
Signaling Switching from Hedgehog-GLI to MAPK Signaling Potentially Serves as a Compensatory Mechanism in Melanoma Cell Lines Resistant to GANT-61

 , ,
, ,  ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Generation of GANT-61 Resistant Cell Lines
2.2. MTT Viability Assay
2.3. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
2.4. Western Blot
2.5. Wound Healing Assay
2.6. Colony Forming Assay
2.7. Immunofluorescence
2.8. Flow Cytometry
2.9. Statistics
3. Results
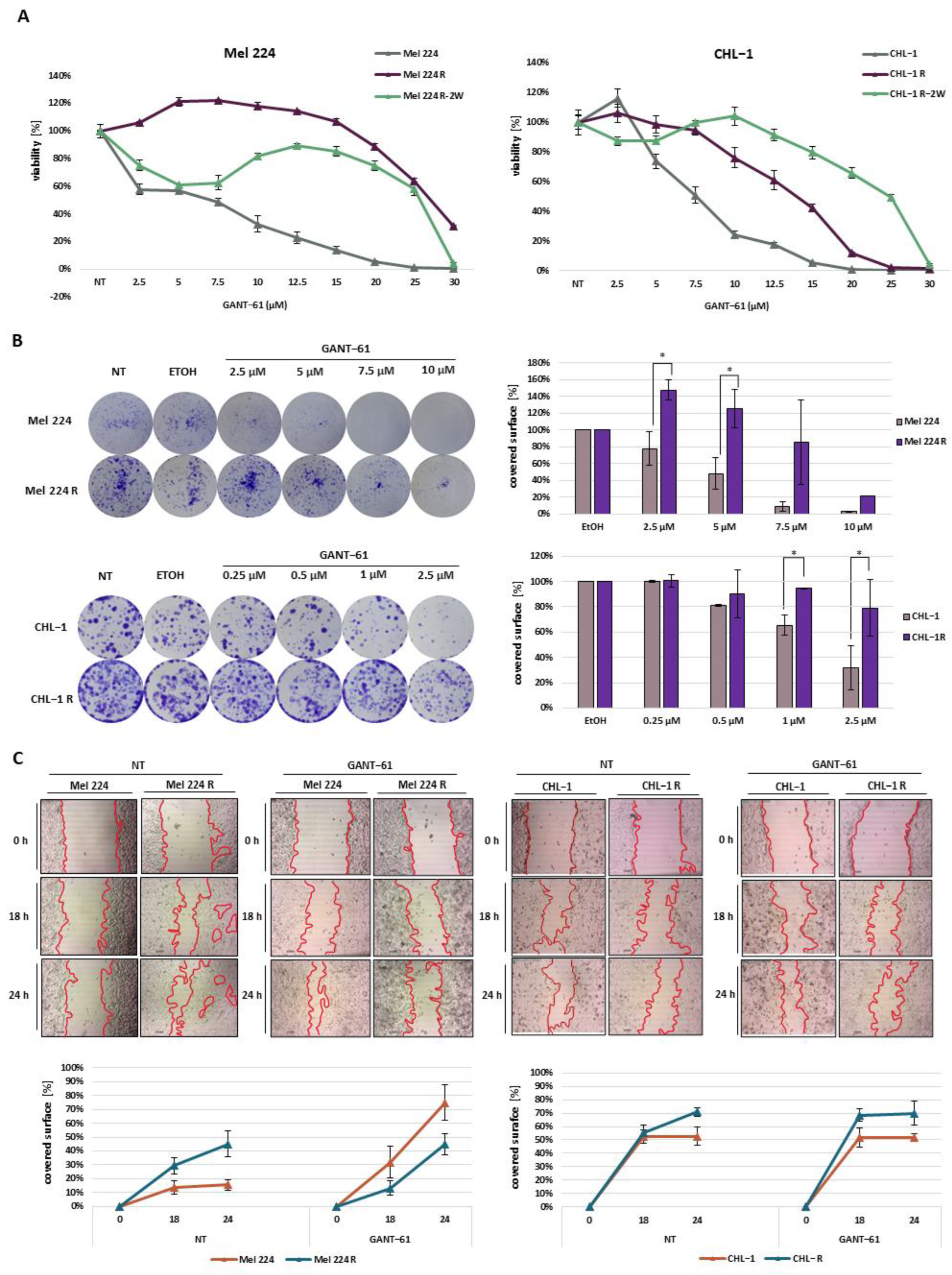
3.1. Establishment of GANT-61-Resistant Mel 224 and CHL-1 Melanoma Cell Lines
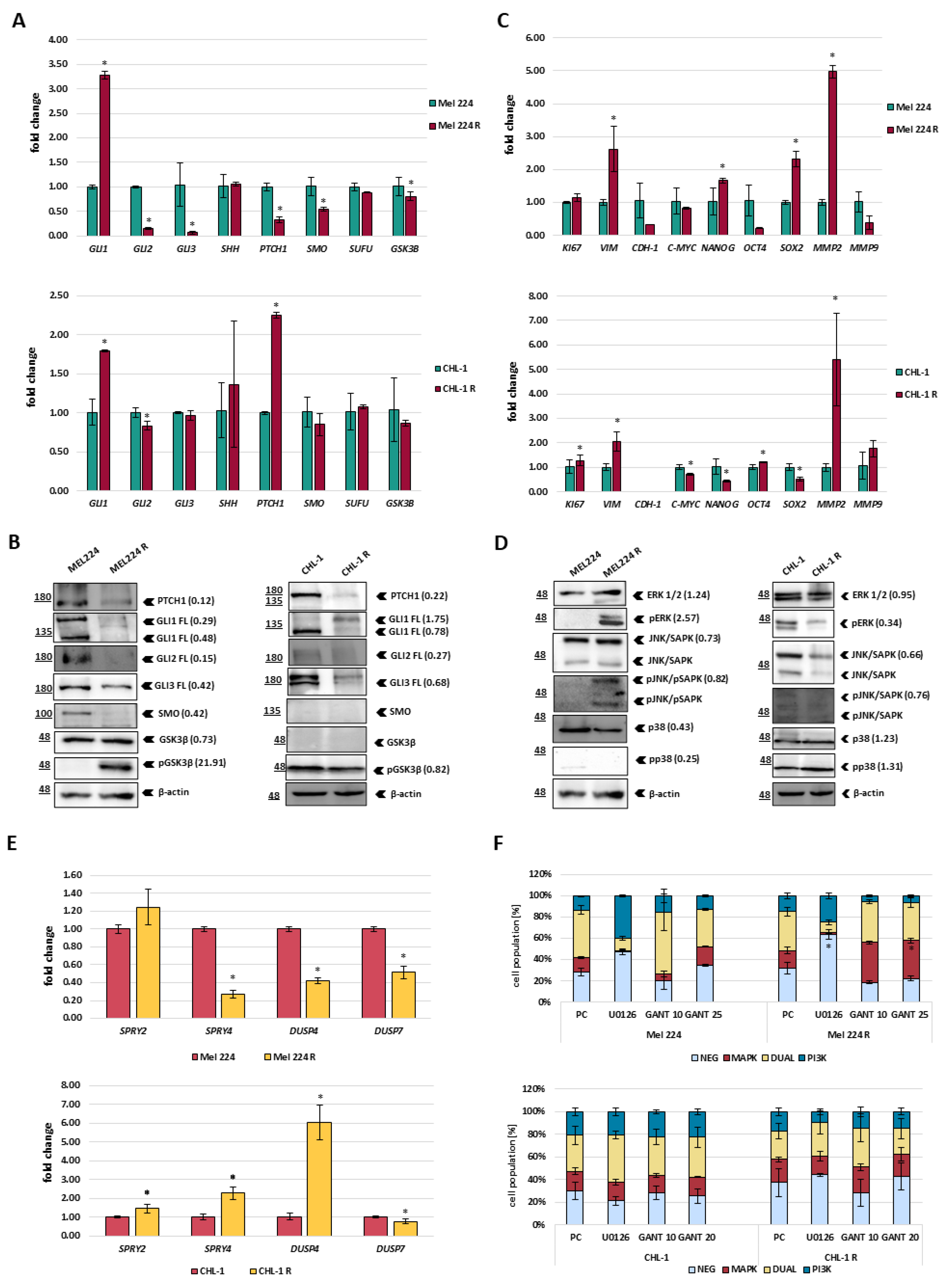
3.2. Mel 224 and CHL-1 Cell Lines Resistant to GANT-61 Exhibit Morphological and Molecular Changes
3.3. Differential Sensitivity of GANT-61 Resistant Cell Lines to HH-GLI and RAS Inhibitors
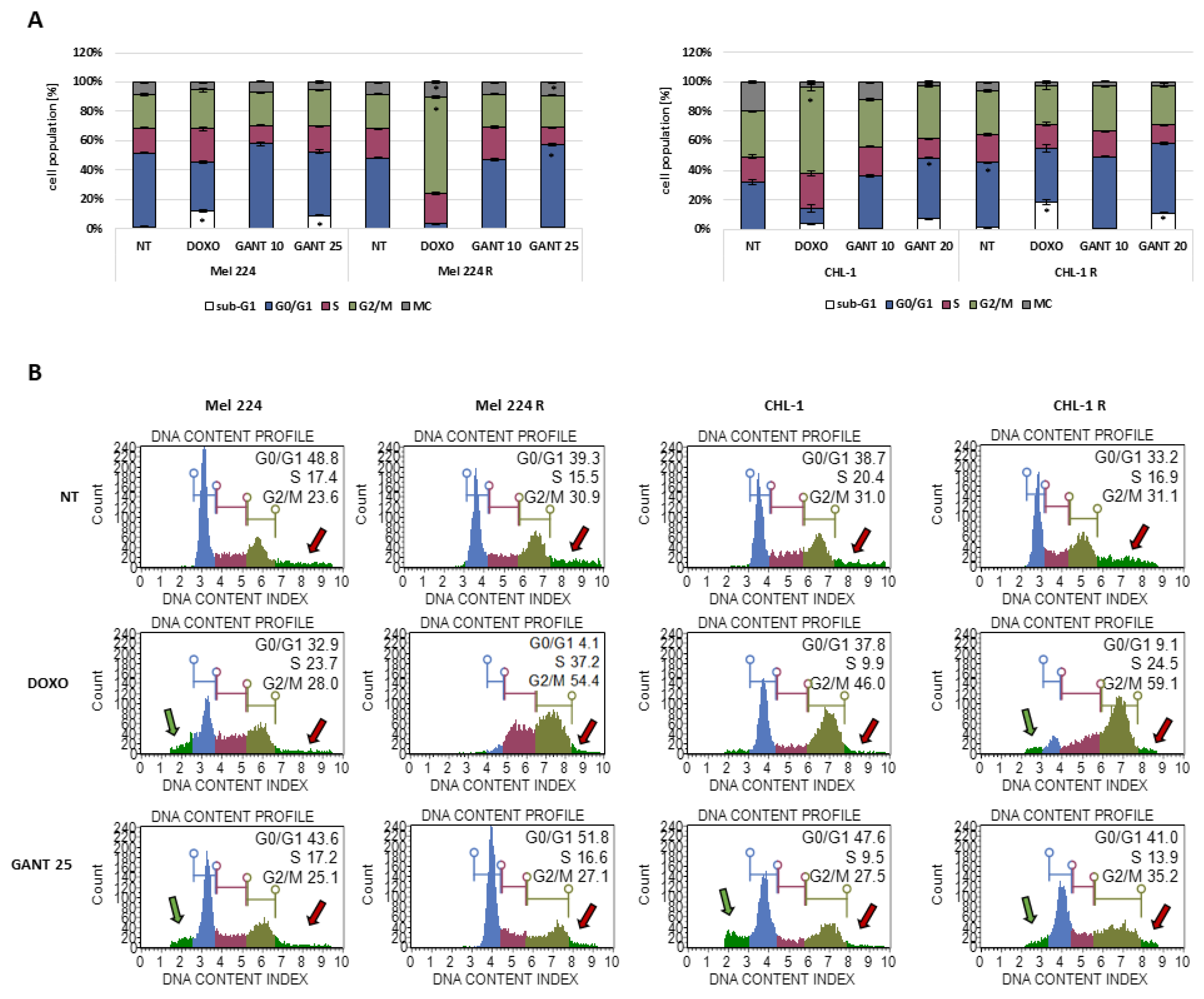
3.4. The Influence of GANT-61 Resistance on Cell Cycle Regulation
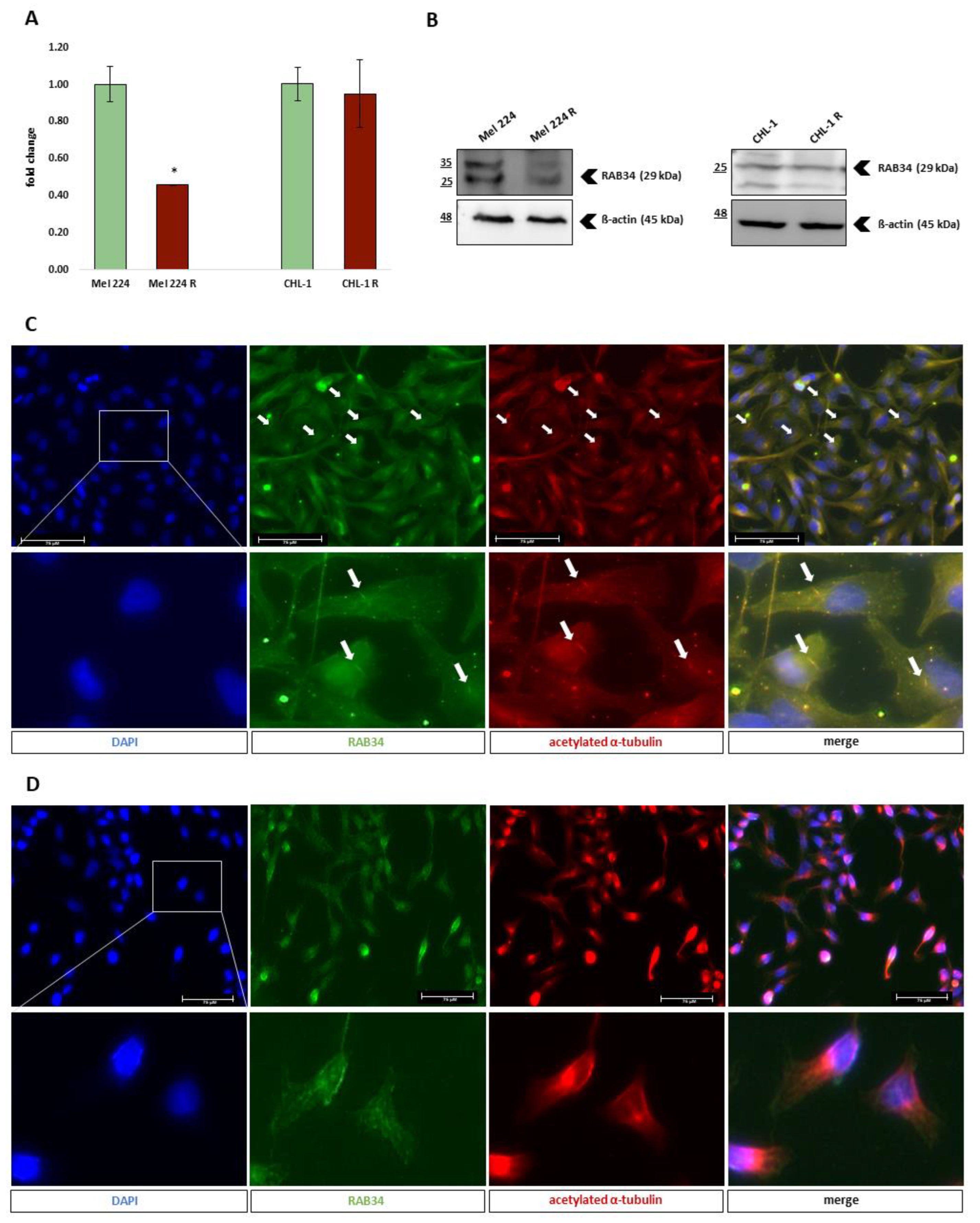
3.5. Primary Cilia Formation in Generated GANT-61 Resistant Cell Lines
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Zhang, G.; Li, G. Targeting MAPK pathway in melanoma therapy. Cancer Metastasis Rev. 2013, 32, 567–584. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Rauch, J.; Kolch, W. Targeting MAPK Signaling in Cancer: Mechanisms of Drug Resistance and Sensitivity. Int. J. Mol. Sci. 2020, 21, 1102. [Google Scholar] [CrossRef]
- Murali, I.; Kasar, S.; Naeem, A.; Tyekucheva, S.; Khalsa, J.K.; Thrash, E.M.; Itchaki, G.; Livitz, D.; Leshchiner, I.; Dong, S.; et al. Activation of the MAPK pathway mediates resistance to PI3K inhibitors in chronic lymphocytic leukemia. Blood 2021, 138, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, A.R.; Robey, R.W.; Luchenko, V.L.; Zhan, Z.; Piekarz, R.L.; Gillet, J.-P.; Kossenkov, A.V.; Wilkerson, J.; Showe, L.C.; Gottesman, M.M.; et al. MAPK pathway activation leads to Bim loss and histone deacetylase inhibitor resistance: Rationale to combine romidepsin with an MEK inhibitor. Blood 2013, 121, 4115–4125. [Google Scholar] [CrossRef]
