee, Y.J.; Shukla, S.D. Pro- and anti-apoptotic roles of c-Jun N-terminal kinase (JNK) in ethanol and acetaldehyde exposed rat hepatocytes. Eur. J. Pharmacol. 2005, 508, 31–45. [Google Scholar] [CrossRef]
- Ohsumi, Y. Historical landmarks of autophagy research. Cell. Res. 2014, 24, 9–23. [Google Scholar] [CrossRef]
- Mizushima, N. A brief history of autophagy from cell biology to physiology and disease. Nat. Cell. Biol. 2018, 20, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Tooze, S.A.; Yoshimori, T. The origin of the autophagosomal membrane. Nat. Cell. Biol. 2010, 12, 831–835. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ding, W.X. Adipose tissue autophagy and homeostasis in alcohol-induced liver injury. Liver Res. 2017, 1, 54–62. [Google Scholar] [CrossRef]
- Yu, L.; Chen, Y.; Tooze, S.A. Autophagy pathway: Cellular and molecular mechanisms. Autophagy 2018, 14, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Panigrahi, D.P.; Patil, S.; Bhutia, S.K. Autophagy in health and disease: A comprehensive review. Biomed. Pharmacother. 2018, 104, 485–495. [Google Scholar] [CrossRef]
- Fan, G.; Li, F.; Wang, P.; Jin, X.; Liu, R. Natural-product-mediated autophagy in the treatment of various liver diseases. Int. J. Mol. Sci. 2022, 23, 15109. [Google Scholar] [CrossRef]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell. Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef]
- Cheng, X.; Ma, X.; Zhu, Q.; Song, D.; Ding, X.; Li, L.; Jiang, X.; Wang, X.; Tian, R.; Su, H.; et al. Pacer is a mediator of mTORC1 and GSK3-TIP60 signaling in regulation of autophagosome maturation and lipid metabolism. Mol Cell. 2019, 73, 788–802. [Google Scholar] [CrossRef]
- Ren, H.; Zhao, F.; Zhang, Q.; Huang, X.; Wang, Z. Autophagy and skin wound healing. Burn. Trauma. 2022, 10, tkac003. [Google Scholar] [CrossRef]
- Tamargo-Gómez, I.; Mariño, G. AMPK: Regulation of metabolic dynamics in the context of autophagy. Int. J. Mol. Sci. 2018, 19, 3812. [Google Scholar] [CrossRef]
- Birgisdottir, Å.B.; Johansen, T. Autophagy and endocytosis-interconnections and interdependencies. J. Cell. Sci. 2020, 133, jcs228114. [Google Scholar] [CrossRef] [PubMed]
- Sheng, J.Q.; Wang, M.R.; Fang, D.; Liu, L.; Huang, W.J.; Tian, D.A.; He, X.X.; Li, P.Y. LncRNA NBR2 inhibits tumorigenesis by regulating autophagy in hepatocellular carcinoma. Biomed. Pharmacother. 2021, 133, 111023. [Google Scholar] [CrossRef] [PubMed]
- Di Malta, C.; Cinque, L.; Settembre, C. Transcriptional regulation of autophagy: Mechanisms and diseases. Front. Cell Dev. Biol. 2019, 7, 114. [Google Scholar] [CrossRef] [PubMed]
- Hashemi, M.; Nadafzadeh, N.; Imani, M.H.; Rajabi, R.; Ziaolhagh, S.; Bayanzadeh, S.D.; Norouzi, R.; Rafiei, R.; Koohpar, Z.K.; Raei, B.; et al. Targeting and regulation of autophagy in hepatocellular carcinoma: Revisiting the molecular interactions and mechanisms for new therapy approaches. Cell Commun. Signal. 2023, 21, 32. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; He, J.; Luo, L.; Wang, K. Targeting the interplay of autophagy and ROS for cancer therapy: An updated overview on phytochemicals. Pharmaceuticals 2023, 16, 92. [Google Scholar] [CrossRef] [PubMed]
- Onishi, M.; Yamano, K.; Sato, M.; Matsuda, N.; Okamoto, K. Molecular mechanisms and physiological functions of mitophagy. EMBO J. 2021, 40, e104705. [Google Scholar] [CrossRef]
- Shibutani, S.T.; Saitoh, T.; Nowag, H.; Münz, C.; Yoshimori, T. Autophagy and autophagy-related proteins in the immune system. Nat. Immunol. 2015, 16, 1014–1024. [Google Scholar] [CrossRef]
- de Lavera, I.; Pavon, A.D.; Paz, M.V.; Oropesa-Avila, M.; de la Mata, M.; Alcocer-Gomez, E.; Garrido-Maraver, J.; Cotan, D.; Alvarez-Cordoba, M.; Sanchez-Alcazar, J.A. The connections among autophagy, inflammasome and mitochondria. Curr. Drug Targets 2017, 18, 1030–1038. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, S.; Xiao, Y.; Zhang, W.; Wu, S.; Qin, T.; Yue, Y.; Qian, W.; Li, L. NLRP3 inflammasome and inflammatory diseases. Oxid. Med. Cell. Longev. 2020, 2020, 4063562. [Google Scholar] [CrossRef]
- Codogno, P.; Meijer, A.J. Autophagy in the liver. J. Hepatol. 2013, 59, 389–391. [Google Scholar] [CrossRef]
- Gual, P.; Gilgenkrantz, H.; Lotersztajn, S. Autophagy in chronic liver diseases: The two faces of Janus. Am. J. Physiol. Cell. Physiol. 2017, 312, 263–273. [Google Scholar] [CrossRef]
- Sun, K.; Guo, X.L.; Zhao, Q.D.; Jing, Y.Y.; Kou, X.R.; Xie, X.Q.; Zhou, Y.; Cai, N.; Gao, L.; Zhao, X.; et al. Paradoxical role of autophagy in the dysplastic and tumor-forming stages of hepatocarcinoma development in rats. Cell. Death Dis. 2013, 4, 501. [Google Scholar] [CrossRef]
- Yazdani, H.O.; Huang, H.; Tsung, A. Autophagy: Dual response in the development of hepatocellular carcinoma. Cells 2019, 8, 91. [Google Scholar] [CrossRef] [PubMed]
- Allaire, M.; Rautou, P.E.; Codogno, P.; Lotersztajn, S. Autophagy in liver diseases: Time for translation? J. Hepatol. 2019, 70, 985–998. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.M.; Chan, H.Y.; Aziz, N.A.; Ramasamy, T.S.; Bong, J.J.; Ch′ng, E.S.; Armon, S.; Peh, S.C.; Teow, S.Y. Interplay of autophagy and cancer stem cells in hepatocellular carcinoma. Mol. Biol. Rep. 2021, 48, 3695–3717. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Kuo, C.F.; Sir, D.; Wang, L.; Govindarajan, S.; Petrovic, L.M.; Ou, J.H. Autophagy inhibits oxidative stress and tumor suppressors to exert its dual effect on hepatocarcinogenesis. Cell Death Differ. 2015, 22, 1025–1034. [Google Scholar] [CrossRef] [PubMed]
- Ni, H.M.; Chao, X.; Yang, H.; Deng, F.; Wang, S.; Bai, Q.; Qian, H.; Cui, Y.; Cui, W.; Shi, Y.; et al. Dual roles of mammalian target of rapamycin in regulating liver injury and tumorigenesis in autophagy-defective mouse liver. Hepatology 2019, 70, 2142–2155. [Google Scholar] [CrossRef]
- Lazova, R.; Camp, R.L.; Klump, V.; Siddiqui, S.F.; Amaravadi, R.K.; Pawelek, J.M. Punctate LC3B expression is a common feature of solid tumors and associated with proliferation, metastasis, and poor outcome. Clin. Cancer Res. 2012, 18, 370–379. [Google Scholar] [CrossRef]
- Wu, D.H.; Jia, C.C.; Chen, J.; Lin, Z.X.; Ruan, D.Y.; Li, X.; Lin, Q.; Dong, M.; Ma, X.K.; Wan, X.B.; et al. Autophagic LC3B overexpression correlates with malignant progression and predicts a poor prognosis in hepatocellular carcinoma. Tumour Biol. 2014, 35, 12225–12233. [Google Scholar] [CrossRef]
- Chava, S.; Lee, C.; Aydin, Y.; Chandra, P.K.; Dash, A.; Chedid, M.; Thung, S.N.; Moroz, K.; Wu, T.; Nayak, N.C.; et al. Chaperone-mediated autophagy compensates for impaired macroautophagy in the cirrhotic liver to promote hepatocellular carcinoma. Oncotarget 2017, 8, 40019–40036. [Google Scholar] [CrossRef]
- Karampa, A.D.; Goussia, A.C.; Glantzounis, G.K.; Mastoridou, E.M.; Anastasopoulos, N.T.; Charchanti, A.V. The role of macroautophagy and chaperone-mediated autophagy in the pathogenesis and management of hepatocellular carcinoma. Cancers 2022, 14, 760. [Google Scholar] [CrossRef]
- Turcios, L.; Chacon, E.; Garcia, C.; Eman, P.; Cornea, V.; Jiang, J.; Spear, B.; Liu, C.; Watt, D.S.; Marti, F.; et al. Autophagic flux modulation by Wnt/β-catenin pathway inhibition in hepatocellular carcinoma. PLoS ONE 2019, 14, 0212538. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Xu, L.; Liu, P.; Jairam, K.; Yin, Y.; Chen, K.; Sprengers, D.; Peppelenbosch, M.P.; Pan, Q.; Smits, R. Blocking Wnt secretion reduces growth of hepatocellular carcinoma cell lines mostly independent of β-catenin signaling. Neoplasia 2016, 18, 711–723. [Google Scholar] [CrossRef]
