

Targeting Mitochondrial DNA Transcription by POLRMT Inhibition or Depletion as a Potential Strategy for Cancer Treatment

Abstract
:1. Mitochondrial Transcription and Metabolism as Targets for New Anti-Cancer Approaches
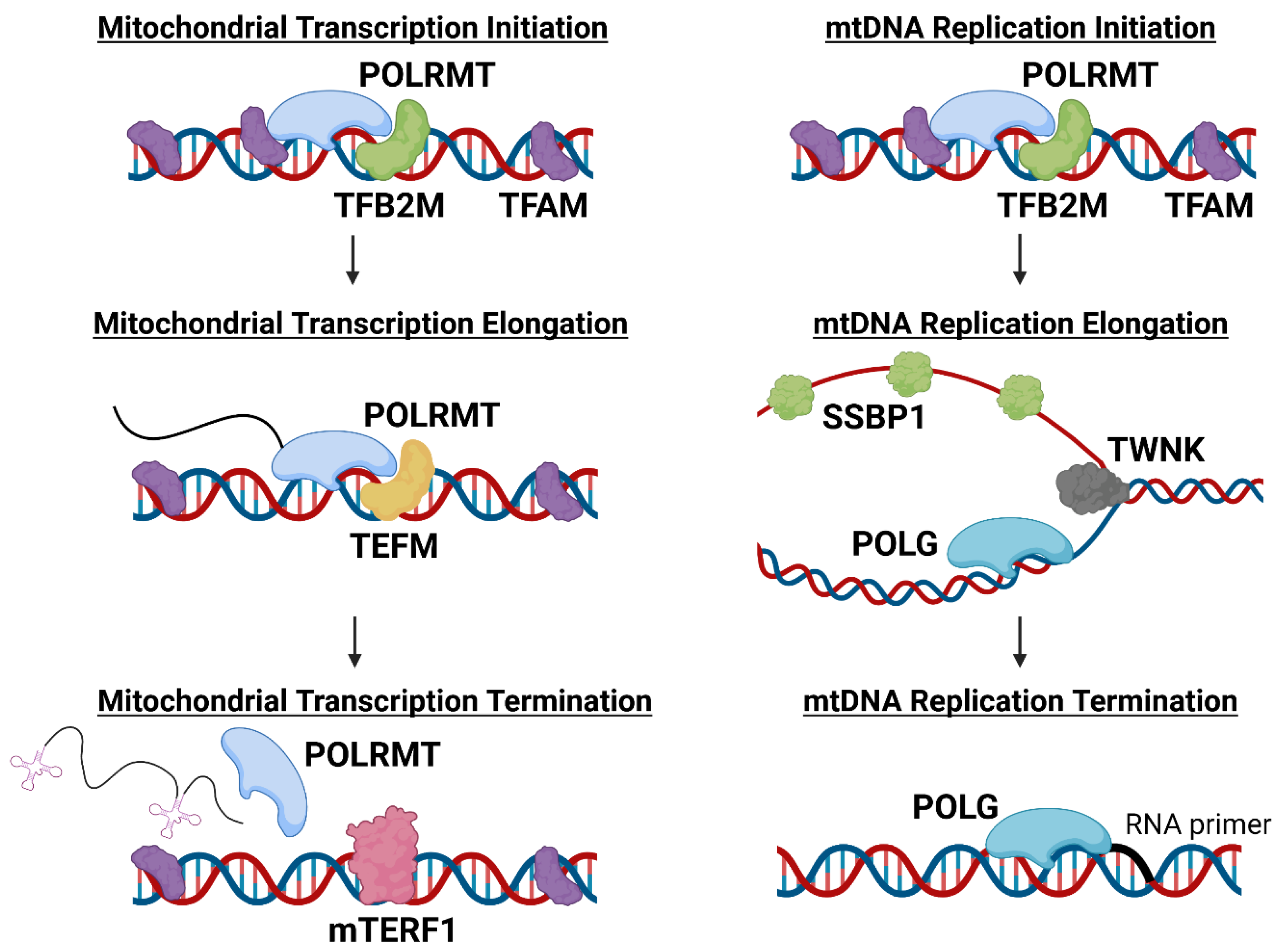
2. Functional Roles of POLRMT in Mitochondria
3. POLRMT Inhibition as an Anti-Cancer Strategy
4. Small Molecule ClpP Agonists as Anti-Cancer Compounds
5. Mechanistic Similarities between ClpP Agonists and POLRMT Inhibitors
5.1. Inhibition of Cell Proliferation
5.2. Cytostatic to Cancer Cells, Harmless to Normal Cells
5.3. Dysregulation of Cancer Cell Metabolic Programs
5.4. Loss of mtDNA Content
5.5. Inhibition of Mitochondrial Transcription
5.6. Summary of Similarities between ClpP Agonists and POLRMT Inhibitors
6. Mechanistic Differences between ClpP Agonists and POLRMT Inhibitors
6.1. Differences in Treatment Response Times
6.2. Known and Unknown Mechanisms of Resistance
6.3. Inhibiting One Protein versus Degrading Many Proteins
7. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chakrabarty, R.P.; Chandel, N.S. Beyond ATP, New Roles of Mitochondria. Biochemist 2022, 44, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Haynes, C.M.; Petrova, K.; Benedetti, C.; Yang, Y.; Ron, D. ClpP Mediates Activation of a Mitochondrial Unfolded Protein Response in C. Elegans. Dev. Cell 2007, 13, 467–480. [Google Scholar] [CrossRef]
- Giampazolias, E.; Zunino, B.; Dhayade, S.; Bock, F.; Cloix, C.; Cao, K.; Roca, A.; Lopez, J.; Ichim, G.; Proïcs, E.; et al. Mitochondrial Permeabilization Engages NF-ΚB-Dependent Anti-Tumour Activity under Caspase Deficiency. Nat. Cell Biol. 2017, 19, 1116–1129. [Google Scholar] [CrossRef] [PubMed]
- Collins, Y.; Chouchani, E.T.; James, A.M.; Menger, K.E.; Cochemé, H.M.; Murphy, M.P. Mitochondrial Redox Signalling at a Glance. J. Cell Sci. 2012, 125, 801–806. [Google Scholar] [CrossRef]
- Riley, J.S.; Tait, S.W. Mitochondrial DNA in Inflammation and Immunity. EMBO Rep. 2020, 21, e49799. [Google Scholar] [CrossRef]
- Qureshi, M.A.; Haynes, C.M.; Pellegrino, M.W. The Mitochondrial Unfolded Protein Response: Signaling from the Powerhouse. J. Biol. Chem. 2017, 292, 13500–13506. [Google Scholar] [CrossRef] [PubMed]
- Bueno, M.J.; Ruiz-Sepulveda, J.L.; Quintela-Fandino, M. Mitochondrial Inhibition: A Treatment Strategy in Cancer? Curr. Oncol. Rep. 2021, 23, 49. [Google Scholar] [CrossRef]
- Rackham, O.; Filipovska, A. Organization and Expression of the Mammalian Mitochondrial Genome. Nat. Rev. Genet. 2022, 23, 606–623. [Google Scholar] [CrossRef]
- Hillen, H.S.; Temiakov, D.; Cramer, P. Structural Basis of Mitochondrial Transcription. Nat. Struct. Mol. Biol. 2018, 25, 754–765. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Kurita, M.; Aiso, S.; Nishimoto, I.; Matsuoka, M. Humanin Inhibits Neuronal Cell Death by Interacting with a Cytokine Receptor Complex or Complexes Involving CNTF Receptor Alpha/WSX-1/Gp130. Mol. Biol. Cell 2009, 20, 2864–2873. [Google Scholar] [CrossRef]