- Brechbiel, J.; Miller-Moslin, K.; Adjei, A.A. Crosstalk between hedgehog and other signaling pathways as a basis for combination therapies in cancer. Cancer Treat. Rev. 2014, 40, 750–759. [Google Scholar] [CrossRef]
- Stecca, B.; Mas, C.; Clement, V.; Zbinden, M.; Correa, R.; Piguet, V.; Beermann, F.; Ruiz i Altaba, A. Melanomas require HEDGEHOG-GLI signaling regulated by interactions between GLI1 and the RAS-MEK/AKT pathways. Proc. Natl. Acad. Sci. USA 2007, 104, 5895–5900. [Google Scholar] [CrossRef]
- Atwood, S.X.; Sarin, K.Y.; Whitson, R.J.; Li, J.R.; Kim, G.; Rezaee, M.; Ally, M.S.; Kim, J.; Yao, C.; Chang, A.L.S.; et al. Smoothened variants explain the majority of drug resistance in basal cell carcinoma. Cancer Cell 2015, 27, 342–353. [Google Scholar] [CrossRef]
- Kurtović, M.; Piteša, N.; Bartoniček, N.; Ozretić, P.; Musani, V.; Čonkaš, J.; Petrić, T.; King, C.; Sabol, M. RNA-seq and ChIP-seq Identification of Unique and Overlapping Targets of GLI Transcription Factors in Melanoma Cell Lines. Cancers 2022, 14, 4540. [Google Scholar] [CrossRef]
- Faião-Flores, F.; Alves-Fernandes, D.K.; Pennacchi, P.C.; Sandri, S.; Vicente, A.L.S.A.; Scapulatempo-Neto, C.; Vazquez, V.L.; Reis, R.M.; Chauhan, J.; Goding, C.R.; et al. Targeting the hedgehog transcription factors GLI1 and GLI2 restores sensitivity to vemurafenib-resistant human melanoma cells. Oncogene 2017, 36, 1849–1861. [Google Scholar] [CrossRef] [PubMed]
- Trnski, D.; Sabol, M.; Gojević, A.; Martinić, M.; Ozretić, P.; Musani, V.; Ramić, S.; Levanat, S. GSK3β and Gli3 play a role in activation of Hedgehog-Gli pathway in human colon cancer—Targeting GSK3β downregulates the signaling pathway and reduces cell proliferation. Biochim. Biophys. Acta 2015, 1852, 2574–2584. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Lauth, M.; Bergström, A.; Shimokawa, T.; Toftgård, R. Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proc. Natl. Acad. Sci. USA 2007, 104, 8455–8460. [Google Scholar] [CrossRef]
