Abstract
In Romania, breast cancer (BC) is the most common malignancy in women. However, there is limited data on the prevalence of predisposing germline mutations in the population in the era of precision medicine, where molecular testing has become an indispensable tool in cancer diagnosis, prognosis, and therapeutics. Therefore, we conducted a retrospective study to determine the prevalence, mutational spectrum, and histopathological prediction factors for hereditary breast cancer (HBC) in Romania. A cohort of 411 women diagnosed with BC selected upon NCCN v.1.2020 guidelines underwent an 84-gene NGS-based panel testing for breast cancer risk assessment during 2018–2022 in the Department of Oncogenetics of the Oncological Institute of Cluj-Napoca, Romania. A total of 135 (33%) patients presented pathogenic mutations in 19 genes. The prevalence of genetic variants was determined, and demographic and clinicopathological characteristics were analyzed. We observed differences among BRCA and non-BRCA carriers regarding family history of cancer, age of onset, and histopathological subtypes. Triple-negative (TN) tumors were more often BRCA1 positive, unlike BRCA2 positive tumors, which were more often the Luminal B subtype. The most frequent non-BRCA mutations were found in CHEK2, ATM, and PALB2, and several recurrent variants were identified for each gene. Unlike other European countries, germline testing for HBC is still limited due to the high costs and is not covered by the National Health System (NSH), thus leading to significant discrepancies related to the screening and prophylaxis of cancer.
1. Introduction
In Romania, 12,000 new BC cases are diagnosed annually, accounting for the second cause of cancer-related deaths after lung cancer [1]. Out of all diagnosed cases, 10% of BC are hereditary (HBC), the consequence of inherited or de novo predisposing mutations that define a group with increased malignancy risk compared to the general population. For the 11 million women in Romania, we do not have epidemiological data on the frequency of predisposing mutations in the general population. In addition, there are no national screening programs or reimbursed genetic testing. Nevertheless, more than 55% of cases are diagnosed in advanced clinical stages, with an overall survival rate below other European countries, defining a significant health concern [2]. BRCA1 and BRCA2 germline mutations are responsible for about 60% of HBCs with an overall 60–80% lifetime risk; the other 40% are associated with other predisposing variants in moderate-to-high penetrance genes such as PALB2, PTEN, TP53, CDH1, CHEK2, ATM, and the MMR group [3,4]. Although BRCA-related HBCs are prevalent, current testing guidelines recommend panel testing to ensure extensive mutation spectrum coverage [5]. Finding the predisposing pathogenic variants is essential for identifying high-risk women in order for them to be further followed in intensive screening programs that allow an early diagnosis or even avoid the onset of the malignancy and provide proper genetic counseling for family members [6,7]. In addition, the current medical practice supports targeted molecular therapy with PARP inhibitors among women with advanced breast neoplasia and triple-negative histology who have BRCA germline mutations and precision treatment [8]. We assume that the incidence of HBC in Romania is the same as that reported in the Caucasian population; however, it is already well-known that there are differences regarding the distribution of pathogenic variants and associations with the malignant phenotype [9,10]. There is very little data related to the distribution and peculiarities of germline variants in women from Romania, as published data only refer to 500 patients [11,12,13].
2. Materials and Methods
The study was conducted in the Department of Oncogenetics of the Oncology Institute of Cluj-Napoca, Romania, between 2018 and 2022. It included patients with at least one NCCN v.2020 molecular testing criteria for HBC susceptibility genes (BC diagnosed before age 50, bilateral BC metachronous or synchronous BC, TNBC before age 60). Based on the initial genetic consult, signing the consent form, and individual financial possibilities, eligible patients were tested using extensive molecular panels in two certified NGS/MLPA laboratories (Invitae and Blueprint) on a panel of 84 high-, moderate-, and low-penetrance genes (AIP, ALK, APC, ATM, AXIN2, BAP1, BARD1, BLM, BMPR1A, BRCA1, BRCA2, BRIP1, CASR, CDC73, CDH1, CDK4, CDKN1B, CDKN1C, CDKN2A, CEBPA, CHEK2, CTNNA1, DICER1, DIS3L2, EGFR, EPCAM, FH, FLCN, GATA2, GPC3, GREM1, HOXB13, HRAS, KIT, MAX, MEN1, MET, MITF, MLH1, MSH2, MSH3* MSH6, MUTYH, NBN, NF1, NF2, NTHL1, PALB2, PDGFRA, PHOX2B, PMS2, POLD1, POLE, POT1, PRKAR1A PTCH1, PTEN, RAD50, RAD51C, RAD51D, RB1, RECQL4, RET, RUNX1, SDHA, SDHAF2, SDHB, SDHC, SDHD, SMAD4, SMARCA4, SMARCB1, SMARCE1, STK11, SUFU, TERC, TERT, TMEM127, TP53, TSC1, TSC2, VHL, WRN, WT1).
Genomic DNA obtained from peripheral blood samples was enriched for targeted regions using a hybridization-based protocol and sequenced using Illumina technology. All targeted regions were sequenced with ≥50× depth and 20 bp of flanking intronic sequence, Reads were aligned to a reference sequence (GRCh37), and sequence changes were identified and interpreted in the context of a single clinically relevant transcript. Promoters, untranslated regions, and other non-coding regions were not interrogated.