- Qian, H.; Yang, Y.; Wang, X. Curcumin enhanced adriamycin-induced human liver-derived Hepatoma G2 cell death through activation of mitochondria-mediated apoptosis and autophagy. Eur. J. Pharm. Sci. 2011, 43, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Li, Y.; Siraj, S.; Jin, H.; Fan, Y.; Yang, X.; Huang, X.; Wang, X.; Wang, J.; Liu, L.; et al. FUN14 domain-containing 1-mediated mitophagy suppresses hepatocarcinogenesis by inhibition of inflammasome activation in mice. Hepatology 2019, 69, 604–621. [Google Scholar] [CrossRef]
- Huang, Q.; Zhan, L.; Cao, H.; Li, J.; Lyu, Y.; Guo, X.; Zhang, J.; Ji, L.; Ren, T.; An, J.; et al. Increased mitochondrial fission promotes autophagy and hepatocellular carcinoma cell survival through the ROS-modulated coordinated regulation of the NFKB and TP53 pathways. Autophagy 2016, 12, 999–1014. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Lee, J.; Kim, J.Y.; Wang, L.; Tian, Y.; Chan, S.T.; Cho, C.; Machida, K.; Chen, D.; Ou, J.J. Mitophagy controls the activities of tumor suppressor p53 to regulate hepatic cancer stem cells. Mol. Cell 2017, 68, 281–292. [Google Scholar] [CrossRef]
- Tang, H.; Da, L.; Mao, Y.; Li, Y.; Li, D.; Xu, Z.; Li, F.; Wang, Y.; Tiollais, P.; Li, T.; et al. Hepatitis B virus X protein sensitizes cells to starvation-induced autophagy via up-regulation of beclin 1 expression. Hepatology 2009, 49, 60–71. [Google Scholar] [CrossRef]
- Sir, D.; Tian, Y.; Chen, W.L.; Ann, D.K.; Yen, T.S.; Ou, J.H. The early autophagic pathway is activated by hepatitis B virus and required for viral DNA replication. Proc. Natl. Acad. Sci. USA 2010, 107, 4383–4388. [Google Scholar] [CrossRef]
- Liu, B.; Fang, M.; Hu, Y.; Huang, B.; Li, N.; Chang, C.; Huang, R.; Xu, X.; Yang, Z.; Chen, Z.; et al. Hepatitis B virus X protein inhibits autophagic degradation by impairing lysosomal maturation. Autophagy 2014, 10, 416–430. [Google Scholar] [CrossRef]
- Lei, Y.; Xu, X.; Liu, H.; Chen, L.; Zhou, H.; Jiang, J.; Yang, Y.; Wu, B. HBx induces hepatocellular carcinogenesis through ARRB1-mediated autophagy to drive the G1/S cycle. Autophagy 2021, 17, 4423–4441. [Google Scholar] [CrossRef] [PubMed]
- Dash, S.; Aydin, Y.; Moroz, K. Chaperone-mediated autophagy in the liver: Good or bad? Cells 2019, 8, 1308. [Google Scholar] [CrossRef] [PubMed]
- Onal, G.; Kutlu, O.; Gozuacik, D.; Dokmeci Emre, S. Lipid droplets in health and disease. Lipids Health Dis. 2017, 16, 128. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, X.; Chu, E.S.H.; Chen, X.; Kang, W.; Wu, F.; To, K.F.; Wong, V.W.S.; Chan, H.L.Y.; Chan, M.T.V.; et al. Defective lysosomal clearance of autophagosomes and its clinical implications in nonalcoholic steatohepatitis. FASEB J. 2018, 32, 37–51. [Google Scholar] [CrossRef]
- Liu, L.; Liao, J.Z.; He, X.X.; Li, P.Y. The role of autophagy in hepatocellular carcinoma: Friend or foe. Oncotarget 2017, 8, 57707–57722. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Yu, F.; Wang, J.; Guo, C.; Fan, X. Autophagy: A new target for nonalcoholic fatty liver disease therapy. Hepat. Med. 2016, 8, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.K.K.; Zhang, L.; Chan, M.T.V. Autophagy, NAFLD and NAFLD-related HCC. Adv. Exp. Med. Biol. 2018, 1061, 127–138. [Google Scholar] [CrossRef]
- Khambu, B.; Huda, N.; Chen, X.; Antoine, D.J.; Li, Y.; Dai, G.; Köhler, U.A.; Zong, W.X.; Waguri, S.; Werner, S.; et al. HMGB1 promotes ductular reaction and tumorigenesis in autophagy-deficient livers. J. Clin. Investig. 2018, 128, 2419–2435. [Google Scholar] [CrossRef]
- Guo, R.; Xu, X.; Babcock, S.A.; Zhang, Y.; Ren, J. Aldehyde dedydrogenase-2 plays a beneficial role in ameliorating chronic alcohol-induced hepatic steatosis and inflammation through regulation of autophagy. J. Hepatol. 2015, 62, 647–656. [Google Scholar] [CrossRef]
- Niture, S.; Gyamfi, M.A.; Lin, M.; Chimeh, U.; Dong, X.; Zheng, W.; Moore, J.; Kumar, D. TNFAIP8 regulates autophagy, cell steatosis, and promotes hepatocellular carcinoma cell proliferation. Cell Death Dis. 2020, 11, 178. [Google Scholar] [CrossRef]
- Chen, K.D.; Lin, C.C.; Tsai, M.C.; Huang, K.T.; Chiu, K.W. Tumor microenvironment mediated by suppression of autophagic flux drives liver malignancy. Biomed. J. 2018, 41, 163–168. [Google Scholar] [CrossRef]
- Chen, W.; Ma, T.; Shen, X.N.; Xia, X.F.; Xu, G.D.; Bai, X.L.; Liang, T.B. Macrophage-induced tumor angiogenesis is regulated by the TSC2-mTOR pathway. Cancer Res. 2012, 72, 1363–1372. [Google Scholar] [CrossRef] [PubMed]
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gélinas, C.; Fan, Y.; et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006, 10, 51–64. [Google Scholar] [CrossRef]
- Booth, L.A.; Roberts, J.L.; Dent, P. The role of cell signaling in the crosstalk between autophagy and apoptosis in the regulation of tumor cell survival in response to sorafenib and neratinib. Semin. Cancer Biol. 2020, 66, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.C.; Zalckvar, E.; Kimchi, A.; Kroemer, G. Self-eating and self-killing: Crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell. Biol. 2007, 8, 741–752. [Google Scholar] [CrossRef]
- Mariño, G.; Niso-Santano, M.; Baehrecke, E.H.; Kroemer, G. Self-consumption: The interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell. Biol. 2014, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Ravanan, P.; Srikumar, I.F.; Talwar, P. Autophagy: The spotlight for cellular stress responses. Life Sci. 2017, 188, 53–67. [Google Scholar] [CrossRef]
- Chen, Z.H.; Lam, H.C.; Jin, Y.; Kim, H.P.; Cao, J.; Lee, S.J.; Ifedigbo, E.; Parameswaran, H.; Ryter, S.W.; Choi, A.M. Autophagy protein microtubule-associated protein 1 light chain-3B (LC3B) activates extrinsic apoptosis during cigarette smoke-induced emphysema. Proc. Natl. Acad. Sci. USA 2010, 107, 18880–18885. [Google Scholar] [CrossRef]
- Yin, S.; Jin, W.; Qiu, Y.; Fu, L.; Wang, T.; Yu, H. Solamargine induces hepatocellular carcinoma cell apoptosis and autophagy via inhibiting LIF/miR-192-5p/CYR61/Akt signaling pathways and eliciting immunostimulatory tumor microenvironment. J. Hematol. Oncol. 2022, 15, 32. [Google Scholar] [CrossRef]
- Guo, L.; Liang, Y.; Wang, S.; Li, L.; Cai, L.; Heng, Y.; Yang, J.; Jin, X.; Zhang, J.; Yuan, S.; et al. Jujuboside B inhibits the proliferation of breast cancer cell lines by inducing apoptosis and autophagy. Front. Pharmacol. 2021, 12, 668887. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Xu, C.L.; Lu, N.Y.; Qiu, F.F.; Zhao, Y.J.; Chang, Y.X.; Wang, J.H.; Zhao, T.J.; Yuan, X.L. Study on mechanism of curcumol against liver fibrosis based on autophagy and apoptosis of hepatic stellate cells. Zhongguo Zhong Yao Za Zhi 2022, 47, 730–736. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Liu, H.; Liu, S.; Wei, M.; Gao, F.; Xue, J.; Sun, L.; Wang, M.; Jiang, H.; Chen, L. Homocysteine induces human mesangial cell apoptosis via the involvement of autophagy and endoplasmic reticulum stress. RSC Adv. 2019, 9, 31720–31727. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.X.; Ni, H.M.; Gao, W.; Hou, Y.F.; Melan, M.A.; Chen, X.; Stolz, D.B.; Shao, Z.M.; Yin, X.M. Differential effects of endoplasmic reticulum stress-induced autophagy on cell survival. J. Biol. Chem. 2007, 282, 4702–4710. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Zou, Q.; Huang, P.; Xiong, L.; Cheng, Y.; Chen, Q.; Li, Y.; He, H.; Yi, W.; Wei, W. Inhibition of autophagy with chloroquine enhanced apoptosis induced by 5-aminolevulinic acid-photodynamic therapy in secondary hyperparathyroidism primary cells and organoids. Biomed. Pharmacother. 2021, 142, 111994. [Google Scholar] [CrossRef] [PubMed]
- Wang, K. Autophagy and apoptosis in liver injury. Cell Cycle 2015, 14, 1631–1642. [Google Scholar] [CrossRef] [PubMed]
- Kouroumalis, E.; Voumvouraki, A.; Augoustaki, A.; Samonakis, D.N. Autophagy in liver diseases. World J. Hepatol. 2021, 13, 6–65. [Google Scholar] [CrossRef]