- Miller, W.H. Molecular targets of arsenic trioxide in malignant cells. Oncologist 2002, 7 (Suppl. S1), 14–19. [Google Scholar] [CrossRef]
- Yang, D.; Cao, F.; Ye, X.; Zhao, H.; Liu, X.; Li, Y.; Shi, C.; Wang, H.; Zhou, J. Arsenic trioxide inhibits the Hedgehog pathway which is aberrantly activated in acute promyelocytic leukemia. Acta Haematol. 2013, 130, 260–267. [Google Scholar] [CrossRef]
- García Ruiz, A.J.; García-Agua Soler, N.; Herrera Acosta, E.; Zalaudek, I.; Malvehy, J. Benefit–risk assessment of sonidegib and vismodegib in the treatment of locally advanced basal cell carcinoma. Drugs Context 2022, 11, 2022-1-2. [Google Scholar] [CrossRef]
- Hs, K.; Ys, L.; Dk, K. Doxorubicin exerts cytotoxic effects through cell cycle arrest and Fas-mediated cell death. Pharmacology 2009, 84, 300–309. [Google Scholar] [CrossRef]
- Zhang, Z.; Hao, C.; Zhang, R.; Pei, X.; Li, J.; Wang, L. A Gli inhibitor GANT61 suppresses cell proliferation, promotes cell apoptosis and induces G1/G0 cycle retardation with a dose- and time-dependent manner through inhibiting Notch pathway in multiple myeloma. Cell Cycle 2020, 19, 2063–2073. [Google Scholar] [CrossRef]
- Plesca, D.; Mazumder, S.; Almasan, A. DNA damage response and apoptosis. Methods Enzym. 2008, 446, 107–122. [Google Scholar] [CrossRef]
- Weihua, Z.; Lin, Q.; Ramoth, A.J.; Fan, D.; Fidler, I.J. Formation of solid tumors by a single multinucleated cancer cell. Cancer 2011, 117, 4092–4099. [Google Scholar] [CrossRef]
- Kim, J.; Dabiri, S.; Seeley, E.S. Primary cilium depletion typifies cutaneous melanoma in situ and malignant melanoma. PLoS ONE 2011, 6, e27410. [Google Scholar] [CrossRef]
- Lang, U.E.; Love, N.R.; Cheung, C.; McCalmont, T.H.; Kim, J. Use of the Ciliation Index to Distinguish Invasive Melanoma From Associated Conventional Melanocytic Nevi. Am. J. Derm. 2020, 42, 11–15. [Google Scholar] [CrossRef]