Only patients with pathogenic and potentially pathogenic mutations were included in the study, although 76 patients presented variants of uncertain significance (VUS) but were not included in the positive group statistical analysis. Data were organized using Microsoft Excel, part of the Microsoft Office 2019 suite (Microsoft Corp., Redmond, WA, USA). Data were then analyzed using R 4.2.2 (R Foundation) [14], RStudio (Posit Software, PBC, Boston, MA, USA) [15]. In addition, the following libraries were loaded in the workspace: stringr [16], readxl [17], and GenVisR [18]. To identify statistically significant differences in quantitative variables between groups, we used the Wilcoxon rank-sum (Mann–Whitney U) test. To identify statistically significant differences in frequencies of qualitative variables between groups, we used either the Chi2 test, or where its assumptions were violated, the Fisher test. Quantitative variables were expressed as mean (standard deviation), and qualitative variables were expressed as percentages.
3. Results
Of 624 women diagnosed with BC, 560 met at least one NCCN v.2020 molecular testing criteria, and 411 women underwent testing.
Among all 411 patients, 135 (32.8%) carried a pathogenic or likely pathogenic heterozygous germline mutation in 19 genes, including high-penetrant breast cancer genes like BRCA1 (43–31.9%), BRCA2 (19–14.1%), PALB2 (12–8.9%), TP53 (3–2.2%), and CDH1 (2–1.5%), and moderate to low-penetrant genes CHEK2 (25–18.5%), ATM (12–8.9%), MMR group PMS2 (2–1.5%), MSH3 (1–0.7%), MSH6 (1–0.7%), MLH1 (1–0.7%), BARD1 (3–2.2%), NF1 (3–2.2%), MUTYH (5–3.7%), EGFR (1–0.7%), SDHB (1–0.7%), RAD50 (3–2.2%), NBN (2–1.5%), and XRCC2 (2–1.5%). We identified 142 defects in all. Of these, 77 were different pathogenic variants, of which the most common defects were frameshift (46–32.4% of all detected defects) and missense (40–28.2%) variants, followed by nonsense (27–19%), deletion/insertion (21–14.8%), and intronic variants (8–5.6%). Seven patients in the group had two pathogenic mutations (Figure 1 and Figure 2).
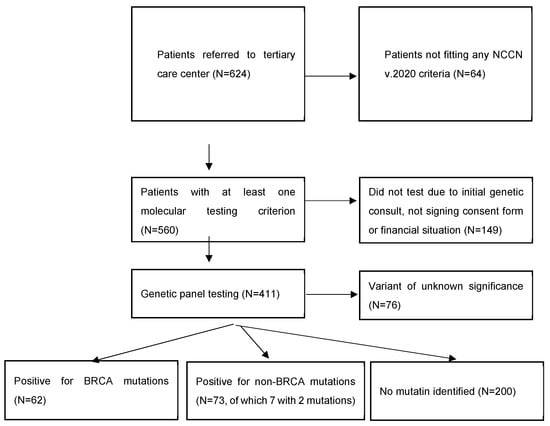
Figure 1.
General characteristics of the cohort.
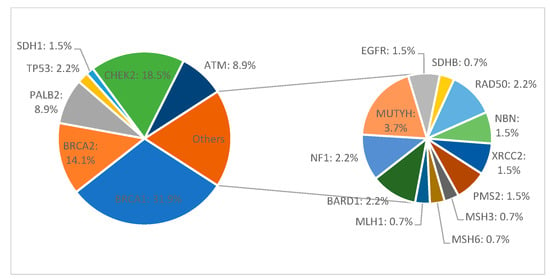
Figure 2.
Mutation frequency in germline-positive patients with hereditary breast cancer.
Three recurrent variants (reported in more than three unrelated patients) c.3607C>T (p.Arg1203Ter), c.181T>G (p.Cys61Gly), and c.5266dupC (p.Gln1756Profs) accounted for (26) 60% of all reported variants were identified in the BRCA1 gene. The c.172_175delTTGT (p.Gln60Argfs) frameshift variant was also recurrent in the PALB2 gene, reported in (4) 33% of carriers, and c.1564_1565delGA(p.Glu522Ilefs) frameshift variant was found in (5) 41% of ATM carriers. Another recurring variant in the CHEK2 gene, c.470T>C (p.Ile157Thr), was reported, accounting for a majority of (18) 72% of the pathogenic variants for this gene and 13% of all non-BRCA pathogenic variants in this cohort (Table 1).

Table 1.
Pathogenic variants reported in breast cancer patients.
The mean age at the primary cancer diagnosis was 41.387 ± 8.084 for BRCA carriers; the mean age at primary cancer diagnosis was significantly higher both for non-BRCA mutation carriers (at 44.466 ± 7.02, Wilcoxon rank sum p = 0.007) and for patients with no mutation (at 45.62 ± 7.367, Wilcoxon rank sum p < 0.001).
The mean age at diagnosis for patients with a positive family history of cancer was 42.798 ± 7.054 versus 45.674 ± 7.607 for patients with no family history of cancer (Wilcoxon rank sum p = 0.002). Among patients with no mutation, 24.5% had positive familial history. In contrast, among patients with any mutation, 49.2% had a positive familial history (54.2% among those with BRCA mutations and 45.2% among those with other types of mutation). There were statistically significant differences between patients with no mutations and both those with BRCA mutations (Chi2 p < 0.001) and those with non-BRCA mutations (Chi2 p < 0.001). At the same time, there were no differences among BRCA and non-BRCA carriers (Chi2 p = 0.302). We did not have information about the family history of 3 patients with BRCA mutations.
The most common tumor histology in the cohort was invasive ductal carcinoma, found in 312 of all tested patients (75.9%), followed by invasive lobular carcinoma, 53 (12.9%), in-situ carcinoma, 30 (7.3%), and other rare histologies, 16 (3.9%). As for the germline-positive patients, the most common tumor histology was invasive ductal carci-noma 102 (75.5%), followed by invasive lobular carcinoma 13 (9.6%) and other rare his-tologies 11 (8.1%). In situ, non-invasive histologies were identified in 9 (6.7%) of diagnosed cases. There were statistically significant differences in the prevalence of tumoral histology types across the mutation types (Fisher exact test p = 0.018). Rare histologies had a lower prevalence in patients with no mutation (1%) than in patients with mutations, while lobular histology was much rarer among patients with BRCA mutations than the others. An interesting finding was that the indication for genetic testing for 42% of patients without familial history of cancer was the TN histology revealing a BRCA1 mutation. A total of 38% of Luminal A cancer patients with an onset <45 years of age and no familial history of cancer revealed BRCA2 or non-BRCA germline variants (Table 2).