- Gómez de Cedrón, M.; Ramírez de Molina, A. Microtargeting cancer metabolism: Opening new therapeutic windows based on lipid metabolism. J. Lipid Res. 2016, 57, 193–206. [Google Scholar] [CrossRef]
- Zhao, T.; Du, H.; Ding, X.; Walls, K.; Yan, C. Activation of mTOR pathway in myeloid-derived suppressor cells stimulates cancer cell proliferation and metastasis in lal(-/-) mice. Oncogene 2015, 34, 1938–1948. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Schlaepfer, I.R.; Bergman, B.C.; Panda, P.K.; Praharaj, P.P.; Naik, P.P.; Agarwal, R.; Bhutia, S.K. ATG14 facilitated lipophagy in cancer cells induce ER stress mediated mitoptosis through a ROS dependent pathway. Free. Radic. Biol. Med. 2017, 104, 199–213. [Google Scholar] [CrossRef]
- Tu, Q.Q.; Zheng, R.Y.; Li, J.; Hu, L.; Chang, Y.X.; Li, L.; Li, M.H.; Wang, R.Y.; Huang, D.D.; Wu, M.C.; et al. Palmitic acid induces autophagy in hepatocytes via JNK2 activation. Acta Pharmacol. Sin. 2014, 35, 504–512. [Google Scholar] [CrossRef]
- Cai, N.; Zhao, X.; Jing, Y.; Sun, K.; Jiao, S.; Chen, X.; Yang, H.; Zhou, Y.; Wei, L. Autophagy protects against palmitate-induced apoptosis in hepatocytes. Cell Biosci. 2014, 4, 28. [Google Scholar] [CrossRef]
- Lou, J.; Wang, Y.; Wang, X.; Jiang, Y. Uncoupling protein 2 regulates palmitic acid-induced hepatoma cell autophagy. Biomed. Res. Int. 2014, 2014, 810401. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Jia, L.; Lin, M.; Shi, Y.; Yin, J.; Liu, Y.; Chen, D.; Meng, Q. ASPP2 attenuates triglycerides to protect against hepatocyte injury by reducing autophagy in a cell and mouse model of non-alcoholic fatty liver disease. J. Cell. Mol. Med. 2015, 19, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Hikita, H.; Tatsumi, T.; Sakamori, R.; Nozaki, Y.; Sakane, S.; Shiode, Y.; Nakabori, T.; Saito, Y.; Hiramatsu, N.; et al. Rubicon inhibits autophagy and accelerates hepatocyte apoptosis and lipid accumulation in nonalcoholic fatty liver disease in mice. Hepatology 2016, 64, 1994–2014. [Google Scholar] [CrossRef] [PubMed]
- Eid, N.; Ito, Y.; Otsuki, Y. Triggering of Parkin mitochondrial translocation in mitophagy: Implications for liver diseases. Front. Pharmacol. 2016, 7, 100. [Google Scholar] [CrossRef]
- Li, S.; Dou, X.; Ning, H.; Song, Q.; Wei, W.; Zhang, X.; Shen, C.; Li, J.; Sun, C.; Song, Z. Sirtuin 3 acts as a negative regulator of autophagy dictating hepatocyte susceptibility to lipotoxicity. Hepatology 2017, 66, 936–952. [Google Scholar] [CrossRef]
- Nikoletopoulou, V.; Markaki, M.; Palikaras, K.; Tavernarakis, N. Crosstalk between apoptosis, necrosis and autophagy. Biochim. Biophys. Acta 2013, 1833, 3448–3459. [Google Scholar] [CrossRef]
- Xie, Z.; Klionsky, D.J. Autophagosome formation: Core machinery and adaptations. Nat. Cell. Biol. 2007, 9, 1102–1109. [Google Scholar] [CrossRef]
- Zeng, X.; Overmeyer, J.H.; Maltese, W.A. Functional specificity of the mammalian Beclin-Vps34 PI 3-kinase complex in macroautophagy versus endocytosis and lysosomal enzyme trafficking. J. Cell. Sci. 2006, 119, 259–270. [Google Scholar] [CrossRef]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef]
- Takacs-Vellai, K.; Vellai, T.; Puoti, A.; Passannante, M.; Wicky, C.; Streit, A.; Kovacs, A.L.; Müller, F. Inactivation of the autophagy gene bec-1 triggers apoptotic cell death in C. elegans. Curr. Biol. 2005, 15, 1513–1517. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.C.; Le Toumelin, G.; Criollo, A.; Rain, J.C.; Gautier, F.; Juin, P.; Tasdemir, E.; Pierron, G.; Troulinaki, K.; Tavernarakis, N.; et al. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J. 2007, 26, 2527–2539. [Google Scholar] [CrossRef] [PubMed]
- Galonek, H.L.; Hardwick, J.M. Upgrading the BCL-2 network. Nat. Cell Biol. 2006, 8, 1317–1319. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.B.; Shi, Y.H.; Zhou, J.; Qiu, S.J.; Xu, Y.; Dai, Z.; Shi, G.M.; Wang, X.Y.; Ke, A.W.; Wu, B.; et al. Association of autophagy defect with a malignant phenotype and poor prognosis of hepatocellular carcinoma. Cancer Res. 2008, 68, 9167–9175. [Google Scholar] [CrossRef] [PubMed]
- Qiu, D.M.; Wang, G.L.; Chen, L.; Xu, Y.Y.; He, S.; Cao, X.L.; Qin, J.; Zhou, J.M.; Zhang, Y.X.; Qun, E. The expression of beclin-1, an autophagic gene, in hepatocellular carcinoma associated with clinical pathological and prognostic significance. BMC Cancer 2014, 14, 327. [Google Scholar] [CrossRef]
- Al-Shenawy, H.A. Expression of Beclin-1, an autophagy-related marker, in chronic hepatitis and hepatocellular carcinoma and its relation with apoptotic markers. APMIS 2016, 124, 229–237. [Google Scholar] [CrossRef]
- Zhang, X.; Jin, L.; Tian, Z.; Wang, J.; Yang, Y.; Liu, J.; Chen, Y.; Hu, C.; Chen, T.; Zhao, Y.; et al. Nitric oxide inhibits autophagy and promotes apoptosis in hepatocellular carcinoma. Cancer Sci. 2019, 110, 1054–1063. [Google Scholar] [CrossRef]
- Tai, W.T.; Shiau, C.W.; Chen, H.L.; Liu, C.Y.; Lin, C.S.; Cheng, A.L.; Chen, P.J.; Chen, K.F. Mcl-1-dependent activation of Beclin 1 mediates autophagic cell death induced by sorafenib and SC-59 in hepatocellular carcinoma cells. Cell. Death. Dis. 2013, 4, 485. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.H.; Ding, Z.B.; Zhou, J.; Hui, B.; Shi, G.M.; Ke, A.W.; Wang, X.Y.; Dai, Z.; Peng, Y.F.; Gu, C.Y.; et al. Targeting autophagy enhances sorafenib lethality for hepatocellular carcinoma via ER stress-related apoptosis. Autophagy 2011, 7, 1159–1172. [Google Scholar] [CrossRef]
- Shimizu, S.; Takehara, T.; Hikita, H.; Kodama, T.; Tsunematsu, H.; Miyagi, T.; Hosui, A.; Ishida, H.; Tatsumi, T.; Kanto, T.; et al. Inhibition of autophagy potentiates the antitumor effect of the multikinase inhibitor sorafenib in hepatocellular carcinoma. Int. J. Cancer 2012, 131, 548–557. [Google Scholar] [CrossRef]
- Chen, S.; Du, Y.; Xu, B.; Li, Q.; Yang, L.; Jiang, Z.; Zeng, Z.; Chen, L. Vaccinia-related kinase 2 blunts sorafenib’s efficacy against hepatocellular carcinoma by disturbing the apoptosis-autophagy balance. Oncogene 2021, 40, 3378–3393. [Google Scholar] [CrossRef] [PubMed]
- Wirawan, E.; Vande Walle, L.; Kersse, K.; Cornelis, S.; Claerhout, S.; Vanoverberghe, I.; Roelandt, R.; De Rycke, R.; Verspurten, J.; Declercq, W.; et al. Caspase-mediated cleavage of Beclin-1 inactivates Beclin-1-induced autophagy and enhances apoptosis by promoting the release of proapoptotic factors from mitochondria. Cell. Death Dis. 2010, 1, 18. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Rubinsztein, D.C. Apoptosis blocks Beclin 1-dependent autophagosome synthesis: An effect rescued by Bcl-xL. Cell Death Differ. 2010, 17, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Yousefi, S.; Perozzo, R.; Schmid, I.; Ziemiecki, A.; Schaffner, T.; Scapozza, L.; Brunner, T.; Simon, H.U. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat. Cell Biol. 2006, 8, 1124–1132. [Google Scholar] [CrossRef]
- Bursch, W.; Karwan, A.; Mayer, M.; Dornetshuber, J.; Fröhwein, U.; Schulte-Hermann, R.; Fazi, B.; Di Sano, F.; Piredda, L.; Piacentini, M.; et al. Cell death and autophagy: Cytokines, drugs, and nutritional factors. Toxicology 2008, 254, 147–157. [Google Scholar] [CrossRef]
- Zou, Z.; Tao, T.; Li, H.; Zhu, X. mTOR signaling pathway and mTOR inhibitors in cancer: Progress and challenges. Cell Biosci. 2020, 10, 31. [Google Scholar] [CrossRef]
- Nie, T.; Yang, S.; Ma, H.; Zhang, L.; Lu, F.; Tao, K.; Wang, R.; Yang, R.; Huang, L.; Mao, Z.; et al. Regulation of ER stress-induced autophagy by GSK3β-TIP60-ULK1 pathway. Cell Death Dis. 2016, 7, 2563. [Google Scholar] [CrossRef]
- Ryu, H.Y.; Kim, L.E.; Jeong, H.; Yeo, B.K.; Lee, J.W.; Nam, H.; Ha, S.; An, H.K.; Park, H.; Jung, S.; et al. GSK3B induces autophagy by phosphorylating ULK1. Exp. Mol. Med. 2021, 53, 369–383. [Google Scholar] [CrossRef]