- Dong, Z.; Wang, Y.; Ding, V.; Yan, X.; Lv, Y.; Zhong, M.; Zhu, F.; Zhao, P.; He, C.; Ding, F.; et al. GLI1 activation is a key mechanism of erlotinib resistance in human non-small cell lung cancer. Oncol. Lett. 2020, 20, 76. [Google Scholar] [CrossRef]
- Devitt, B.; Liu, W.; Salemi, R.; Wolfe, R.; Kelly, J.; Tzen, C.-Y.; Dobrovic, A.; McArthur, G. Clinical outcome and pathological features associated with NRAS mutation in cutaneous melanoma. Pigment. Cell Melanoma Res. 2011, 24, 666–672. [Google Scholar] [CrossRef]
- Lochter, A.; Galosy, S.; Muschler, J.; Freedman, N.; Werb, Z.; Bissell, M.J. Matrix metalloproteinase stromelysin-1 triggers a cascade of molecular alterations that leads to stable epithelial-to-mesenchymal conversion and a premalignant phenotype in mammary epithelial cells. J. Cell Biol. 1997, 139, 1861–1872. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.-J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef]
- Hirschmann-Jax, C.; Foster, A.E.; Wulf, G.G.; Nuchtern, J.G.; Jax, T.W.; Gobel, U.; Goodell, M.A.; Brenner, M.K. A distinct “side population” of cells with high drug efflux capacity in human tumor cells. Proc. Natl. Acad. Sci. USA 2004, 101, 14228–14233. [Google Scholar] [CrossRef]
- Amable, L.; Gavin, E.; Kudo, K.; Meng, E.; Rocconi, R.P.; Shevde, L.A.; Reed, E. GLI1 upregulates C-JUN through a specific 130-kDa isoform. Int. J. Oncol. 2014, 44, 655–661. [Google Scholar] [CrossRef]
- Shimokawa, T.; Tostar, U.; Lauth, M.; Palaniswamy, R.; Kasper, M.; Toftgård, R.; Zaphiropoulos, P.G. Novel Human Glioma-associated Oncogene 1 (GLI1) Splice Variants Reveal Distinct Mechanisms in the Terminal Transduction of the Hedgehog Signal. J. Biol. Chem. 2008, 283, 14345–14354. [Google Scholar] [CrossRef] [PubMed]