Table 2.
Histopathological and molecular characteristics for the cohort of 411 patients. † = 3 patients with BRCA mutations lacked information regarding family history of breast cancer and were excluded from analysis.
The most common molecular subtype for germline-positive patients was Luminal B (77–57%), followed by triple negative (33–24.4%) and Luminal A (25–18%) subtypes. Luminal B histology was predominant in patients with no mutation compared to mutation carriers (Chi2 p < 0.001, OR = 3.68 CI 95% 2.23–6.08). TN histology was predominant among BRCA mutations, compared to the other groups (Chi2 p < 0.001, OR = 6.71 CI 95% 3.57–12.65). The distributions of the molecular types in breast cancer patients with the BRCA and non-BRCA mutations differed significantly depending on the gene involved. The Luminal A subtype was prevalent in tumors positive for moderate-to-low penetrance mutations. The BRCA1-associated cancers were significantly more often TN than tumors harboring other mutations (Chi2 p < 0.001, OR = 9.69 CI 95% 4.83–19.45). Luminal B subtypes, particularly Luminal B HER2-positive subtypes, were reported more frequently in BRCA2 tumors but with no statistical significance compared to the other BRCA1 and non-BRCA tumors (p = 0.158). The Luminal A subtype was more frequently associated with CHEK2-positive tumors than other mutations (p = 0.013).
The mean Ki67 index value was 45.194 ± 23.717 for BRCA carriers, significantly higher than for non-BRCA mutation carriers (35.808 ± 20.427, Wilcoxon rank sum p = 0.016) but not compared to the mean value of Ki67 index in patients with no mutations (42.175 ± 19.681, Wilcoxon rank sum p = 0.366) (Figure 3).
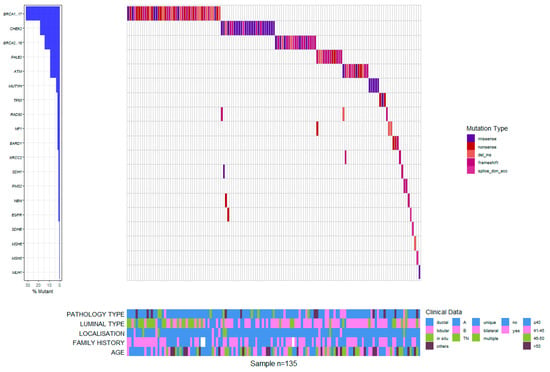
Figure 3.
Waterfall plot of mutation profiles and clinical data for the Romanian cohort.
4. Discussion
4.1. Mutation Prevalence
In countries such as Romania, Bulgaria, Ukraine, Malta, Albania, Serbia, Bosnia, and Herzegovina, Macedonia, and Montenegro, surveys on BRCA1 and BRCA2 mutations have yet to be conducted, and no data are available. From Croatia, no conclusive data about the founder BRCA1/2 mutation pattern is available, since only some individual mutations and benign variants were reported in one study [19].
It is complicated to estimate the prevalence of pathogenic mutations in the general population, considering that until now, there have only been two studies published about the Romanian population. One, published in 2022, studying 250 women with breast cancer and 240 with ovarian cancer who underwent germline molecular testing for the detection of pathogenic BRCA1 and BRCA2 mutations, revealed that the most common variants identified were 5266delC, followed by 4218delG and c.68_69delAG for BRCA1, and c.9371A>T and c.1528G>T for BRCA2 [11]. The other study conducted on 130 breast cancer patients tested by multigene panel analysis (BRCA1, BRCA2, TP53, STK11, CDH1, PTEN, PALB2, CHEK2, ATM), highlighted BRCA1 c.3607C>T as the most common variant in the group, prevalent in triple-negative invasive carcinomas [12].
As expected, the most common mutations in our group were found in BRCA1 and BRCA2 genes, exceeding half of the reported mutations in this study. Frameshift and missense mutations leading to a complete or partial loss of tumor suppressor effect in BRCA genes were the most common defects. The c.3607C>T, c.181T>G missense variants, and c.5266dupC and c.68_69delAG(p.GluValfs) frameshift variant were recurrent in BRCA1 carriers. The c.181T>G is one of the most common causes of breast and ovarian cancer in patients with Eastern European and Polish heritage and in Sicily [20,21,22], and therefore expected to be a common variant in our study. An older study in a Nord-Eastern region of Romania revealed that the BRCA1 5382insC mutation was not observed in any of the 120 breast and 50 ovarian cancer patients, contradictory when compared to reported data for the Romanian and Eastern European populations [23].
Interestingly, only one BRCA2 mutation was found in 3 patients, the c.9371A>T (p.Asn3124Ile) missense variant, a founder defect in the eastern European population.
Only three other studies in the same demographics account for 320 patients with pathogenic mutations related to breast cancer. One study including 107 patients diagnosed with breast or ovarian cancer revealed that the c.5266dupC Ashkenazi founder mutation was the most common BRCA1 pathogenic variant reported in 36.67% of cases, closely followed by c.3607C>T in 30% of cases. Only one BRCA2 variant, c.9371A>T, was recurrent in this study [11]. Another smaller study, including 44 breast cancer patients revealed the same pattern in the BRCA1 mutational prevalence; BRCA1 c.5266dupC and c.3607C>T, BRCA2 c.9371A>T were also prevalent in this group [13]. The previous study, including 56 patients, revealed a higher prevalence for BRCA1 c.3607C>T variant than c.5266dupC and a recurrent BRCA2 mutation, c.8755-1G>A [12].