- Ro, S.; Ma, H.-Y.; Park, C.; Ortogero, N.; Song, R.; Hennig, G.W.; Zheng, H.; Lin, Y.-M.; Moro, L.; Hsieh, J.-T.; et al. The Mitochondrial Genome Encodes Abundant Small Noncoding RNAs. Cell Res. 2013, 23, 759–774. [Google Scholar] [CrossRef] [PubMed]
- Kummer, E.; Ban, N. Mechanisms and Regulation of Protein Synthesis in Mitochondria. Nat. Rev. Mol. Cell Biol. 2021, 22, 307–325. [Google Scholar] [CrossRef] [PubMed]
- Sainero-Alcolado, L.; Liaño-Pons, J.; Ruiz-Pérez, M.V.; Arsenian-Henriksson, M. Targeting Mitochondrial Metabolism for Precision Medicine in Cancer. Cell Death Differ. 2022, 29, 1304–1317. [Google Scholar] [CrossRef] [PubMed]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.S.; et al. Metformin Inhibits Mitochondrial Complex I of Cancer Cells to Reduce Tumorigenesis. eLife 2014, 3, e02242. [Google Scholar] [CrossRef]
- Moullan, N.; Mouchiroud, L.; Wang, X.; Ryu, D.; Williams, E.G.; Mottis, A.; Jovaisaite, V.; Frochaux, M.V.; Quiros, P.M.; Deplancke, B.; et al. Tetracyclines Disturb Mitochondrial Function across Eukaryotic Models: A Call for Caution in Biomedical Research. Cell Rep. 2015, 10, 1681–1691. [Google Scholar] [CrossRef]
- Dijk, S.N.; Protasoni, M.; Elpidorou, M.; Kroon, A.M.; Taanman, J.-W. Mitochondria as Target to Inhibit Proliferation and Induce Apoptosis of Cancer Cells: The Effects of Doxycycline and Gemcitabine. Sci. Rep. 2020, 10, 4363. [Google Scholar] [CrossRef]
- Rivas, M.O.G.; Stuart, S.D.; Thach, D.; Dahan, M.; Shorr, R.; Zachar, Z.; Bingham, P.M. Evidence for a Novel, Effective Approach to Targeting Carcinoma Catabolism Exploiting the First-in-Class, Anti-Cancer Mitochondrial Drug, CPI-613. PLoS ONE 2022, 17, e0269620. [Google Scholar] [CrossRef]
- Delaunay, S.; Pascual, G.; Feng, B.; Klann, K.; Behm, M.; Hotz-Wagenblatt, A.; Richter, K.; Zaoui, K.; Herpel, E.; Münch, C.; et al. Mitochondrial RNA Modifications Shape Metabolic Plasticity in Metastasis. Nature 2022, 607, 593–603. [Google Scholar] [CrossRef]
- Bergbrede, T.; Hoberg, E.; Larsson, N.-G.; Falkenberg, M.; Gustafsson, C.M. An Adaptable High-Throughput Technology Enabling the Identification of Specific Transcription Modulators. SLAS Discov. 2017, 22, 378–386. [Google Scholar] [CrossRef]
- Bonekamp, N.A.; Peter, B.; Hillen, H.S.; Felser, A.; Bergbrede, T.; Choidas, A.; Horn, M.; Unger, A.; Di Lucrezia, R.; Atanassov, I.; et al. Small-Molecule Inhibitors of Human Mitochondrial DNA Transcription. Nature 2020, 588, 712–716. [Google Scholar] [CrossRef]
- Reznik, E.; Miller, M.L.; Şenbabaoğlu, Y.; Riaz, N.; Sarungbam, J.; Tickoo, S.K.; Al-Ahmadie, H.A.; Lee, W.; Seshan, V.E.; Hakimi, A.A.; et al. Mitochondrial DNA Copy Number Variation across Human Cancers. eLife 2016, 5, e10769. [Google Scholar] [CrossRef] [PubMed]
- van Osch, F.H.M.; Voets, A.M.; Schouten, L.J.; Gottschalk, R.W.H.; Simons, C.C.J.M.; van Engeland, M.; Lentjes, M.H.F.M.; van den Brandt, P.A.; Smeets, H.J.M.; Weijenberg, M.P. Mitochondrial DNA Copy Number in Colorectal Cancer: Between Tissue Comparisons, Clinicopathological Characteristics and Survival. Carcinogenesis 2015, 36, 1502–1510. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Ju, Y.S.; Kim, Y.; Li, J.; Wang, Y.; Yoon, C.J.; Yang, Y.; Martincorena, I.; Creighton, C.J.; Weinstein, J.N.; et al. Comprehensive Molecular Characterization of Mitochondrial Genomes in Human Cancers. Nat. Genet. 2020, 52, 342–352. [Google Scholar] [CrossRef]