- Pecoraro, C.; Faggion, B.; Balboni, B.; Carbone, D.; Peters, G.J.; Diana, P.; Assaraf, Y.G.; Giovannetti, E. GSK3β as a novel promising target to overcome chemoresistance in pancreatic cancer. Drug. Resist. Updat. 2021, 58, 100779. [Google Scholar] [CrossRef] [PubMed]
- Stiehl, T.; Baran, N.; Ho, A.D.; Marciniak-Czochra, A. Clonal selection and therapy resistance in acute leukaemias: Mathematical modelling explains different proliferation patterns at diagnosis and relapse. J. R. Soc. Interface 2014, 11, 20140079. [Google Scholar] [CrossRef]
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug Resistance in Cancer: An Overview. Cancers 2014, 6, 1769–1792. [Google Scholar] [CrossRef] [PubMed]
- Jänne, P.A.; Gray, N.; Settleman, J. Factors underlying sensitivity of cancers to small-molecule kinase inhibitors. Nat. Rev. Drug. Discov. 2009, 8, 709–723. [Google Scholar] [CrossRef]
- Pua, L.J.W.; Mai, C.-W.; Chung, F.F.-L.; Khoo, A.S.-B.; Leong, C.-O.; Lim, W.-M.; Hii, L.-W. Functional Roles of JNK and p38 MAPK Signaling in Nasopharyngeal Carcinoma. Int. J. Mol. Sci. 2022, 23, 1108. [Google Scholar] [CrossRef]
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef]
- Guo, X.; Ma, N.; Wang, J.; Song, J.; Bu, X.; Cheng, Y.; Sun, K.; Xiong, H.; Jiang, G.; Zhang, B.; et al. Increased p38-MAPK is responsible for chemotherapy resistance in human gastric cancer cells. BMC Cancer 2008, 8, 375. [Google Scholar] [CrossRef]
- Kuonen, F.; Huskey, N.E.; Shankar, G.; Jaju, P.; Whitson, R.J.; Rieger, K.E.; Atwood, S.X.; Sarin, K.Y.; Oro, A.E. Loss of primary cilia drives switching from Hedgehog to Ras/MAPK pathway in resistant basal cell carcinoma. J. Investig. Dermatol. 2019, 139, 1439. [Google Scholar] [CrossRef]
- Balko, J.M.; Schwarz, L.J.; Bhola, N.E.; Kurupi, R.; Owens, P.; Miller, T.W.; Gómez, H.; Cook, R.S.; Arteaga, C.L. Activation of MAPK pathways due to DUSP4 loss promotes cancer stem cell-like phenotypes in basal-like breast cancer. Cancer Res. 2013, 73, 6346–6358. [Google Scholar] [CrossRef]