Mutations in the PALB2 gene, a high-penetrant gene associated with a 40–60% lifetime risk of BC, were reported as the third most common high-risk gene in this study. One particular variant, c.172_175delTTGT (p.Gln60Argfs), was recurrent in PALB2 carriers and was reported in other studies to be associated with hereditary breast and pancreatic cancer [24,25]. There are no available data on PALB2 mutation prevalence in the Romanian population. Only one study included East European and, thus, Romanian cancer-diagnosed patients, but with no detailed description of PALB2 mutation [26]. With a frequency ranging from 0.5 to 1.0%, the truncating PALB2 variant c.172_175delTTGT has recently been discussed in Central and Eastern Europe [27], Poland, Belarus, Germany, and Russia [28,29,30]. Considering the aforementioned, c.172_175delTTGT could be considered a PALB2 founder mutation for the Romanian population. Another mutation, the c.2257C>T (p.Arg753Ter), also seems to be recurrent for the PALB2 gene and has previously been reported in Poland and the Eastern-European population [31].
Germline testing for breast cancer should also include the PALB2 gene, considering the frequency of pathogenic defects in the general population and because PALB2 heterozygotes should be considered for the same therapeutic regimens and clinical trials as those for BRCA1 and BRCA2 carriers.
ATM is a moderately penetrant gene associated with a 20–40% risk for breast cancer. The recurrent Slavic founder mutation c.1564_1565delGA (p.Glu522Ilefs) frameshift variant was found in 41% of ATM carriers and previously reported in the Romanian population [14].
Consistent with currently available data, pathogenic variants in CHEK2 were the most frequently identified after pathogenic variants in BRCA1 or BRCA2 genes. Debates on the impact of CHEK2 pathogenic mutations on breast cancer risk are ongoing, with emerging data to classify pathogenic mutations of CHEK2 in moderate to low-penetrant variants [32]. The c.470T>C (p.Ile157Thr) variant was recurrent in our cohort, with more than half of CHEK2 mutated patients carrying this mutation. Compared to other CHEK2 pathogenic variants, c.470T>C has an attenuated association with BC, was not associated with non-breast cancers [33,34], and is probably modulated by other genetic factors or non-genetic risk factors to increase BC risk. Along with hormone-related risk factors [35], it has been shown that a family history of BC correlates with higher risks for women with CHEK2 pathogenic variants [36,37,38]. Surprisingly, the common c.1100del CHEK2 variant was reported only in one patient. On the other hand, two out of three patients carrying the c.902delT CHEK2 moderate-risk variant had other germline pathogenic mutations in the BRCA1 gene.
One unexpected finding in our study is related to the MUTYH gene. The association between MUTYH mutations and HBC risk is controversial, as there a higher level of evidence that carriers homozygous for MUTYH pathogenic variants have an increased risk of BC [39]. However, a higher frequency of heterozygous MUTYH mutations in families with breast and colorectal cancer has also been reported compared to the general population [40,41]. We identified the c.650G>A recurrent heterozygous mutation in three patients in our cohort. Two patients had invasive lobular carcinoma and intestinal polyps, and the third had a parent diagnosed with colon cancer. Among other variants, the c.650G>A mutation is reported in ClinVar to be associated with invasive Luminal B breast carcinoma [42].
4.2. Molecular Subtypes Associations
Invasive carcinoma of the breast is considered a heterogeneous group of malignant epithelial tumors. Current data describes a wide range of morphological phenotypes and specific histopathological and molecular subtypes among sporadic and hereditary types but with significant differences between genotypes of germline mutations-associated tumors [43,44]. Most BRCA1-associated breast cancers are invasive ductal carcinomas of non-special type and fall into the “basal-like” intrinsic molecular subtype [45]. These triple-negative (TN) tumors lack estrogen, progesterone receptors, and human epidermal growth factor receptor 2 expressions. BRCA2-associated breast cancers are more likely to be found in the “luminal type” and share common characteristics with sporadic tumors [46]. Luminal A tumors have Ki67 ≤ 14% and lack HER2 protein overexpression. Luminal B tumors have higher Ki67 values and are HER2-positive [47]. In our study, basal histology was a common finding in BRCA1 patients, and luminal types were more commonly identified in BRCA2 and non-BRCA carriers. Ductal in situ carcinoma (DISC) was a rare histological finding in our cohort as most diagnosed cases were symptomatic and therefore T2/T3, N/M > 1 at diagnosis. Less than 5% of cases were diagnosed as DISC, all part of annual screening due to a positive family history of breast cancer.
Ki-67 proliferative marker is considered an essential prognostic in breast cancer. It has a significantly higher expression in BRCA-positive breast cancer [48,49]. It implies its potential as an efficient prognostic factor in BRCA-positive breast cancer and future therapeutic implications in the context of emerging data suggest it might be advantageous to promote, rather than hinder, cell proliferation for immunotherapy to be optimally effective [50,51].
4.3. Cohort Particularities
We consider that our study group is representative of Romania since the women enrolled came from different geographic areas and ethnicities (multidimensional scaling analysis supported the genetic similarity of the Wallachia, Moldavia, and Dobrudja groups with the Balkans, while the Transylvanian population was closely related to Central European groups) [52].