- Tseng, L.-M.; Yin, P.-H.; Chi, C.-W.; Hsu, C.-Y.; Wu, C.-W.; Lee, L.-M.; Wei, Y.-H.; Lee, H.-C. Mitochondrial DNA Mutations and Mitochondrial DNA Depletion in Breast Cancer. Genes Chromosom. Cancer 2006, 45, 629–638. [Google Scholar] [CrossRef]
- Wallace, D.C. Mitochondria and Cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef] [PubMed]
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and Cancer. Cell 2016, 166, 555–566. [Google Scholar] [CrossRef]
- Zong, W.-X.; Rabinowitz, J.D.; White, E. Mitochondria and Cancer. Mol. Cell 2016, 61, 667–676. [Google Scholar] [CrossRef]
- Filograna, R.; Mennuni, M.; Alsina, D.; Larsson, N.-G. Mitochondrial DNA Copy Number in Human Disease: The More the Better? FEBS Lett. 2021, 595, 976–1002. [Google Scholar] [CrossRef]
- Grandhi, S.; Bosworth, C.; Maddox, W.; Sensiba, C.; Akhavanfard, S.; Ni, Y.; LaFramboise, T. Heteroplasmic Shifts in Tumor Mitochondrial Genomes Reveal Tissue-Specific Signals of Relaxed and Positive Selection. Hum. Mol. Genet. 2017, 26, 2912–2922. [Google Scholar] [CrossRef]
- Lax, N.Z.; Turnbull, D.M.; Reeve, A.K. Mitochondrial Mutations. Neuroscientist 2011, 17, 645–658. [Google Scholar] [CrossRef]
- Van Gisbergen, M.W.; Voets, A.M.; Starmans, M.H.W.; De Coo, I.F.M.; Yadak, R.; Hoffmann, R.F.; Boutros, P.C.; Smeets, H.J.M.; Dubois, L.; Lambin, P. How Do Changes in the MtDNA and Mitochondrial Dysfunction Influence Cancer and Cancer Therapy? Challenges, Opportunities and Models. Mutat. Res. Rev. Mutat. Res. 2015, 764, 16–30. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Ou, L.; Zhang, Y.; Shen, F.; Chen, Y. A First-in-Class POLRMT Specific Inhibitor IMT1 Suppresses Endometrial Carcinoma Cell Growth. Cell Death Dis. 2023, 14, 152. [Google Scholar] [CrossRef] [PubMed]
- Graves, P.R.; Aponte-Collazo, L.J.; Fennell, E.M.J.; Graves, A.C.; Hale, A.E.; Dicheva, N.; Herring, L.E.; Gilbert, T.S.K.; East, M.P.; McDonald, I.M.; et al. Mitochondrial Protease ClpP Is a Target for the Anticancer Compounds ONC201 and Related Analogues. ACS Chem. Biol. 2019, 14, 1020–1029. [Google Scholar] [CrossRef] [PubMed]
- Fennell, E.M.J.; Aponte-Collazo, L.J.; Wynn, J.D.; Drizyte-Miller, K.; Leung, E.; Greer, Y.E.; Graves, P.R.; Iwanowicz, A.A.; Ashamalla, H.; Holmuhamedov, E.; et al. Characterization of TR-107, a Novel Chemical Activator of the Human Mitochondrial Protease ClpP. Pharmacol. Res. Perspect. 2022, 10, e00993. [Google Scholar] [CrossRef]
- Mabanglo, M.F.; Wong, K.S.; Barghash, M.M.; Leung, E.; Chuang, S.H.W.; Ardalan, A.; Majaesic, E.M.; Wong, C.J.; Zhang, S.; Lang, H.; et al. Potent ClpP Agonists with Anticancer Properties Bind with Improved Structural Complementarity and Alter the Mitochondrial N-Terminome. Structure 2022, 31, 185–200. [Google Scholar] [CrossRef]