- Castedo, M.; Ferri, K.F.; Kroemer, G. Mammalian target of rapamycin (mTOR): Pro- and anti-apoptotic. Cell. Death Differ. 2002, 9, 99–100. [Google Scholar] [CrossRef]
- Germain, M.; Nguyen, A.P.; Le Grand, J.N.; Arbour, N.; Vanderluit, J.L.; Park, D.S.; Opferman, J.T.; Slack, R.S. MCL-1 is a stress sensor that regulates autophagy in a developmentally regulated manner. EMBO J. 2011, 30, 395–407. [Google Scholar] [CrossRef]
- Xu, G.; Ma, T.; Zhou, C.; Zhao, F.; Peng, K.; Li, B. β-Carotene attenuates apoptosis and autophagy via PI3K/AKT/mTOR signaling pathway in necrotizing enterocolitis model cells IEC-6. Evid. Based Complement Alternat. Med. 2022, 2022, 2502263. [Google Scholar] [CrossRef] [PubMed]
- Porta, C.; Paglino, C.; Mosca, A. Targeting PI3K/Akt/mTOR signaling in cancer. Front. Oncol. 2014, 4, 64. [Google Scholar] [CrossRef] [PubMed]
- Matter, M.S.; Decaens, T.; Andersen, J.B.; Thorgeirsson, S.S. Targeting the mTOR pathway in hepatocellular carcinoma: Current state and future trends. J. Hepatol. 2014, 60, 855–865. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Pi, C.; Wang, G. Inhibition of PI3K/Akt/mTOR pathway by apigenin induces apoptosis and autophagy in hepatocellular carcinoma cells. Biomed. Pharmacother. 2018, 103, 699–707. [Google Scholar] [CrossRef] [PubMed]
- Ye, R.; Dai, N.; He, Q.; Guo, P.; Xiang, Y.; Zhang, Q.; Hong, Z.; Zhang, Q. Comprehensive anti-tumor effect of Brusatol through inhibition of cell viability and promotion of apoptosis caused by autophagy via the PI3K/Akt/mTOR pathway in hepatocellular carcinoma. Biomed. Pharmacother. 2018, 105, 962–973. [Google Scholar] [CrossRef]
- Wu, Y.; Zhang, Y.; Qin, X.; Geng, H.; Zuo, D.; Zhao, Q. PI3K/AKT/mTOR pathway-related long non-coding RNAs: Roles and mechanisms in hepatocellular carcinoma. Pharmacol. Res. 2020, 160, 105195. [Google Scholar] [CrossRef]
- Guo, M.; Li, N.; Zheng, J.; Wang, W.; Wu, Y.; Han, X.; Guo, J.; Chen, W.; Bai, Z.; Bai, W.; et al. Epigenetic regulation of hepatocellular carcinoma progression through the mTOR signaling pathway. Can. J. Gastroenterol. Hepatol. 2021, 2021, 5596712. [Google Scholar] [CrossRef]
- Wei, L.; Wang, X.; Lv, L.; Liu, J.; Xing, H.; Song, Y.; Xie, M.; Lei, T.; Zhang, N.; Yang, M. The emerging role of microRNAs and long noncoding RNAs in drug resistance of hepatocellular carcinoma. Mol. Cancer 2019, 18, 147. [Google Scholar] [CrossRef] [PubMed]
- Hong, F.; Gao, Y.; Li, Y.; Zheng, L.; Xu, F.; Li, X. Inhibition of HIF1A-AS1 promoted starvation-induced hepatocellular carcinoma cell apoptosis by reducing HIF-1α/mTOR-mediated autophagy. World J. Surg. Oncol. 2020, 18, 113. [Google Scholar] [CrossRef]
- Sun, R.; Zhai, R.; Ma, C.; Miao, W. Combination of aloin and metformin enhances the antitumor effect by inhibiting the growth and invasion and inducing apoptosis and autophagy in hepatocellular carcinoma through PI3K/AKT/mTOR pathway. Cancer Med. 2020, 9, 1141–1151. [Google Scholar] [CrossRef]
- Ferrín, G.; Guerrero, M.; Amado, V.; Rodríguez-Perálvarez, M.; De la Mata, M. Activation of mTOR signaling pathway in hepatocellular carcinoma. Int. J. Mol. Sci. 2020, 21, 1266. [Google Scholar] [CrossRef] [PubMed]
- Besson, A.; Dowdy, S.F.; Roberts, J.M. CDK inhibitors: Cell cycle regulators and beyond. Dev. Cell 2008, 14, 159–169. [Google Scholar] [CrossRef] [PubMed]
- McKay, L.K.; White, J.P. The AMPK/p27Kip1 pathway as a novel target to promote autophagy and resilience in aged cells. Cells 2021, 10, 1430. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Shao, S.H.; Xu, Z.X.; Hennessy, B.; Ding, Z.; Larrea, M.; Kondo, S.; Dumont, D.J.; Gutterman, J.U.; Walker, C.L.; et al. The energy sensing LKB1-AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nat. Cell. Biol. 2007, 9, 218–224. [Google Scholar] [CrossRef]
- White, J.P.; Billin, A.N.; Campbell, M.E.; Russell, A.J.; Huffman, K.M.; Kraus, W.E. The AMPK/p27Kip1 axis regulates autophagy/apoptosis decisions in aged skeletal muscle stem cells. Stem Cell Rep. 2018, 11, 425–439. [Google Scholar] [CrossRef]
- Wang, H.; Luo, J.; Tian, X.; Xu, L.; Zhai, Z.; Cheng, M.; Chen, L.; Luo, S. DNAJC5 promotes hepatocellular carcinoma cells proliferation though regulating SKP2 mediated p27 degradation. Biochim. Biophys. Acta Mol. Cell. Res. 2021, 1868, 118994. [Google Scholar] [CrossRef]
- Luo, Y.; Fu, Z.; Wu, P.; Zheng, D.; Zhang, X. The clinicopathological and prognostic significance of P27kip in hepatocellular carcinoma patients: A systemic review and meta-analysis. Gene 2020, 734, 144351. [Google Scholar] [CrossRef]
- Krueger, A.; Baumann, S.; Krammer, P.H.; Kirchhoff, S. FLICE-inhibitory proteins: Regulators of death receptor-mediated apoptosis. Mol. Cell. Biol. 2001, 21, 8247–8254. [Google Scholar] [CrossRef]
- Lee, A.R.; Park, Y.K.; Dezhbord, M.; Kim, K.H. Interaction between the hepatitis B virus and cellular FLIP variants in viral replication and the innate immune system. Viruses 2022, 14, 373. [Google Scholar] [CrossRef] [PubMed]
- Nakagiri, S.; Murakami, A.; Takada, S.; Akiyama, T.; Yonehara, S. Viral FLIP enhances Wnt signaling downstream of stabilized beta-catenin, leading to control of cell growth. Mol. Cell. Biol. 2005, 25, 9249–9258. [Google Scholar] [CrossRef]
- Bélanger, C.; Gravel, A.; Tomoiu, A.; Janelle, M.E.; Gosselin, J.; Tremblay, M.J.; Flamand, L. Human herpesvirus 8 viral FLICE-inhibitory protein inhibits Fas-mediated apoptosis through binding and prevention of procaspase-8 maturation. J. Hum. Virol. 2001, 4, 62–73. [Google Scholar]
- Feoktistova, M.; Geserick, P.; Kellert, B.; Dimitrova, D.P.; Langlais, C.; Hupe, M.; Cain, K.; MacFarlane, M.; Häcker, G.; Leverkus, M. cIAPs block ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol. Cell 2011, 43, 449–463. [Google Scholar] [CrossRef]
- Lee, J.S.; Li, Q.; Lee, J.Y.; Lee, S.H.; Jeong, J.H.; Lee, H.R.; Chang, H.; Zhou, F.C.; Gao, S.J.; Liang, C.; et al. FLIP-mediated autophagy regulation in cell death control. Nat. Cell. Biol. 2009, 11, 1355–1362. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Seong, B.L. Pro-apoptotic function of HBV X protein is mediated by interaction with c-FLIP and enhancement of death-inducing signal. EMBO J. 2003, 22, 2104–2116. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.R.; Lim, K.H.; Park, E.S.; Kim, D.H.; Park, Y.K.; Park, S.; Kim, D.S.; Shin, G.C.; Kang, H.S.; Won, J.; et al. Multiple functions of cellular FLIP are essential for replication of hepatitis B virus. J. Virol. 2018, 92, 18. [Google Scholar] [CrossRef]
- Saito, K.; Meyer, K.; Warner, R.; Basu, A.; Ray, R.B.; Ray, R. Hepatitis C virus core protein inhibits tumor necrosis factor alpha-mediated apoptosis by a protective effect involving cellular FLICE inhibitory protein. J. Virol. 2006, 80, 4372–4379. [Google Scholar] [CrossRef]
- Park, J.; Kang, W.; Ryu, S.W.; Kim, W.I.; Chang, D.Y.; Lee, D.H.; Park, D.Y.; Choi, Y.H.; Choi, K.; Shin, E.C.; et al. Hepatitis C virus infection enhances TNFα-induced cell death via suppression of NF-κB. Hepatology 2012, 56, 831–840. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.W.; Li, H.Y.; Lau, W.Y.; Cao, L.Q.; Li, Y.; Jiang, X.F.; Yang, X.W.; Xue, P. Gli2 silencing enhances TRAIL-induced apoptosis and reduces tumor growth in human hepatoma cells in vivo. Cancer Biol. Ther. 2014, 15, 1667–1676. [Google Scholar] [CrossRef]
- Rubinstein, A.D.; Eisenstein, M.; Ber, Y.; Bialik, S.; Kimchi, A. The autophagy protein Atg12 associates with antiapoptotic Bcl-2 family members to promote mitochondrial apoptosis. Mol. Cell 2011, 44, 698–709. [Google Scholar] [CrossRef] [PubMed]
- Su, M.; Mei, Y.; Sinha, S. Role of the crosstalk between autophagy and apoptosis in cancer. J. Oncol. 2013, 2013, 102735. [Google Scholar] [CrossRef]
- Geering, B. Death-associated protein kinase 2: Regulator of apoptosis, autophagy and inflammation. Int. J. Biochem. Cell. Biol. 2015, 65, 151–154. [Google Scholar] [CrossRef]