An expected finding was that we had a higher mutation prevalence than other studies. Most patients addressing genetic counseling services had at least two or three NCCN-based testing criteria. We noticed that most patients had at least two or more NCCN-based eligibility criteria for genetic testing. This particularity implicitly associates a higher positivity rate than other groups where patients were tested with only one criterion. The over-selection is mainly due to inappropriate screening and testing guidelines in hereditary cancers. Among the factors contributing to the underutilization of genetic testing services in Romanians, we mention lower awareness of testing among patients and medical staff and support for obtaining genetic counseling and testing, particularly in resource-limited settings representing 60% of the general population. Genetic counseling, testing, and after-testing discussions are often described as complicated and inaccessible for many women in Romania. Paradoxically, approximately 40% of patients with eligibility testing criteria did not have the genetic test because of objective financial impediments. Genetic testing in Romania must be supported by the government or reimbursed by health insurance to avoid significant discrepancies between socio-economic groups.
Recurrent mutations in our study were c.3607C>T, c.181T>G, c.5266dupC in the BRCA1 gene, c.9371A>T in the BRCA2 gene, c.172_175delTTGT in the PALB2 gene, c.1564_1565delGA in the ATM gene, c.470T>C in CHEK2 gene, and c.650G>A in the MUTYH gene. For high-penetrant genes such as BRCA1, BRCA2, PALB2, and moderate-high penetrant genes such as ATM, recurrent mutation frequency converges to already published data for Eastern European populations. We observed a high frequency for CHEK2 variants, the most frequent moderate-risk breast cancer predisposition gene. Despite contradictory data on the c.407T>C pathogenicity, this variant may have more than a polygenic role model in breast carcinogenesis and deserves further large-data analysis. An unexpected finding in our cohort was the presence of heterozygous MUTYH pathogenic variants, among which c.650G>A was recurrent and already in the attention of various investigators for the association with invasive breast carcinomas [41].
We also evaluated the attitude regarding surgical prophylaxis among women with a high risk of bilateralization. If most patients with pathogenic mutations in genes with increased penetrance opted for prophylactic mastectomy in favor of conservative imaging methods, paradoxically, more patients with pathogenic mutations in genes with low or moderate penetrance requested genetic or surgical consult to perform radical surgical prophylaxis. In this regard, prophylactic mastectomy recommendations were always preceded by the Tumour Board assessment and psychological counseling to avoid unnecessary interventions.
Genetic counseling has been challenging due to insufficient genetic screening programs and medical education. For example, an extensive pre-pandemic report revealed that even in developed countries, 50% of women diagnosed with breast cancer do not receive genetic counseling [53]. If genetic counseling was available without significant impediments for educated patients younger than 40 years of age, in older women or women with low education, we confronted issues related to understanding the information, advantages, and the medical use of genetic testing. In this case, more than one genetic counseling session or integration of another family member was necessary. Since most of our subjects were submitted to genetic testing at diagnosis, a critical timing in patients’ medical management also focused on identifying subjects at higher risk of psychological distress to address them for psychological support and give them the appropriate coping strategy. We observed that a positive genetic test still creates a significant emotional stigma for the patient and other family members and therefore requires professional assistance to avoid further emotional distress.
5. Conclusions
The genetic characteristics of HBC in Romania are similar to those reported for the East Caucasian and Slavic populations. BRCA carriers and patients with a family history of cancer are diagnosed with breast cancer earlier than carriers of other mutations. TN tumors are associated with BRCA1 mutations, while Luminal B subtype tumors are associated with BRCA2 mutations. Further attention is recommended for moderate-penetrant genes like CHEK2 to determine the role of pathogenic variants in assessing BC predisposition. Romania, part of the EU, requires the implementation of genetic screening programs and proper genetic counseling services to reduce the number of women with hereditary genetic components and late diagnosis.
Author Contributions