- Wang, P.; Zhang, T.; Wang, X.; Xiao, H.; Li, H.; Zhou, L.-L.; Yang, T.; Wei, B.; Zhu, Z.; Zhou, L.; et al. Aberrant Human ClpP Activation Disturbs Mitochondrial Proteome Homeostasis to Suppress Pancreatic Ductal Adenocarcinoma. Cell Chem. Biol. 2022, 29, 1396–1408.e8. [Google Scholar] [CrossRef]
- Allen, J.E.; Krigsfeld, G.; Patel, L.; Mayes, P.A.; Dicker, D.T.; Wu, G.S.; El-Deiry, W.S. Identification of TRAIL-Inducing Compounds Highlights Small Molecule ONC201/TIC10 as a Unique Anti-Cancer Agent That Activates the TRAIL Pathway. Mol. Cancer 2015, 14, 99. [Google Scholar] [CrossRef]
- Fennell, E.M.J.; Aponte-Collazo, L.J.; Pathmasiri, W.; Rushing, B.R.; Barker, N.K.; Partridge, M.C.; Li, Y.-Y.; White, C.A.; Greer, Y.E.; Herring, L.E.; et al. Multi-Omics Analyses Reveal ClpP Activators Disrupt Essential Mitochondrial Pathways in Triple-Negative Breast Cancer. Front. Pharmacol. 2023, 14, 1136317. [Google Scholar] [CrossRef]
- Kline, C.L.B.; Van den Heuvel, A.P.J.; Allen, J.E.; Prabhu, V.V.; Dicker, D.T.; El-Deiry, W.S. ONC201 Kills Solid Tumor Cells by Triggering an Integrated Stress Response Dependent on ATF4 Activation by Specific EIF2α Kinases. Sci. Signal. 2016, 9, ra18. [Google Scholar] [CrossRef]
- Ray, J.E.; Ralff, M.D.; Jhaveri, A.; Zhou, L.; Dicker, D.T.; Ross, E.A.; El-Deiry, W.S. Antitumorigenic Effect of Combination Treatment with ONC201 and TRAIL in Endometrial Cancer in Vitro and in Vivo. Cancer Biol. Ther. 2021, 22, 554–563. [Google Scholar] [CrossRef]
- Yuan, X.; Kho, D.; Xu, J.; Gajan, A.; Wu, K.; Wu, G.S. ONC201 Activates ER Stress to Inhibit the Growth of Triple-Negative Breast Cancer Cells. Oncotarget 2017, 8, 21626–21638. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.E.; Crowder, R.; El-Deiry, W.S. First-in-Class Small Molecule ONC201 Induces DR5 and Cell Death in Tumor but Not Normal Cells to Provide a Wide Therapeutic Index as an Anti-Cancer Agent. PLoS ONE 2015, 10, e0143082. [Google Scholar] [CrossRef] [PubMed]
- Hillen, H.S.; Morozov, Y.I.; Sarfallah, A.; Temiakov, D.; Cramer, P. Structural Basis of Mitochondrial Transcription Initiation. Cell 2017, 171, 1072–1081.e10. [Google Scholar] [CrossRef] [PubMed]
- Shutt, T.E.; Bestwick, M.; Shadel, G.S. The Core Human Mitochondrial Transcription Initiation Complex. Transcription 2011, 2, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, B.A.; Durisic, N.; Mativetsky, J.M.; Costantino, S.; Hancock, M.A.; Grutter, P.; Shoubridge, E.A. The Mitochondrial Transcription Factor TFAM Coordinates the Assembly of Multiple DNA Molecules into Nucleoid-like Structures. Mol. Biol. Cell 2007, 18, 3225–3236. [Google Scholar] [CrossRef]
- Kukat, C.; Wurm, C.A.; Spåhr, H.; Falkenberg, M.; Larsson, N.-G.; Jakobs, S. Super-Resolution Microscopy Reveals That Mammalian Mitochondrial Nucleoids Have a Uniform Size and Frequently Contain a Single Copy of MtDNA. Proc. Natl. Acad. Sci. USA 2011, 108, 13534–13539. [Google Scholar] [CrossRef]