- Raveh, T.; Droguett, G.; Horwitz, M.S.; DePinho, R.A.; Kimchi, A. DAP kinase activates a p19ARF/p53-mediated apoptotic checkpoint to suppress oncogenic transformation. Nat. Cell. Biol. 2001, 3, 1–7. [Google Scholar] [CrossRef]
- Jang, C.W.; Chen, C.H.; Chen, C.C.; Chen, J.Y.; Su, Y.H.; Chen, R.H. TGF-beta induces apoptosis through Smad-mediated expression of DAP-kinase. Nat. Cell. Biol. 2002, 4, 51–58. [Google Scholar] [CrossRef]
- Li, H.; Ray, G.; Yoo, B.H.; Erdogan, M.; Rosen, K.V. Down-regulation of death-associated protein kinase-2 is required for beta-catenin-induced anoikis resistance of malignant epithelial cells. J. Biol. Chem. 2009, 284, 2012–2022. [Google Scholar] [CrossRef] [PubMed]
- Schlegel, C.R.; Fonseca, A.V.; Stöcker, S.; Georgiou, M.L.; Misterek, M.B.; Munro, C.E.; Carmo, C.R.; Seckl, M.J.; Costa-Pereira, A.P. DAPK2 is a novel modulator of TRAIL-induced apoptosis. Cell. Death Differ. 2014, 21, 1780–1791. [Google Scholar] [CrossRef]
- Ber, Y.; Shiloh, R.; Gilad, Y.; Degani, N.; Bialik, S.; Kimchi, A. DAPK2 is a novel regulator of mTORC1 activity and autophagy. Cell. Death Differ. 2015, 22, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Zalckvar, E.; Berissi, H.; Mizrachy, L.; Idelchuk, Y.; Koren, I.; Eisenstein, M.; Sabanay, H.; Pinkas-Kramarski, R.; Kimchi, A. DAP-kinase-mediated phosphorylation on the BH3 domain of beclin 1 promotes dissociation of beclin 1 from Bcl-XL and induction of autophagy. EMBO Rep. 2009, 10, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Gilad, Y.; Shiloh, R.; Ber, Y.; Bialik, S.; Kimchi, A. Discovering protein-protein interactions within the programmed cell death network using a protein-fragment complementation screen. Cell Rep. 2014, 8, 909–921. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, G.G.; Zhang, Z.; Chun, S.; Leung, B.C.; Lai, P.B. Induction of autophagy in hepatocellular carcinoma cells by SB203580 requires activation of AMPK and DAPK but not p38 MAPK. Apoptosis 2012, 17, 325–334. [Google Scholar] [CrossRef]
- Huang, Y.; Wang, C.; Li, K.; Ye, Y.; Shen, A.; Guo, L.; Chen, P.; Meng, C.; Wang, Q.; Yang, X.; et al. Death-associated protein kinase 1 suppresses hepatocellular carcinoma cell migration and invasion by upregulation of DEAD-box helicase 20. Cancer Sci. 2020, 111, 2803–2813. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, L.L.; Tian, Y.; Chen, Y.; Zha, W.H.; Li, Y.; Wu, F.J. Upregulation of DAPK2 ameliorates oxidative damage and apoptosis of placental cells in hypertensive disorder complicating pregnancy by suppressing human placental microvascular endothelial cell autophagy through the mTOR signaling pathway. Int. J. Biol. Macromol. 2019, 121, 488–497. [Google Scholar] [CrossRef]
- Li, T.; Wu, Y.N.; Wang, H.; Ma, J.Y.; Zhai, S.S.; Duan, J. Dapk1 improves inflammation, oxidative stress and autophagy in LPS-induced acute lung injury via p38MAPK/NF-κB signaling pathway. Mol. Immunol. 2020, 120, 13–22. [Google Scholar] [CrossRef]
- Li, Y.; Huang, H.; Yu, H.; Mo, T.; Wei, T.; Li, G.; Jia, Y.; Huang, X.; Tu, M.; Yan, X.; et al. Differential gene expression analysis after DAPK1 knockout in hepatocellular carcinoma cells. PeerJ 2022, 10, 13711. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J. p53, the cellular gatekeeper for growth and division. Cell 1997, 88, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Kuribayashi, K.; El-Deiry, W.S. Regulation of programmed cell death by the p53 pathway. Adv. Exp. Med. Biol. 2008, 615, 201–221. [Google Scholar] [CrossRef] [PubMed]
- Brady, C.A.; Jiang, D.; Mello, S.S.; Johnson, T.M.; Jarvis, L.A.; Kozak, M.M.; Kenzelmann Broz, D.; Basak, S.; Park, E.J.; McLaughlin, M.E.; et al. Distinct p53 transcriptional programs dictate acute DNA-damage responses and tumor suppression. Cell 2011, 145, 571–583. [Google Scholar] [CrossRef]
- Riley, T.; Sontag, E.; Chen, P.; Levine, A. Transcriptional control of human p53-regulated genes. Nat. Rev. Mol. Cell. Biol. 2008, 9, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Kroemer, G. Cytoplasmic functions of the tumour suppressor p53. Nature 2009, 458, 1127–1130. [Google Scholar] [CrossRef]
- Vaseva, A.V.; Marchenko, N.D.; Ji, K.; Tsirka, S.E.; Holzmann, S.; Moll, U.M. p53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell 2012, 149, 1536–1548. [Google Scholar] [CrossRef]
- Vaseva, A.V.; Moll, U.M. The mitochondrial p53 pathway. Biochim. Biophys. Acta 2009, 1787, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018, 25, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Zhang, H.; Levine, A.J.; Jin, S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc. Natl. Acad. Sci. USA 2005, 102, 8204–8209. [Google Scholar] [CrossRef]
- Tasdemir, E.; Maiuri, M.C.; Galluzzi, L.; Vitale, I.; Djavaheri-Mergny, M.; D’Amelio, M.; Criollo, A.; Morselli, E.; Zhu, C.; Harper, F.; et al. Regulation of autophagy by cytoplasmic p53. Nat. Cell. Biol. 2008, 10, 676–687. [Google Scholar] [CrossRef] [PubMed]
- Crighton, D.; Wilkinson, S.; O′Prey, J.; Syed, N.; Smith, P.; Harrison, P.R.; Gasco, M.; Garrone, O.; Crook, T.; Ryan, K.M. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 2006, 126, 121–134. [Google Scholar] [CrossRef]
- Thomas, A.; Giesler, T.; White, E. p53 mediates bcl-2 phosphorylation and apoptosis via activation of the Cdc42/JNK1 pathway. Oncogene 2000, 19, 5259–5269. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Shukla, N.; Singh, S.S.; Kushwaha, S.; Shrivastava, R. Mechanism of interaction between autophagy and apoptosis in cancer. Apoptosis 2021, 26, 512–533. [Google Scholar] [CrossRef] [PubMed]
- Livesey, K.M.; Kang, R.; Vernon, P.; Buchser, W.; Loughran, P.; Watkins, S.C.; Zhang, L.; Manfredi, J.J.; Zeh, H.J., III; Li, L.; et al. p53/HMGB1 complexes regulate autophagy and apoptosis. Cancer Res. 2012, 72, 1996–2005. [Google Scholar] [CrossRef]
- Zhang, X.; Zheng, Q.; Yue, X.; Yuan, Z.; Ling, J.; Yuan, Y.; Liang, Y.; Sun, A.; Liu, Y.; Li, H.; et al. ZNF498 promotes hepatocellular carcinogenesis by suppressing p53-mediated apoptosis and ferroptosis via the attenuation of p53 Ser46 phosphorylation. J. Exp. Clin. Cancer Res. 2022, 41, 79. [Google Scholar] [CrossRef]
- Pratt, M.A.; White, D.; Kushwaha, N.; Tibbo, E.; Niu, M.Y. Cytoplasmic mutant p53 increases Bcl-2 expression in estrogen receptor-positive breast cancer cells. Apoptosis 2007, 12, 657–669. [Google Scholar] [CrossRef]
- Marques, M.A.; de Andrade, G.C.; Silva, J.L.; de Oliveira, G.A.P. Protein of a thousand faces: The tumor-suppressive and oncogenic responses of p53. Front. Mol. Biosci. 2022, 9, 944955. [Google Scholar] [CrossRef]
- Yang, C.; Huang, X.; Li, Y.; Chen, J.; Lv, Y.; Dai, S. Prognosis and personalized treatment prediction in TP53-mutant hepatocellular carcinoma: An in silico strategy towards precision oncology. Brief Bioinform. 2021, 22, 164. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Ge, W.; Zhou, J.; Gao, B.; Qian, X.; Wang, W. The role of tumor associated macrophages in hepatocellular carcinoma. J. Cancer 2021, 12, 1284–1294. [Google Scholar] [CrossRef] [PubMed]
- Ngabire, D.; Kim, G.D. Autophagy and inflammatory response in the tumor microenvironment. Int. J. Mol. Sci. 2017, 18, 2016. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Xu, L.; Jing, Y.; Han, Z.; Chen, X.; Cai, C.; Zhao, P.; Zhao, X.; Yang, L.; Wei, L. Autophagy-deficient Kupffer cells promote tumorigenesis by enhancing mtROS-NF-κB-IL1α/β-dependent inflammation and fibrosis during the preneoplastic stage of hepatocarcinogenesis. Cancer Lett. 2017, 388, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.P.; Su, Y.C.; Lee, P.H.; Lei, H.Y. Targeting NFKB by autophagy to polarize hepatoma-associated macrophage differentiation. Autophagy 2013, 9, 619–621. [Google Scholar] [CrossRef]