- Yao, Y.; Luo, J.; Bian, Y.; Sun, Y.; Shi, M.; Xia, D.; Niu, M.; Zhao, K.; Zeng, L.; Chen, W.; et al. Sprouty2 regulates proliferation and survival of multiple myeloma by inhibiting activation of the ERK1/2 pathway in vitro and in vivo. Exp. Hematol. 2016, 44, 474–482.e2. [Google Scholar] [CrossRef]
- Kumar, R.; Njauw, C.-N.; Reddy, B.Y.; Ji, Z.; Rajadurai, A.; Klebanov, N.; Tsao, H. Growth suppression by dual BRAF(V600E) and NRAS(Q61) oncogene expression is mediated by SPRY4 in melanoma. Oncogene 2019, 38, 3504–3520. [Google Scholar] [CrossRef]
- Chen, J.K.; Taipale, J.; Cooper, M.K.; Beachy, P.A. Inhibition of Hedgehog signaling by direct binding of cyclopamine to Smoothened. Genes. Dev. 2002, 16, 2743–2748. [Google Scholar] [CrossRef]
- Wang, C.; Wu, H.; Evron, T.; Vardy, E.; Han, G.W.; Huang, X.-P.; Hufeisen, S.J.; Mangano, T.J.; Urban, D.J.; Katritch, V.; et al. Structural basis for Smoothened receptor modulation and chemoresistance to anticancer drugs. Nat. Commun. 2014, 5, 4355. [Google Scholar] [CrossRef]
- Thompson, C.L.; Wiles, A.; Poole, C.A.; Knight, M.M. Lithium chloride modulates chondrocyte primary cilia and inhibits Hedgehog signaling. FASEB J. 2016, 30, 716–726. [Google Scholar] [CrossRef]
- Wang, X.; Fang, Z.; Wang, A.; Luo, C.; Cheng, X.; Lu, M. Lithium Suppresses Hedgehog Signaling via Promoting ITCH E3 Ligase Activity and Gli1–SUFU Interaction in PDA Cells. Front. Pharm. 2017, 8, 820. [Google Scholar] [CrossRef]
- Duda, P.; Akula, S.M.; Abrams, S.L.; Steelman, L.S.; Martelli, A.M.; Cocco, L.; Ratti, S.; Candido, S.; Libra, M.; Montalto, G.; et al. Targeting GSK3 and Associated Signaling Pathways Involved in Cancer. Cells 2020, 9, 1110. [Google Scholar] [CrossRef]
- Dummer, R.; Ascierto, P.A.; Basset-Seguin, N.; Dréno, B.; Garbe, C.; Gutzmer, R.; Hauschild, A.; Krattinger, R.; Lear, J.T.; Malvehy, J.; et al. Sonidegib and vismodegib in the treatment of patients with locally advanced basal cell carcinoma: A joint expert opinion. J. Eur. Acad. Derm. Venereol. 2020, 34, 1944–1956. [Google Scholar] [CrossRef]