- Bonekamp, N.A.; Jiang, M.; Motori, E.; Villegas, R.G.; Koolmeister, C.; Atanassov, I.; Mesaros, A.; Park, C.B.; Larsson, N.-G. High Levels of TFAM Repress Mammalian Mitochondrial DNA Transcription in Vivo. Life Sci. Alliance 2021, 4, 11. [Google Scholar] [CrossRef]
- Lodeiro, M.F.; Uchida, A.; Bestwick, M.; Moustafa, I.M.; Arnold, J.J.; Shadel, G.S.; Cameron, C.E. Transcription from the Second Heavy-Strand Promoter of Human MtDNA Is Repressed by Transcription Factor A in Vitro. Proc. Natl. Acad. Sci. USA 2012, 109, 6513–6518. [Google Scholar] [CrossRef]
- Kühl, I.; Miranda, M.; Posse, V.; Milenkovic, D.; Mourier, A.; Siira, S.J.; Bonekamp, N.A.; Neumann, U.; Filipovska, A.; Polosa, P.L.; et al. POLRMT Regulates the Switch between Replication Primer Formation and Gene Expression of Mammalian MtDNA. Sci. Adv. 2016, 2, e1600963. [Google Scholar] [CrossRef]
- Falabella, M.; Fernandez, R.J.; Johnson, F.B.; Kaufman, B.A. Potential Roles for G-Quadruplexes in Mitochondria. Curr. Med. Chem. 2019, 26, 2918–2932. [Google Scholar] [CrossRef]
- Falkenberg, M. Mitochondrial DNA Replication in Mammalian Cells: Overview of the Pathway. Essays Biochem. 2018, 62, 287–296. [Google Scholar] [CrossRef]
- Jiang, S.; Koolmeister, C.; Misic, J.; Siira, S.; Kühl, I.; Silva Ramos, E.; Miranda, M.; Jiang, M.; Posse, V.; Lytovchenko, O.; et al. TEFM Regulates Both Transcription Elongation and RNA Processing in Mitochondria. EMBO Rep. 2019, 20, e48101. [Google Scholar] [CrossRef] [PubMed]
- Nadalutti, C.A.; Ayala-Peña, S.; Santos, J.H. Mitochondrial DNA Damage as Driver of Cellular Outcomes. Am. J. Physiol. -Cell Physiol. 2022, 322, C136–C150. [Google Scholar] [CrossRef] [PubMed]
- Salem, A.F.; Whitaker-Menezes, D.; Howell, A.; Sotgia, F.; Lisanti, M.P. Mitochondrial Biogenesis in Epithelial Cancer Cells Promotes Breast Cancer Tumor Growth and Confers Autophagy Resistance. Cell Cycle 2012, 11, 4174–4180. [Google Scholar] [CrossRef] [PubMed]
- Sotgia, F.; Whitaker-Menezes, D.; Martinez-Outschoorn, U.E.; Salem, A.F.; Tsirigos, A.; Lamb, R.; Sneddon, S.; Hulit, J.; Howell, A.; Lisanti, M.P. Mitochondria “Fuel” Breast Cancer Metabolism: Fifteen Markers of Mitochondrial Biogenesis Label Epithelial Cancer Cells, but Are Excluded from Adjacent Stromal Cells. Cell Cycle 2012, 11, 4390–4401. [Google Scholar] [CrossRef]
- Han, Q.-C.; Zhang, X.-Y.; Yan, P.-H.; Chen, S.-F.; Liu, F.-F.; Zhu, Y.-R.; Tian, Q. Identification of Mitochondrial RNA Polymerase as a Potential Therapeutic Target of Osteosarcoma. Cell Death Discov. 2021, 7, 393. [Google Scholar] [CrossRef]
- Zhou, T.; Sang, Y.-H.; Cai, S.; Xu, C.; Shi, M. The Requirement of Mitochondrial RNA Polymerase for Non-Small Cell Lung Cancer Cell Growth. Cell Death Dis. 2021, 12, 751. [Google Scholar] [CrossRef]
- Wang, Y.; Ou, L.; Li, X.; Zheng, T.; Zhu, W.; Li, P.; Wu, L.; Zhao, T. The Mitochondrial RNA Polymerase POLRMT Promotes Skin Squamous Cell Carcinoma Cell Growth. Cell Death Discov. 2022, 8, 347. [Google Scholar] [CrossRef]
- Sarrazin, C.; Hézode, C.; Zeuzem, S.; Pawlotsky, J.-M. Antiviral Strategies in Hepatitis C Virus Infection. J. Hepatol. 2012, 56, S88–S100. [Google Scholar] [CrossRef]