- Lin, H.; Yan, J.; Wang, Z.; Hua, F.; Yu, J.; Sun, W.; Li, K.; Liu, H.; Yang, H.; Lv, Q.; et al. Loss of immunity-supported senescence enhances susceptibility to hepatocellular carcinogenesis and progression in Toll-like receptor 2-deficient mice. Hepatology 2013, 57, 171–182. [Google Scholar] [CrossRef]
- Tan, H.Y.; Wang, N.; Man, K.; Tsao, S.W.; Che, C.M.; Feng, Y. Autophagy-induced RelB/p52 activation mediates tumour-associated macrophage repolarisation and suppression of hepatocellular carcinoma by natural compound baicalin. Cell Death Dis. 2015, 6, 1942. [Google Scholar] [CrossRef]
- Zhao, M.; Finlay, D.; Liddington, R.; Vuori, K. SRC plays a specific role in the cross-talk between apoptosis and autophagy via phosphorylation of a novel regulatory site on AMPK. Autophagy Rep. 2022, 1, 38–41. [Google Scholar] [CrossRef]
- Xu, H.; Ye, D.; Ren, M.; Zhang, H.; Bi, F. Ferroptosis in the tumor microenvironment: Perspectives for immunotherapy. Trends Mol Med. 2021, 27, 856–867. [Google Scholar] [CrossRef]
- Gu, X.; Liu, Y.; Dai, X.; Yang, Y.G.; Zhang, X. Deciphering the potential roles of ferroptosis in regulating tumor immunity and tumor immunotherapy. Front Immunol. 2023, 14, 1137107. [Google Scholar] [CrossRef]
- Cai, H.; Ren, Y.; Chen, S.; Wang, Y.; Chu, L. Ferroptosis and tumor immunotherapy: A promising combination therapy for tumors. Front Oncol. 2023, 13, 1119369. [Google Scholar] [CrossRef]
- Guo, J.Y.; Chen, H.Y.; Mathew, R.; Fan, J.; Strohecker, A.M.; Karsli-Uzunbas, G.; Kamphorst, J.J.; Chen, G.; Lemons, J.M.; Karantza, V.; et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011, 25, 460–470. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Sheng, J.; Shen, L.; Su, J.; Xu, Y.; Xie, Q.; Wu, Y.; Zhang, X.; Sun, L. Autophagy inhibitor chloroquine increases sensitivity to cisplatin in QBC939 cholangiocarcinoma cells by mitochondrial ROS. PLoS ONE 2017, 12, 0173712. [Google Scholar] [CrossRef] [PubMed]
- Hori, Y.S.; Hosoda, R.; Akiyama, Y.; Sebori, R.; Wanibuchi, M.; Mikami, T.; Sugino, T.; Suzuki, K.; Maruyama, M.; Tsukamoto, M.; et al. Chloroquine potentiates temozolomide cytotoxicity by inhibiting mitochondrial autophagy in glioma cells. J. Neurooncol. 2015, 122, 11–20. [Google Scholar] [CrossRef]
- Goldsmith, J.; Levine, B.; Debnath, J. Autophagy and cancer metabolism. Methods Enzymol. 2014, 542, 25–57. [Google Scholar] [CrossRef] [PubMed]
- Shetty, S.; Kumar, R.; Bharati, S. Mito-TEMPO, a mitochondria-targeted antioxidant, prevents N-nitrosodiethylamine-induced hepatocarcinogenesis in mice. Free Radic. Biol. Med. 2019, 136, 76–86. [Google Scholar] [CrossRef]
- Shetty, S.; Anushree, U.; Kumar, R.; Bharati, S. Mitochondria-targeted antioxidant, mito-TEMPO mitigates initiation phase of N-Nitrosodiethylamine-induced hepatocarcinogenesis. Mitochondrion 2021, 58, 123–130. [Google Scholar] [CrossRef]
- Li, J.; Jiang, R.; Cong, X.; Zhao, Y. UCP2 gene polymorphisms in obesity and diabetes, and the role of UCP2 in cancer. FEBS Lett. 2019, 593, 2525–2534. [Google Scholar] [CrossRef]
- Fernández-Tussy, P.; Rodríguez-Agudo, R.; Fernández-Ramos, D.; Barbier-Torres, L.; Zubiete-Franco, I.; Davalillo, S.L.; Herraez, E.; Goikoetxea-Usandizaga, N.; Lachiondo-Ortega, S.; Simón, J.; et al. Anti-miR-518d-5p overcomes liver tumor cell death resistance through mitochondrial activity. Cell Death Dis. 2021, 12, 555. [Google Scholar] [CrossRef]
- Hou, Z.L.; Han, F.Y.; Lou, L.L.; Zhao, W.Y.; Huang, X.X.; Yao, G.D.; Song, S.J. The nature compound dehydrocrenatidine exerts potent antihepatocellular carcinoma by destroying mitochondrial complexes in vitro and in vivo. Phytother. Res. 2022, 36, 1353–1371. [Google Scholar] [CrossRef]
- Yao, J.; Wang, J.; Xu, Y.; Guo, Q.; Sun, Y.; Liu, J.; Li, S.; Guo, Y.; Wei, L. CDK9 inhibition blocks the initiation of PINK1-PRKN-mediated mitophagy by regulating the SIRT1-FOXO3-BNIP3 axis and enhances the therapeutic effects involving mitochondrial dysfunction in hepatocellular carcinoma. Autophagy 2022, 18, 1879–1897. [Google Scholar] [CrossRef]
- Abate, M.; Festa, A.; Falco, M.; Lombardi, A.; Luce, A.; Grimaldi, A.; Zappavigna, S.; Sperlongano, P.; Irace, C.; Caraglia, M.; et al. Mitochondria as playmakers of apoptosis, autophagy and senescence. Semin. Cell. Dev. Biol. 2020, 98, 139–153. [Google Scholar] [CrossRef]
- Zhang, C.; Zhao, Y.; Yu, M.; Qin, J.; Ye, B.; Wang, Q. Mitochondrial dysfunction and chronic liver disease. Curr. Issues Mol. Biol. 2022, 44, 3156–3165. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Liu, D.; Chen, L.; He, Y.; Tian, X.; Qi, L.; Chen, L.; Luo, Y.; Chen, Z.; Hu, X.; et al. PNO1 regulates autophagy and apoptosis of hepatocellular carcinoma via the MAPK signaling pathway. Cell Death Dis. 2021, 12, 552. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef] [PubMed]
- Latunde-Dada, G.O. Ferroptosis: Role of lipid peroxidation, iron and ferritinophagy. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 1893–1900. [Google Scholar] [CrossRef]
- Chen, X.; Comish, P.B.; Tang, D.; Kang, R. Characteristics and biomarkers of ferroptosis. Front. Cell. Dev. Biol. 2021, 9, 637162. [Google Scholar] [CrossRef]
- Zheng, J.; Conrad, M. The metabolic underpinnings of ferroptosis. Cell Metab. 2020, 32, 920–937. [Google Scholar] [CrossRef] [PubMed]
- Koppula, P.; Zhuang, L.; Gan, B. Cystine transporter SLC7A11/xCT in cancer: Ferroptosis, nutrient dependency, and cancer therapy. Protein Cell 2021, 12, 599–620. [Google Scholar] [CrossRef]
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.J., III; Kang, R.; Tang, D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016, 12, 1425–1428. [Google Scholar] [CrossRef]
- Sun, X.; Ou, Z.; Chen, R.; Niu, X.; Chen, D.; Kang, R.; Tang, D. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 2016, 63, 173–184. [Google Scholar] [CrossRef]
- Tang, D.; Chen, X.; Kang, R.; Kroemer, G. Ferroptosis: Molecular mechanisms and health implications. Cell Res. 2021, 31, 107–125. [Google Scholar] [CrossRef] [PubMed]
- Dodson, M.; de la Vega, M.R.; Cholanians, A.B.; Schmidlin, C.J.; Chapman, E.; Zhang, D.D. Modulating NRF2 in disease: Timing is everything. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 555–575. [Google Scholar] [CrossRef]
- Song, X.; Zhu, S.; Chen, P.; Hou, W.; Wen, Q.; Liu, J.; Xie, Y.; Liu, J.; Klionsky, D.J.; Kroemer, G.; et al. AMPK-mediated BECN1 phosphorylation promotes ferroptosis by directly blocking system Xc-activity. Curr. Biol. 2018, 28, 2388–2399. [Google Scholar] [CrossRef]
- Zhou, B.; Liu, J.; Kang, R.; Klionsky, D.J.; Kroemer, G.; Tang, D. Ferroptosis is a type of autophagy-dependent cell death. Semin. Cancer Biol. 2020, 66, 89–100. [Google Scholar] [CrossRef]
- Hong, S.H.; Lee, D.H.; Lee, Y.S.; Jo, M.J.; Jeong, Y.A.; Kwon, W.T.; Choudry, H.A.; Bartlett, D.L.; Lee, Y.J. Molecular crosstalk between ferroptosis and apoptosis: Emerging role of ER stress-induced p53-independent PUMA expression. Oncotarget 2017, 8, 115164–115178. [Google Scholar] [CrossRef]
- Huang, C.; Yang, M.; Deng, J.; Li, P.; Su, W.; Jiang, R. Upregulation and activation of p53 by erastin-induced reactive oxygen species contribute to cytotoxic and cytostatic effects in A549 lung cancer cells. Oncol. Rep. 2018, 40, 2363–2370. [Google Scholar] [CrossRef] [PubMed]
- Kew, M.C. Hepatic iron overload and hepatocellular carcinoma. Liver Cancer 2014, 3, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Torti, S.V.; Manz, D.H.; Paul, B.T.; Blanchette-Farra, N.; Torti, F.M. Iron and cancer. Annu. Rev. Nutr. 2018, 38, 97–125. [Google Scholar] [CrossRef]
- Liao, H.; Shi, J.; Wen, K.; Lin, J.; Liu, Q.; Shi, B.; Yan, Y.; Xiao, Z. Molecular targets of ferroptosis in hepatocellular carcinoma. J. Hepatocell. Carcinoma 2021, 8, 985–996. [Google Scholar] [CrossRef]