Conceptualization, P.A.A.-C., N.Z.A. and B.F.; methodology, M.S.M.; software, V.F.C.; investigation, A.C., G.B.-M., C.L., D.L.M., M.M. and A.P.T.; Resources, G.B.-M.; writing—original draft preparation, E.K.; writing—review and editing, A.C. and F.P.; supervision, A.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki, and approved by the Institutional Ethics Committee of I. Chiricuta Oncology Institute of Cluj-Napoca, Romania (ONCOGEN/1.03.2018).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
Data is available on request to the corresponding author.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Furtunescu, F.; Bohiltea, R.E.; Voinea, S.; Georgescu, T.A.; Munteanu, O.; Neacsu, A.; Pop, C.S. Breast cancer mortality gaps in Romanian women compared to the EU after 10 years of accession: Is breast cancer screening a priority for action in Romania? (Review of the Statistics). Exp. Ther. Med. 2021, 21, 268. [Google Scholar] [CrossRef]
- Motoi, G.; Niţă, A.M. The efficiency of public policies and programs for breast cancer prevention. Socio-medical perspectives within a Romania–France comparison. Rom. J. Morphol. Embryol. 2021, 62, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Yao, Q.; Xu, Y.; Yu, C.; Zhang, J.; Wang, Q.; Li, J.; Shi, D.; Yu, B.; Zeng, Y.; et al. Characteristics of Germline Non-BRCA Mutation Status of High-Risk Breast Cancer Patients in China and Correlation with High-Risk Factors and Multigene Testing Suggestions. Front. Genet. 2021, 12, 674094. [Google Scholar] [CrossRef]
- Bono, M.; Fanale, D.; Incorvaia, L.; Cancelliere, D.; Fiorino, A.; Calò, V.; Dimino, A.; Filorizzo, C.; Corsini, L.; Brando, C.; et al. Impact of deleterious variants in other genes beyond BRCA1/2 detected in breast/ovarian and pancreatic cancer patients by NGS-based multi-gene panel testing: Looking over the hedge. ESMO Open 2021, 6, 100235. [Google Scholar] [CrossRef]
- LaDuca, H.; Polley, E.C.; Yussuf, A.; Hoang, L.; Gutierrez, S.; Hart, S.N.; Yadav, S.; Hu, C.; Na, J.; Goldgar, D.E.; et al. A clinical guide to hereditary cancer panel testing: Evaluation of gene-specific cancer associations and sensitivity of genetic testing criteria in a cohort of 165,000 high-risk patients. Genet. Med. 2020, 22, 407–415. [Google Scholar] [CrossRef]
- Manahan, E.R.; Kuerer, H.M.; Sebastian, M.; Hughes, K.S.; Boughey, J.C.; Euhus, D.M.; Boolbol, S.K.; Taylor, W.A. Consensus Guidelines on Genetic’ Testing for Hereditary Breast Cancer from the American Society of Breast Surgeons. Ann. Surg. Oncol. 2019, 26, 3025–3031. [Google Scholar] [CrossRef] [PubMed]
- Forbes, C.; Fayter, D.; de Kock, S.; Quek, R.G. A systematic review of international guidelines and recommendations for the genetic screening, diagnosis, genetic counseling, and treatment of BRCA-mutated breast cancer. Cancer Manag. Res. 2019, 11, 2321–2337. [Google Scholar] [CrossRef] [PubMed]
- Tung, N.; Garber, J.E. PARP inhibition in breast cancer: Progress made and future hopes. Npj Breast Cancer 2022, 8, 47. [Google Scholar] [CrossRef] [PubMed]
- Lilyquist, J.; Ruddy, K.J.; Vachon, C.M.; Couch, F.J. Common Genetic Variation and Breast Cancer Risk-Past, Present, and Future. Cancer Epidemiol. Biomark. Prev. 2018, 27, 380–394. [Google Scholar] [CrossRef]
- Hlavac, V.; Kovacova, M.; Elsnerova, K.; Brynychova, V.; Kozevnikovova, R.; Raus, K.; Kopeckova, K.; Mestakova, S.; Vrana, D.; Gatek, J.; et al. Use of Germline Genetic Variability for Prediction of Chemoresistance and Prognosis of Breast Cancer Patients. Cancers 2018, 10, 511. [Google Scholar] [CrossRef]
- Vidra, R.; Ciuleanu, T.E.; Nemeș, A.; Pascu, O.; Heroiu, A.M.; Antone, N.; Vidrean, A.I.; Oprean, C.M.; Pop, L.A.; Berindan-Neagoe, I.; et al. Spectrum of BRCA1/2 Mutations in Romanian Breast and Ovarian Cancer Patients. Int. J. Environ. Res. Public Health 2022, 19, 4314. [Google Scholar] [CrossRef]
- Goidescu, I.G.; Caracostea, G.; Eniu, D.T.; Stamatian, F.V. Prevalence of deleterious mutations among patients with breast cancer referred for multigene panel testing in a Romanian population. Med. Pharm. Rep. 2018, 91, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Eniu, A.; Pop, L.; Stoian, A.; Dronca, E.; Matei, R.; Ligtenberg, M.; Ouchene, H.; Onisim, A.; Rotaru, O.; Eniu, R.; et al. Understanding BRCA1 and BRCA2 mutated breast cancer cases in Romania: First report on founder mutations in Romanians. Ann. Oncol. 2017, 28, v60–v61. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2022; Available online: https://www.R-project.org/ (accessed on 27 April 2023).
- RStudio: Integrated Development Environment for R; RStudio, PBC: Boston, MA, USA, 2022; Available online: http://www.rstudio.com (accessed on 27 April 2023).
- Wickham, H. Stringr: Simple, Consistent Wrappers for Common String Operations 2022. Available online: https://CRAN.R-project.org/package=stringr (accessed on 27 April 2023).
- Wickham, H.; Bryan, J. Readxl: Read Excel Files. 2023. Available online: https://CRAN.R-project.org/package=readxl (accessed on 27 April 2023).