- Arnold, J.J.; Smidansky, E.D.; Moustafa, I.M.; Cameron, C.E. Human Mitochondrial RNA Polymerase: Structure–Function, Mechanism and Inhibition. Biochim. Et Biophys. Acta (BBA)—Gene Regul. Mech. 2012, 1819, 948–960. [Google Scholar] [CrossRef]
- Wedam, R.; Greer, Y.E.; Wisniewski, D.J.; Weltz, S.; Kundu, M.; Voeller, D.; Lipkowitz, S. Targeting Mitochondria with ClpP Agonists as a Novel Therapeutic Opportunity in Breast Cancer. Cancers 2023, 15, 1936. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.S.; Houry, W.A. Recent Advances in Targeting Human Mitochondrial AAA+ Proteases to Develop Novel Cancer Therapeutics. Adv. Exp. Med. Biol. 2019, 1158, 119–142. [Google Scholar] [CrossRef] [PubMed]
- Home—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ (accessed on 27 January 2023).
- Arrillaga-Romany, I.; Odia, Y.; Prabhu, V.V.; Tarapore, R.S.; Merdinger, K.; Stogniew, M.; Oster, W.; Allen, J.E.; Mehta, M.; Batchelor, T.T.; et al. Biological Activity of Weekly ONC201 in Adult Recurrent Glioblastoma Patients. Neuro Oncol. 2020, 22, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Stein, M.N.; Bertino, J.R.; Kaufman, H.L.; Mayer, T.; Moss, R.; Silk, A.; Chan, N.; Malhotra, J.; Rodriguez, L.; Aisner, J.; et al. First-in-Human Clinical Trial of Oral ONC201 in Patients with Refractory Solid Tumors. Clin. Cancer Res. 2017, 23, 4163–4169. [Google Scholar] [CrossRef]
- Fei, X.; Bell, T.A.; Jenni, S.; Stinson, B.M.; Baker, T.A.; Harrison, S.C.; Sauer, R.T. Structures of the ATP-Fueled ClpXP Proteolytic Machine Bound to Protein Substrate. eLife 2020, 9, e52774. [Google Scholar] [CrossRef] [PubMed]
- Szczepanowska, K.; Trifunovic, A. Mitochondrial Matrix Proteases: Quality Control and Beyond. FEBS J. 2022, 289, 7128–7146. [Google Scholar] [CrossRef]
- Ishizawa, J.; Zarabi, S.F.; Davis, R.E.; Halgas, O.; Nii, T.; Jitkova, Y.; Zhao, R.; St-Germain, J.; Heese, L.E.; Egan, G.; et al. Mitochondrial ClpP-Mediated Proteolysis Induces Selective Cancer Cell Lethality. Cancer Cell 2019, 35, 721–737.e9. [Google Scholar] [CrossRef]
- Mabanglo, M.F.; Houry, W.A. Recent Structural Insights into the Mechanism of ClpP Protease Regulation by AAA+ Chaperones and Small Molecules. J. Biol. Chem. 2022, 298, 101781. [Google Scholar] [CrossRef]
- Di Cristofano, F.R.; Fong, M.W.; Huntington, K.E.; Carneiro, B.A.; Zhou, L.; El-Deiry, W.S. Synergistic Activity of ABT-263 and ONC201/TIC10 against Solid Tumor Cell Lines Is Associated with Suppression of Anti-Apoptotic Mcl-1, BAG3, PAkt, and Upregulation of pro-Apoptotic Noxa and Bax Cleavage during Apoptosis. Am. J. Cancer Res. 2023, 13, 307–325. [Google Scholar]
- Zhang, J.; Luo, B.; Sui, J.; Qiu, Z.; Huang, J.; Yang, T.; Luo, Y. IMP075 Targeting ClpP for Colon Cancer Therapy in Vivo and in Vitro. Biochem. Pharmacol. 2022, 204, 115232. [Google Scholar] [CrossRef]
- Lev, A.; Lulla, A.R.; Ross, B.C.; Ralff, M.D.; Makhov, P.B.; Dicker, D.T.; El-Deiry, W.S. ONC201 Targets AR and AR-V7 Signaling, Reduces PSA, and Synergizes with Everolimus in Prostate Cancer. Mol. Cancer Res. 2018, 16, 754–766. [Google Scholar] [CrossRef] [PubMed]