- Jennis, M.; Kung, C.P.; Basu, S.; Budina-Kolomets, A.; Leu, J.I.; Khaku, S.; Scott, J.P.; Cai, K.Q.; Campbell, M.R.; Porter, D.K.; et al. An African-specific polymorphism in the TP53 gene impairs p53 tumor suppressor function in a mouse model. Genes Dev. 2016, 30, 918–930. [Google Scholar] [CrossRef]
- Nie, J.; Lin, B.; Zhou, M.; Wu, L.; Zheng, T. Role of ferroptosis in hepatocellular carcinoma. J. Cancer Res. Clin. Oncol. 2018, 144, 2329–2337. [Google Scholar] [CrossRef]
- Sun, X.; Niu, X.; Chen, R.; He, W.; Chen, D.; Kang, R.; Tang, D. Metallothionein-1G facilitates sorafenib resistance through inhibition of ferroptosis. Hepatology 2016, 64, 488–500. [Google Scholar] [CrossRef] [PubMed]
- Shang, Y.; Luo, M.; Yao, F.; Wang, S.; Yuan, Z.; Yang, Y. Ceruloplasmin suppresses ferroptosis by regulating iron homeostasis in hepatocellular carcinoma cells. Cell. Signal. 2020, 72, 109633. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Lu, P.Z.; Zhu, G.Z.; Hooi, S.C.; Wu, Y.; Huang, X.W.; Dai, H.Q.; Chen, P.H.; Li, Z.J.; Su, W.J.; et al. ACSL4 is a predictive biomarker of sorafenib sensitivity in hepatocellular carcinoma. Acta Pharmacol. Sin. 2021, 42, 160–170. [Google Scholar] [CrossRef]
- Sun, X.J.; Xu, G.L. Overexpression of Acyl-CoA Ligase 4 (ACSL4) in patients with hepatocellular carcinoma and its prognosis. Med. Sci. Monit. 2017, 23, 4343–4350. [Google Scholar] [CrossRef] [PubMed]
- Quan, J.; Bode, A.M.; Luo, X. ACSL family: The regulatory mechanisms and therapeutic implications in cancer. Eur. J. Pharmacol. 2021, 909, 174397. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, J.; Kang, R.; Klionsky, D.J.; Tang, D. Ferroptosis: Machinery and regulation. Autophagy 2021, 17, 2054–2081. [Google Scholar] [CrossRef]
- Chen, J.; Ding, C.; Chen, Y.; Hu, W.; Yu, C.; Peng, C.; Feng, X.; Cheng, Q.; Wu, W.; Lu, Y.; et al. ACSL4 reprograms fatty acid metabolism in hepatocellular carcinoma via c-Myc/SREBP1 pathway. Cancer Lett. 2021, 502, 154–165. [Google Scholar] [CrossRef]
- Li, H.; Song, J.; He, Y.; Liu, Y.; Liu, Z.; Sun, W.; Hu, W.; Lei, Q.Y.; Hu, X.; Chen, Z.; et al. CRISPR/Cas9 screens reveal that hexokinase 2 enhances cancer stemness and tumorigenicity by activating the ACSL4-fatty acid β-oxidation pathway. Adv. Sci. 2022, 9, e2105126. [Google Scholar] [CrossRef]
- Han, Y.M.; Jeong, M.; Park, J.M.; Kim, M.Y.; Go, E.J.; Cha, J.Y.; Kim, K.J.; Hahm, K.B. The ω-3 polyunsaturated fatty acids prevented colitis-associated carcinogenesis through blocking dissociation of β-catenin complex, inhibiting COX-2 through repressing NF-κB, and inducing 15-prostaglandin dehydrogenase. Oncotarget 2016, 7, 63583–63595. [Google Scholar] [CrossRef] [PubMed]
- Ou, W.; Mulik, R.S.; Anwar, A.; McDonald, J.G.; He, X.; Corbin, I.R. Low-density lipoprotein docosahexaenoic acid nanoparticles induce ferroptotic cell death in hepatocellular carcinoma. Free Radic. Biol. Med. 2017, 112, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Weylandt, K.H.; Krause, L.F.; Gomolka, B.; Chiu, C.Y.; Bilal, S.; Nadolny, A.; Waechter, S.F.; Fischer, A.; Rothe, M.; Kang, J.X. Suppressed liver tumorigenesis in fat-1 mice with elevated omega-3 fatty acids is associated with increased omega-3 derived lipid mediators and reduced TNF-α. Carcinogenesis 2011, 32, 897–903. [Google Scholar] [CrossRef]
- Lim, L.J.; Wong, S.Y.S.; Huang, F.; Lim, S.; Chong, S.S.; Ooi, L.L.; Kon, O.L.; Lee, C.G. Roles and regulation of long noncoding RNAs in hepatocellular carcinoma. Cancer Res. 2019, 79, 5131–5139. [Google Scholar] [CrossRef]
- Qi, W.; Li, Z.; Xia, L.; Dai, J.; Zhang, Q.; Wu, C.; Xu, S. LncRNA GABPB1-AS1 and GABPB1 regulate oxidative stress during erastin-induced ferroptosis in HepG2 hepatocellular carcinoma cells. Sci. Rep. 2019, 9, 16185. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Peng, B.; Liang, Q.; Chen, X.; Cai, Y.; Zeng, S.; Gao, K.; Wang, X.; Yi, Q.; Gong, Z.; et al. Construction of a ferroptosis-related nine-lncRNA signature for predicting prognosis and immune response in hepatocellular carcinoma. Front. Immunol. 2021, 12, 719175. [Google Scholar] [CrossRef]
- Xiong, Y.; Ouyang, Y.; Fang, K.; Sun, G.; Tu, S.; Xin, W.; Wei, Y.; Xiao, W. Prediction of prognosis and molecular mechanism of ferroptosis in hepatocellular carcinoma based on bioinformatics methods. Comput. Math. Methods Med. 2022, 2022, 4558782. [Google Scholar] [CrossRef]
- Lyu, N.; Zeng, Y.; Kong, Y.; Chen, Q.; Deng, H.; Ou, S.; Bai, Y.; Tang, H.; Wang, X.; Zhao, M. Ferroptosis is involved in the progression of hepatocellular carcinoma through the circ0097009/miR-1261/SLC7A11 axis. Ann. Transl. Med. 2021, 9, 675. [Google Scholar] [CrossRef]
- Zhang, B.; Zhao, J.; Liu, B.; Shang, Y.; Chen, F.; Zhang, S.; He, J.; Fan, Y.; Tan, K. Development and validation of a novel ferroptosis-related gene signature for prognosis and immunotherapy in hepatocellular carcinoma. Front. Mol. Biosci. 2022, 9, 940575. [Google Scholar] [CrossRef]
- Liang, J.Y.; Wang, D.S.; Lin, H.C.; Chen, X.X.; Yang, H.; Zheng, Y.; Li, Y.H. A novel ferroptosis-related gene signature for overall survival prediction in patients with hepatocellular carcinoma. Int. J. Biol. Sci. 2020, 16, 2430–2441. [Google Scholar] [CrossRef] [PubMed]
- Pan, F.; Lin, X.; Hao, L.; Wang, T.; Song, H.; Wang, R. The critical role of ferroptosis in hepatocellular carcinoma. Front. Cell Dev. Biol. 2022, 10, 882571. [Google Scholar] [CrossRef]
- Huang, Z.; Xia, H.; Cui, Y.; Yam, J.W.P.; Xu, Y. Ferroptosis: From basic research to clinical therapeutics in hepatocellular carcinoma. J. Clin. Transl. Hepatol. 2023, 11, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Brunetti, O.; Gnoni, A.; Licchetta, A.; Longo, V.; Calabrese, A.; Argentiero, A.; Delcuratolo, S.; Solimando, A.G.; Casadei-Gardini, A.; Silvestris, N. Predictive and prognostic factors in HCC patients treated with sorafenib. Medicina 2019, 55, 707. [Google Scholar] [CrossRef] [PubMed]
- Facciorusso, A.; Abd El Aziz, M.A.; Singh, S.; Pusceddu, S.; Milione, M.; Giacomelli, L.; Sacco, R. Statin use decreases the incidence of hepatocellular carcinoma: An updated meta-analysis. Cancers 2020, 12, 874. [Google Scholar] [CrossRef]
- Solimando, A.G.; Susca, N.; Argentiero, A.; Brunetti, O.; Leone, P.; De Re, V.; Fasano, R.; Krebs, M.; Petracci, E.; Azzali, I.; et al. Second-line treatments for advanced hepatocellular carcinoma: A systematic review and Bayesian network meta-analysis. Clin. Exp. Med. 2022, 22, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Zhu, J.; Li, J.; Fan, K.; Gao, Y.; Cheng, S.; Kong, C.; Zheng, L.; Wu, F.; Weng, Q.; et al. The ferroptosis and iron-metabolism signature robustly predicts clinical diagnosis, prognosis and immune microenvironment for hepatocellular carcinoma. Cell Commun. Signal. 2020, 18, 174. [Google Scholar] [CrossRef]
- Brun, S.; Bestion, E.; Raymond, E.; Bassissi, F.; Jilkova, Z.M.; Mezouar, S.; Rachid, M.; Novello, M.; Tracz, J.; Hamaï, A.; et al. GNS561, a clinical-stage PPT1 inhibitor, is efficient against hepatocellular carcinoma via modulation of lysosomal functions. Autophagy 2022, 18, 678–694. [Google Scholar] [CrossRef]
- Wang, W.; Green, M.; Choi, J.E.; Gijón, M.; Kennedy, P.D.; Johnson, J.K.; Liao, P.; Lang, X.; Kryczek, I.; Sell, A.; et al. CD8+ T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 2019, 569, 270–274. [Google Scholar] [CrossRef]
- Zheng, C.; Peng, Y.; Wang, H.; Wang, Y.; Liu, L.; Zhao, Q. Identification and validation of ferroptosis-related subtypes and a predictive signature in hepatocellular carcinoma. Pharmgenomics Pers. Med. 2023, 16, 39–58. [Google Scholar] [CrossRef]
- Fan, G.; Wei, X.; Xu, X. Is the era of sorafenib over? A review of the literature. Ther. Adv. Med. Oncol. 2020, 12, 1758835920927602. [Google Scholar] [CrossRef]