- Skidmore, Z.L.; Wagner, A.H.; Lesurf, R.; Campbell, K.M.; Kunisaki, J.; Griffith, O.L.; Griffith, M. GenVisR: Genomic Visualizations in R. Bioinformatics 2016, 32, 3012–3014. [Google Scholar] [CrossRef] [PubMed]
- Janavičius, R. Founder BRCA1/2 mutations in the Europe: Implications for hereditary breast-ovarian cancer prevention and control. EPMA J. 2010, 1, 397–412. [Google Scholar] [CrossRef]
- Górski, B.; Byrski, T.; Huzarski, T.; Jakubowska, A.; Menkiszak, J.; Gronwald, J.; Płużańska, A.; Bębenek, M.; Fischer-Maliszewska, Ł.; Grzybowska, E.; et al. Founder Mutations in the BRCA1 Gene in Polish Families with Breast-Ovarian Cancer. Am. J. Hum. Genet. 2000, 66, 1963–1968. [Google Scholar] [CrossRef]
- Kaufman, B.; Laitman, Y.; Gronwald, J.; Lubinski, J.; Friedman, E. Haplotype of the C61G BRCA1 Mutation in Polish and Jewish Individuals. Genet. Test. Mol. Biomark. 2009, 13, 465–469. [Google Scholar] [CrossRef]
- Bogdanova, N.; Antonenkova, N.; Rogov, Y.; Karstens, J.; Hillemanns, P.; Dörk, T. High frequency and allele-specific differences of BRCA1 founder mutations in breast cancer and ovarian cancer patients from Belarus. Clin. Genet. 2010, 78, 364–372. [Google Scholar] [CrossRef]
- Negură, L.; Duşa, C.P.; Balmuş, M.I.; Azoicăi, D.; Negură, A.M.; Marinca, M.V.; Miron, L. BRCA1 5382insC founder mutation has not a significative recurrent presence in Northeastern Romanian cancer patients. Rom. J. Morphol. Embryol. 2015, 56, 379–385. [Google Scholar]
- Janatova, M.; Kleibl, Z.; Stribrna, J.; Panczak, A.; Vesela, K.; Zimovjanova, M.; Kleiblova, P.; Dundr, P.; Soukupova, J.; Pohlreich, P. The PALB2 gene is a strong candidate for clinical testing in BRCA1- and BRCA2-negative hereditary breast cancer. Cancer Epidemiol. Biomark. Prev. 2013, 22, 2323–2332. [Google Scholar] [CrossRef]
- Wojcik, P.; Jasiowka, M.; Strycharz, E.; Sobol, M.; Hodorowicz-Zaniewska, D.; Skotnicki, P.; Byrski, T.; Blecharz, P.; Marczyk, E.; Cedrych, I.; et al. Recurrent mutations of BRCA1, BRCA2 and PALB2 in the population of breast and ovarian cancer patients in Southern Poland. Hered. Cancer Clin. Pract. 2016, 14, 5. [Google Scholar] [CrossRef] [PubMed]
- Tsaousis, G.N.; Papadopoulou, E.; Apessos, A.; Agiannitopoulos, K.; Pepe, G.; Kampouri, S.; Diamantopoulos, N.; Floros, T.; Iosifidou, R.; Katopodi, O.; et al. Analysis of hereditary cancer syndromes by using a panel of genes: Novel and multiple pathogenic mutations. BMC Cancer 2019, 19, 535. [Google Scholar] [CrossRef] [PubMed]
- Hilz, P.; Heinrihsone, R.; Pätzold, L.A.; Qi, Q.; Trofimovics, G.; Gailite, L.; Irmejs, A.; Gardovskis, J.; Miklasevics, E.; Daneberga, Z. Allelic variants of breast cancer susceptibility genes PALB2 and RECQL in the Latvian population. Hered. Cancer Clin. Pract. 2019, 17, 17. [Google Scholar] [CrossRef] [PubMed]
- Cybulski, C.; Kluźniak, W.; Huzarski, T.; Wokołorczyk, D.; Kashyap, A.; Jakubowska, A.; Szwiec, M.; Byrski, T.; Dębniak, T.; Górski, B.; et al. Clinical outcomes in women with breast cancer and a PALB2 mutation: A prospective cohort analysis. Lancet Oncol. 2015, 16, 638–644. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef]
- Suspitsin, E.; Sokolenko, A.; Bizin, I.; Tumakova, A.; Guseva, M.; Sokolova, N.; Vakhlyarskaya, S.; Kondratenko, I.; Imyanitov, E. ATM mutation spectrum in Russian children with ataxia-telangiectasia. Eur. J. Med. Genet. 2020, 63, 103630. [Google Scholar] [CrossRef] [PubMed]
- Kluska, A.; Balabas, A.; Piatkowska, M.; Czarny, K.; Paczkowska, K.; Nowakowska, D.; Mikula, M.; Ostrowski, J. PALB2 mutations in BRCA1/2-mutation negative breast and ovarian cancer patients from Poland. BMC Med. Genomics 2017, 10, 14. [Google Scholar] [CrossRef]
- Neben, C.L.; Zimmer, A.D.; Stedden, W.; van den Akker, J.; O’Connor, R.; Chan, R.C.; Chen, E.; Tan, Z.; Leon, A.; Ji, J.; et al. Multi-Gene Panel Testing of 23,179 Individuals for Hereditary Cancer Risk Identifies Pathogenic Variant Carriers Missed by Current Genetic Testing Guidelines. J. Mol. Diagn. 2019, 21, 646–657. [Google Scholar] [CrossRef]
- Boonen, R.A.; Wiegant, W.W.; Celosse, N.; Vroling, B.; Heijl, S.; Kote-Jarai, Z.; Mijuskovic, M.; Cristea, S.; Solleveld-Westerink, N.; van Wezel, T.; et al. Functional Analysis Identifies Damaging CHEK2 Missense Variants Associated with Increased Cancer Risk. Cancer Res. 2022, 82, 615–631. [Google Scholar] [CrossRef]
- Bychkovsky, B.L.; Agaoglu, N.B.; Horton, C.; Zhou, J.; Yussuf, A.; Hemyari, P.; Richardson, M.E.; Young, C.; LaDuca, H.; McGuinness, D.L.; et al. Differences in Cancer Phenotypes Among Frequent CHEK2 Variants and Implications for Clinical Care—Checking CHEK2. JAMA Oncol. 2022, 8, 1598–1606. [Google Scholar] [CrossRef]
- Ivanov, M.; Sharova, M.; Olsen, A.; Lebedeva, A.; Ignatova, E.; Mouse, G.; Mileyko, V. Letter to the Editor: CHEK2 I157T-Pluto Among Numerous Low-Risk Genetic Factors Requiring Discharge From a Range of Pathogenic Variants? J. Natl. Compr. Cancer Netw. 2022, 20. [Google Scholar] [CrossRef]