- Ralff, M.D.; Kline, C.L.B.; Küçükkase, O.C.; Wagner, J.; Lim, B.; Dicker, D.T.; Prabhu, V.V.; Oster, W.; El-Deiry, W.S. ONC201 Demonstrates Antitumor Effects in Both Triple-Negative and Non–Triple-Negative Breast Cancers through TRAIL-Dependent and TRAIL-Independent Mechanisms. Mol. Cancer Ther. 2017, 16, 1290–1298. [Google Scholar] [CrossRef] [PubMed]
- Mishukov, A.; Odinokova, I.; Mndlyan, E.; Kobyakova, M.; Abdullaev, S.; Zhalimov, V.; Glukhova, X.; Galat, V.; Galat, Y.; Senotov, A.; et al. ONC201-Induced Mitochondrial Dysfunction, Senescence-like Phenotype, and Sensitization of Cultured BT474 Human Breast Cancer Cells to TRAIL. Int. J. Mol. Sci. 2022, 23, 15551. [Google Scholar] [CrossRef] [PubMed]
- Greer, Y.E.; Porat-Shliom, N.; Nagashima, K.; Stuelten, C.; Crooks, D.; Koparde, V.N.; Gilbert, S.F.; Islam, C.; Ubaldini, A.; Ji, Y.; et al. ONC201 Kills Breast Cancer Cells in Vitro by Targeting Mitochondria. Oncotarget 2018, 9, 18454–18479. [Google Scholar] [CrossRef] [PubMed]
- Mabanglo, M.F.; Bhandari, V.; Houry, W.A. Substrates and Interactors of the ClpP Protease in the Mitochondria. Curr. Opin. Chem. Biol. 2022, 66, 102078. [Google Scholar] [CrossRef]
- Fan, Y.; Wang, J.; Fang, Z.; Pierce, S.R.; West, L.; Staley, A.; Tucker, K.; Yin, Y.; Sun, W.; Kong, W.; et al. Anti-Tumor and Anti-Invasive Effects of ONC201 on Ovarian Cancer Cells and a Transgenic Mouse Model of Serous Ovarian Cancer. Front. Oncol. 2022, 12, 789450. [Google Scholar] [CrossRef]
- Greer, Y.E.; Hernandez, L.; Fennell, E.M.J.; Kundu, M.; Voeller, D.; Chari, R.; Gilbert, S.F.; Gilbert, T.S.K.; Ratnayake, S.; Tang, B.; et al. Mitochondrial Matrix Protease ClpP Agonists Inhibit Cancer Stem Cell Function in Breast Cancer Cells by Disrupting Mitochondrial Homeostasis. Cancer Res. Commun. 2022, 2, 1144–1161. [Google Scholar] [CrossRef]
- Ishizawa, J.; Kojima, K.; Chachad, D.; Ruvolo, P.; Ruvolo, V.; Jacamo, R.O.; Borthakur, G.; Mu, H.; Zeng, Z.; Tabe, Y.; et al. ATF4 Induction through an Atypical Integrated Stress Response to ONC201 Triggers P53-Independent Apoptosis in Hematological Malignancies. Sci. Signal. 2016, 9, ra17. [Google Scholar] [CrossRef]
- Jhaveri, A.V.; Zhou, L.; Ralff, M.D.; Lee, Y.S.; Navaraj, A.; Carneiro, B.A.; Safran, H.; Prabhu, V.V.; Ross, E.A.; Lee, S.; et al. Combination of ONC201 and TLY012 Induces Selective, Synergistic Apoptosis in Vitro and Significantly Delays PDAC Xenograft Growth in Vivo. Cancer Biol. 2021, 22, 607–618. [Google Scholar] [CrossRef]
- Rumman, M.; Buck, S.; Polin, L.; Dzinic, S.; Boerner, J.; Winer, I.S. ONC201 Induces the Unfolded Protein Response (UPR) in High- and Low-Grade Ovarian Carcinoma Cell Lines and Leads to Cell Death Regardless of Platinum Sensitivity. Cancer Med. 2021, 10, 3373–3387. [Google Scholar] [CrossRef]
- Tu, Y.; He, J.; Liu, H.; Lee, H.C.; Wang, H.; Ishizawa, J.; Allen, J.E.; Andreeff, M.; Orlowski, R.Z.; Davis, R.E.; et al. The Imipridone ONC201 Induces Apoptosis and Overcomes Chemotherapy Resistance by Up-Regulation of Bim in Multiple Myeloma. Neoplasia 2017, 19, 772–780. [Google Scholar] [CrossRef]