- Louandre, C.; Ezzoukhry, Z.; Godin, C.; Barbare, J.C.; Mazière, J.C.; Chauffert, B.; Galmiche, A. Iron-dependent cell death of hepatocellular carcinoma cells exposed to sorafenib. Int. J. Cancer 2013, 133, 1732–1742. [Google Scholar] [CrossRef]
- Gao, R.; Kalathur, R.K.R.; Coto-Llerena, M.; Ercan, C.; Buechel, D.; Shuang, S.; Piscuoglio, S.; Dill, M.T.; Camargo, F.D.; Christofori, G.; et al. YAP/TAZ and ATF4 drive resistance to Sorafenib in hepatocellular carcinoma by preventing ferroptosis. EMBO Mol. Med. 2021, 13, e14351. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Yao, Y.; Rao, Y.; Huang, X.; Wei, L.; You, Z.; Zheng, G.; Hou, X.; Su, Y.; Varghese, Z.; et al. Cholesterol sensor SCAP contributes to sorafenib resistance by regulating autophagy in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2022, 41, 116. [Google Scholar] [CrossRef]
- Jing, Z.; Ye, X.; Ma, X.; Hu, X.; Yang, W.; Shi, J.; Chen, G.; Gong, L. SNGH16 regulates cell autophagy to promote Sorafenib resistance through suppressing miR-23b-3p via sponging EGR1 in hepatocellular carcinoma. Cancer Med. 2020, 9, 4324–4338. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Chan, Y.T.; Tan, H.Y.; Zhang, C.; Guo, W.; Xu, Y.; Sharma, R.; Chen, Z.S.; Zheng, Y.C.; Wang, N.; et al. Epigenetic regulation of ferroptosis via ETS1/miR-23a-3p/ACSL4 axis mediates sorafenib resistance in human hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2022, 41, 3. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.P.; Liu, J.P.; Feng, J.F.; Zhu, C.P.; Yang, Y.; Zhou, W.P.; Ding, J.; Huang, C.K.; Cui, Y.L.; Ding, C.H.; et al. miR-541 potentiates the response of human hepatocellular carcinoma to sorafenib treatment by inhibiting autophagy. Gut 2020, 69, 1309–1321. [Google Scholar] [CrossRef]
- Fondevila, F.; Méndez-Blanco, C.; Fernández-Palanca, P.; Payo-Serafín, T.; van Pelt, J.; Verslype, C.; González-Gallego, J.; Mauriz, J.L. Autophagy-related chemoprotection against sorafenib in human hepatocarcinoma: Role of FOXO3 upregulation and modulation by regorafenib. Int. J. Mol. Sci. 2021, 22, 11770. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Lv, Z.; Wang, M.; Zhang, D.; Liu, D.; Zhu, F. HBV enhances sorafenib resistance in hepatocellular carcinoma by reducing ferroptosis via SRSF2-mediated abnormal PCLAF splicing. Int. J. Mol. Sci. 2023, 24, 3263. [Google Scholar] [CrossRef]
- Sun, J.; Zhou, C.; Zhao, Y.; Zhang, X.; Chen, W.; Zhou, Q.; Hu, B.; Gao, D.; Raatz, L.; Wang, Z.; et al. Quiescin sulfhydryl oxidase 1 promotes sorafenib-induced ferroptosis in hepatocellular carcinoma by driving EGFR endosomal trafficking and inhibiting NRF2 activation. Redox Biol. 2021, 41, 101942. [Google Scholar] [CrossRef]
- Shao, W.Q.; Zhu, W.W.; Luo, M.J.; Fan, M.H.; Li, Q.; Wang, S.H.; Lin, Z.F.; Zhao, J.; Zheng, Y.; Dong, Q.Z.; et al. Cholesterol suppresses GOLM1-dependent selective autophagy of RTKs in hepatocellular carcinoma. Cell Rep. 2022, 39, 110712. [Google Scholar] [CrossRef]
- Li, Y.; Xu, B.; Ren, X.; Wang, L.; Xu, Y.; Zhao, Y.; Yang, C.; Yuan, C.; Li, H.; Tong, X.; et al. Inhibition of CISD2 promotes ferroptosis through ferritinophagy-mediated ferritin turnover and regulation of p62-Keap1-NRF2 pathway. Cell. Mol. Biol. Lett. 2022, 27, 81. [Google Scholar] [CrossRef]
- Li, H.; Zhao, J.; Zhong, X.L.; Xu, P.Y.; Du, L.J.; Fang, P.; Tan, L.J.; Li, M.J.; Zhang, C.F.; Cao, T.S. CPLX2 regulates ferroptosis and apoptosis through NRF2 pathway in human hepatocellular carcinoma cells. Appl. Biochem. Biotechnol. 2023, 195, 597–609. [Google Scholar] [CrossRef] [PubMed]
- Bai, T.; Wang, S.; Zhao, Y.; Zhu, R.; Wang, W.; Sun, Y. Haloperidol, a sigma receptor 1 antagonist, promotes ferroptosis in hepatocellular carcinoma cells. Biochem. Biophys. Res. Commun. 2017, 491, 919–925. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Feng, G.; Zhu, J.; Su, Z.; Guo, R.; Liu, J.; Zhang, H.; Zhai, Y. 3-Methyladenine-enhanced susceptibility to sorafenib in hepatocellular carcinoma cells by inhibiting autophagy. Anti-Cancer Drugs 2021, 32, 386–393. [Google Scholar] [CrossRef]
- Wu, H.; Wang, T.; Liu, Y.; Li, X.; Xu, S.; Wu, C.; Zou, H.; Cao, M.; Jin, G.; Lang, J.; et al. Mitophagy promotes sorafenib resistance through hypoxia-inducible ATAD3A dependent Axis. J. Exp. Clin. Cancer Res. 2020, 39, 274. [Google Scholar] [CrossRef]
- Zheng, Y.; Huang, C.; Lu, L.; Yu, K.; Zhao, J.; Chen, M.; Liu, L.; Sun, Q.; Lin, Z.; Zheng, J.; et al. STOML2 potentiates metastasis of hepatocellular carcinoma by promoting PINK1-mediated mitophagy and regulates sensitivity to lenvatinib. J. Hematol. Oncol. 2021, 14, 16. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, H.N.; Wang, K.; Zhang, L.; Huang, Z.; Liu, J.; Zhang, Z.; Luo, M.; Lei, Y.; Peng, Y.; et al. Ketoconazole exacerbates mitophagy to induce apoptosis by downregulating cyclooxygenase-2 in hepatocellular carcinoma. J. Hepatol. 2019, 70, 66–77. [Google Scholar] [CrossRef]
- Wang, Z.; Zhu, Q.; Li, X.; Ren, X.; Li, J.; Zhang, Y.; Zeng, S.; Xu, L.; Dong, X.; Zhai, B. TOP2A inhibition reverses drug resistance of hepatocellular carcinoma to regorafenib. Am. J. Cancer Res. 2022, 12, 4343–4360. [Google Scholar]
- Chang, W.T.; Bow, Y.D.; Fu, P.J.; Li, C.Y.; Wu, C.Y.; Chang, Y.H.; Teng, Y.N.; Li, R.N.; Lu, M.C.; Liu, Y.C.; et al. A marine terpenoid, heteronemin, induces both the apoptosis and ferroptosis of hepatocellular carcinoma cells and involves the ROS and MAPK pathways. Oxid. Med. Cell. Longev. 2021, 2021, 7689045. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Zhang, J.; Hou, J.; Hui, M.; Qi, H.; Lei, T.; Zhang, X.; Zhao, L.; Du, H. Induction of autophagy via the PI3K/Akt/mTOR signaling pathway by Pueraria flavonoids improves non-alcoholic fatty liver disease in obese mice. Biomed. Pharmacother. 2023, 157, 114005. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Shang, L.; Jiang, W.; Wu, W. Shikonin induces apoptosis and autophagy via downregulation of pyrroline-5-carboxylate reductase1 in hepatocellular carcinoma cells. Bioengineered 2022, 13, 7904–7918. [Google Scholar] [CrossRef] [PubMed]
- Ji, A.; Hu, L.; Ma, D.; Qiang, G.; Yan, D.; Zhang, G.; Jiang, C. Myricetin induces apoptosis and protective autophagy through endoplasmic reticulum stress in hepatocellular carcinoma. Evid. Based Complement. Altern. Med. 2022, 2022, 3115312. [Google Scholar] [CrossRef]
- Jiang, Z.; Gao, L.; Liu, C.; Wang, J.; Han, Y.; Pan, J. Sarmentosin induces autophagy-dependent apoptosis via activation of Nrf2 in hepatocellular carcinoma. J. Clin. Transl. Hepatol. 2023. [Google Scholar] [CrossRef]
- Oura, K.; Morishita, A.; Hamaya, S.; Fujita, K.; Masaki, T. The roles of epigenetic regulation and the tumor microenvironment in the mechanism of resistance to systemic therapy in hepatocellular carcinoma. Int. J. Mol. Sci. 2023, 24, 2805. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kouroumalis, E.; Tsomidis, I.; Voumvouraki, A. Pathogenesis of Hepatocellular Carcinoma: The Interplay of Apoptosis and Autophagy. Biomedicines 2023, 11, 1166. https://doi.org/10.3390/biomedicines11041166
Kouroumalis E, Tsomidis I, Voumvouraki A. Pathogenesis of Hepatocellular Carcinoma: The Interplay of Apoptosis and Autophagy. Biomedicines. 2023; 11(4):1166. https://doi.org/10.3390/biomedicines11041166
Chicago/Turabian StyleKouroumalis, Elias, Ioannis Tsomidis, and Argyro Voumvouraki. 2023. "Pathogenesis of Hepatocellular Carcinoma: The Interplay of Apoptosis and Autophagy" Biomedicines 11, no. 4: 1166. https://doi.org/10.3390/biomedicines11041166
APA StyleKouroumalis, E., Tsomidis, I., & Voumvouraki, A. (2023). Pathogenesis of Hepatocellular Carcinoma: The Interplay of Apoptosis and Autophagy. Biomedicines, 11(4), 1166. https://doi.org/10.3390/biomedicines11041166