- Malhi, V.; Colburn, D.; Williams, S.J.; Hop, C.E.C.A.; Dresser, M.J.; Chandra, P.; Graham, R.A. A clinical drug–drug interaction study to evaluate the effect of a proton-pump inhibitor, a combined P-glycoprotein/cytochrome 450 enzyme (CYP)3A4 inhibitor, and a CYP2C9 inhibitor on the pharmacokinetics of vismodegib. Cancer Chemother. Pharm. 2016, 78, 41–49. [Google Scholar] [CrossRef]
- Cabanos, H.F.; Hata, A.N. Emerging Insights into Targeted Therapy-Tolerant Persister Cells in Cancer. Cancers 2021, 13, 2666. [Google Scholar] [CrossRef]
- Rotblat, B.; Ehrlich, M.; Haklai, R.; Kloog, Y. The Ras inhibitor farnesylthiosalicylic acid (Salirasib) disrupts the spatiotemporal localization of active Ras: A potential treatment for cancer. Methods Enzym. 2008, 439, 467–489. [Google Scholar] [CrossRef]
- Yang, G.-L.; Tao, H.-R.; Wang, H.-W.; Sun, Y.; Zhang, L.-D.; Zhang, C.; He, W.; Xu, M.-H.; Zhao, J.-M.; Gao, F.-H. Ara-C increases gastric cancer cell invasion by upregulating CD-147-MMP-2/MMP-9 via the ERK signaling pathway. Oncol. Rep. 2015, 33, 2045–2051. [Google Scholar] [CrossRef] [PubMed]
- Yano, S.; Miwa, S.; Mii, S.; Hiroshima, Y.; Uehara, F.; Yamamoto, M.; Kishimoto, H.; Tazawa, H.; Bouvet, M.; Fujiwara, T.; et al. Invading cancer cells are predominantly in G0/G1 resulting in chemoresistance demonstrated by real-time FUCCI imaging. Cell Cycle 2014, 13, 953–960. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.E.; Kirk, J.; Ji, Y.; Tang, D.G. Tumor Dormancy and Slow-Cycling Cancer Cells. Adv. Exp. Med. Biol. 2019, 1164, 199–206. [Google Scholar] [CrossRef]
- Xu, S.; Liu, Y.; Meng, Q.; Wang, B. Rab34 small GTPase is required for Hedgehog signaling and an early step of ciliary vesicle formation in mouse. J. Cell. Sci. 2018, 131, jcs213710. [Google Scholar] [CrossRef]