- Park, B.; Hopper, J.L.; Win, A.K.; Dowty, J.G.; Sung, H.K.; Ahn, C.; Kim, S.-W.; Lee, M.H.; Lee, J.; Lee, J.W.; et al. Reproductive factors as risk modifiers of breast cancer in BRCA mutation carriers and high-risk non-carriers. Oncotarget 2017, 8, 102110–102118. [Google Scholar] [CrossRef]
- Cybulski, C.; Wokołorczyk, D.; Jakubowska, A.; Huzarski, T.; Byrski, T.; Gronwald, J.; Masojć, B.; Dębniak, T.; Górski, B.; Blecharz, P.; et al. Risk of Breast Cancer in Women with a CHEK2 Mutation with and Without a Family History of Breast Cancer. J. Clin. Oncol. 2011, 29, 3747–3752. [Google Scholar] [CrossRef]
- Gallagher, S.; Hughes, E.; Kurian, A.W.; Domchek, S.M.; Garber, J.; Probst, B.; Morris, B.; Tshiaba, P.; Meek, S.; Rosenthal, E.; et al. Comprehensive Breast Cancer Risk Assessment for CHEK2 and ATM Pathogenic Variant Carriers Incorporating a Polygenic Risk Score and the Tyrer-Cuzick Model. JCO Precis. Oncol. 2021, 5, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Win, A.K.; Reece, J.C.; Dowty, J.G.; Buchanan, D.D.; Clendenning, M.; Rosty, C.; Southey, M.C.; Young, J.P.; Cleary, S.P.; Kim, H.; et al. Risk of extracolonic cancers for people with biallelic and monoallelic mutations in MUTYH: Extracolonic cancer risks for people with biallelic and monoallelic MUTYH mutations. Int. J. Cancer 2016, 139, 1557–1563. [Google Scholar] [CrossRef]
- Wasielewski, M.; Out, A.A.; Vermeulen, J.; Nielsen, M.; Ouweland, A.V.D.; Tops, C.M.J.; Wijnen, J.T.; Vasen, H.; Weiss, M.M.; Klijn, J.G.M.; et al. Increased MUTYH mutation frequency among Dutch families with breast cancer and colorectal cancer. Breast Cancer Res. Treat. 2010, 124, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Rennert, G.; Lejbkowicz, F.; Cohen, I.; Pinchev, M.; Rennert, H.S.; Barnett-Griness, O. MutYH mutation carriers have increased breast cancer risk. Cancer 2012, 118, 1989–1993. [Google Scholar] [CrossRef]
- VCV000140877.35—ClinVar—NCBI. Available online: https://www.ncbi.nlm.nih.gov/clinvar/variation/140877/?new_evidence=true (accessed on 18 February 2023).
- Honrado, E.; Benítez, J.; Palacios, J. The Pathology of Hereditary Breast Cancer. Hered. Cancer Clin. Pract. 2004, 2, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Stoppa-Lyonnet, D. The biological effects and clinical implications of BRCA mutations: Where do we go from here? Eur. J. Hum. Genet. 2016, 24 (Suppl. 1), S3–S9. [Google Scholar] [CrossRef]
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; Van De Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Krammer, J.; Pinker-Domenig, K.; Robson, M.E.; Gönen, M.; Bernard-Davila, B.; Morris, E.A.; Mangino, D.A.; Jochelson, M.S. Breast cancer detection and tumor characteristics in BRCA1 and BRCA2 mutation carriers. Breast Cancer Res. Treat. 2017, 163, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Mavaddat, N.; Barrowdale, D.; Andrulis, I.L.; Domchek, S.M.; Eccles, D.; Nevanlinna, H.; Ramus, S.J.; Spurdle, A.; Robson, M.; Sherman, M.; et al. Pathology of Breast and Ovarian Cancers among BRCA1 and BRCA2 Mutation Carriers: Results from the Consortium of Investigators of Modifiers of BRCA1/2 (CIMBA). Cancer Epidemiol. Biomark. Prev. 2012, 21, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, T.O.; Leung, S.C.Y.; Rimm, D.L.; Dodson, A.; Acs, B.; Badve, S.; Denkert, C.; Ellis, M.J.; Fineberg, S.; Flowers, M.; et al. Assessment of Ki67 in Breast Cancer: Updated Recommendations From the International Ki67 in Breast Cancer Working Group. JNCI J. Natl. Cancer Inst. 2020, 113, 808–819. [Google Scholar] [CrossRef]
- Lakhani, S.R.; Van De Vijver, M.J.; Jacquemier, J.; Anderson, T.J.; Osin, P.P.; McGuffog, L.; Easton, D.F. The Pathology of Familial Breast Cancer: Predictive Value of Immunohistochemical Markers Estrogen Receptor, Progesterone Receptor, HER-2, and p53 in Patients with Mutations in BRCA1 and BRCA2. J. Clin. Oncol. 2002, 20, 2310–2318. [Google Scholar] [CrossRef] [PubMed]
- Hacking, S.M.; Wang, Y. Practical Issues of Ki-67 Evaluation in Breast Cancer Clinical Practice. J. Clin. Transl. Pathol. 2022. [Google Scholar] [CrossRef]
- Mrouj, K.; Andrés-Sánchez, N.; Dubra, G.; Singh, P.; Sobecki, M.; Chahar, D.; Al Ghoul, E.; Aznar, A.B.; Prieto, S.; Pirot, N.; et al. Ki-67 regulates global gene expression and promotes sequential stages of carcinogenesis. Proc. Natl. Acad. Sci. USA 2021, 118, e2026507118. [Google Scholar] [CrossRef]
- Cocoş, R.; Schipor, S.; Hervella, M.; Cianga, P.; Popescu, R.; Bănescu, C.; Constantinescu, M.; Martinescu, A.; Raicu, F. Genetic affinities among the historical provinces of Romania and Central Europe as revealed by an mtDNA analysis. BMC Genet. 2017, 18, 20. [Google Scholar] [CrossRef] [PubMed]
- Katz, S.J.; Ward, K.C.; Hamilton, A.S.; McLeod, M.C.; Wallner, L.P.; Morrow, M.; Jagsi, R.; Hawley, S.T.; Kurian, A.W. Gaps in Receipt of Clinically Indicated Genetic Counseling After Diagnosis of Breast Cancer. J. Clin. Oncol. 2018, 36, 1218–1224. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).