- Radford, R.; Slattery, C.; Jennings, P.; Blacque, O.; Pfaller, W.; Gmuender, H.; Van Delft, J.; Ryan, M.P.; McMorrow, T. Carcinogens induce loss of the primary cilium in human renal proximal tubular epithelial cells independently of effects on the cell cycle. Am. J. Physiol. Ren. Physiol. 2012, 302, F905–F916. [Google Scholar] [CrossRef]
- Zhang, G.; Wang, S. Proteomic approach to substrates related to MAPK pathway in 293T cells. Cell. Biol. Int. 2007, 31, 1–10. [Google Scholar] [CrossRef]
- Hassounah, N.B.; Nagle, R.; Saboda, K.; Roe, D.J.; Dalkin, B.L.; McDermott, K.M. Primary cilia are lost in preinvasive and invasive prostate cancer. PLoS ONE 2013, 8, e68521. [Google Scholar] [CrossRef]
- Choudhury, A.; Neumann, N.M.; Raleigh, D.R.; Lang, U.E. Clinical Implications of Primary Cilia in Skin Cancer. Dermatol. Ther. 2020, 10, 233–248. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piteša, N.; Kurtović, M.; Bartoniček, N.; Gkotsi, D.S.; Čonkaš, J.; Petrić, T.; Musani, V.; Ozretić, P.; Riobo-Del Galdo, N.A.; Sabol, M. Signaling Switching from Hedgehog-GLI to MAPK Signaling Potentially Serves as a Compensatory Mechanism in Melanoma Cell Lines Resistant to GANT-61. Biomedicines 2023, 11, 1353. https://doi.org/10.3390/biomedicines11051353
Piteša N, Kurtović M, Bartoniček N, Gkotsi DS, Čonkaš J, Petrić T, Musani V, Ozretić P, Riobo-Del Galdo NA, Sabol M. Signaling Switching from Hedgehog-GLI to MAPK Signaling Potentially Serves as a Compensatory Mechanism in Melanoma Cell Lines Resistant to GANT-61. Biomedicines. 2023; 11(5):1353. https://doi.org/10.3390/biomedicines11051353
Chicago/Turabian StylePiteša, Nikolina, Matea Kurtović, Nenad Bartoniček, Danai S. Gkotsi, Josipa Čonkaš, Tina Petrić, Vesna Musani, Petar Ozretić, Natalia A. Riobo-Del Galdo, and Maja Sabol. 2023. "Signaling Switching from Hedgehog-GLI to MAPK Signaling Potentially Serves as a Compensatory Mechanism in Melanoma Cell Lines Resistant to GANT-61" Biomedicines 11, no. 5: 1353. https://doi.org/10.3390/biomedicines11051353