- Prabhu, V.V.; Morrow, S.; Rahman Kawakibi, A.; Zhou, L.; Ralff, M.; Ray, J.; Jhaveri, A.; Ferrarini, I.; Lee, Y.; Parker, C.; et al. ONC201 and Imipridones: Anti-Cancer Compounds with Clinical Efficacy. Neoplasia 2020, 22, 725–744. [Google Scholar] [CrossRef] [PubMed]
- Cole, A.; Wang, Z.; Coyaud, E.; Voisin, V.; Gronda, M.; Jitkova, Y.; Mattson, R.; Hurren, R.; Babovic, S.; Maclean, N.; et al. Inhibition of the Mitochondrial Protease ClpP as a Therapeutic Strategy for Human Acute Myeloid Leukemia. Cancer Cell 2015, 27, 864–876. [Google Scholar] [CrossRef] [PubMed]
- Lycan, T.; Pardee, T.; Petty, W.; Bonomi, M.; Alistar, A.; Lamar, Z.; Isom, S.; Chan, M.; Miller, A.; Ruiz, J. A Phase II Clinical Trial of CPI-613 in Patients with Relapsed or Refractory Small Cell Lung Carcinoma. PLoS ONE 2016, 11, e0164244. [Google Scholar] [CrossRef]
- Brown, J.R.; Chan, D.K.; Shank, J.J.; Griffith, K.A.; Fan, H.; Szulawski, R.; Yang, K.; Reynolds, R.K.; Johnston, C.; McLean, K.; et al. Phase II Clinical Trial of Metformin as a Cancer Stem Cell–Targeting Agent in Ovarian Cancer. JCI Insight 2020, 5, e133247. [Google Scholar] [CrossRef] [PubMed]
- Mihaylova, M.M.; Shaw, R.J. The AMP-Activated Protein Kinase (AMPK) Signaling Pathway Coordinates Cell Growth, Autophagy, & Metabolism. Nat. Cell Biol. 2011, 13, 1016–1023. [Google Scholar] [CrossRef]
- TeSlaa, T.; Teitell, M.A. Techniques to Monitor Glycolysis. Methods Enzym. 2014, 542, 91–114. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does It Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Chandel, N.S. We Need to Talk about the Warburg Effect. Nat. Metab. 2020, 2, 127–129. [Google Scholar] [CrossRef]
- Gatenby, R.A.; Gillies, R.J. Why Do Cancers Have High Aerobic Glycolysis? Nat. Rev. Cancer 2004, 4, 891–899. [Google Scholar] [CrossRef]
- Elstrom, R.L.; Bauer, D.E.; Buzzai, M.; Karnauskas, R.; Harris, M.H.; Plas, D.R.; Zhuang, H.; Cinalli, R.M.; Alavi, A.; Rudin, C.M.; et al. Akt Stimulates Aerobic Glycolysis in Cancer Cells. Cancer Res. 2004, 64, 3892–3899. [Google Scholar] [CrossRef] [PubMed]
- Evans, K.W.; Yuca, E.; Scott, S.S.; Zhao, M.; Paez Arango, N.; Cruz Pico, C.X.; Saridogan, T.; Shariati, M.; Class, C.A.; Bristow, C.A.; et al. Oxidative Phosphorylation Is a Metabolic Vulnerability in Chemotherapy-Resistant Triple-Negative Breast Cancer. Cancer Res. 2021, 81, 5572–5581. [Google Scholar] [CrossRef] [PubMed]
- Illangeswaran, R.S.S.; Jebanesan, D.Z.P.; Sivakumar, K.K.; Vidhyadharan, R.T.; Rajamani, B.M.; Janet, N.B.; David, E.; Velayudhan, S.R.; Mathews, V.; Balasubramanian, P. Chemotherapeutic Drugs Elicit Stemness and Metabolic Alteration to Mediate Acquired Drug-Resistant Phenotype in Acute Myeloid Leukemia Cell Lines. Leuk. Res. 2023, 128, 107054. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.L.; Inguva, A.; Jordan, C.T. Targeting Energy Metabolism in Cancer Stem Cells: Progress and Challenges in Leukemia and Solid Tumors. Cell Stem Cell 2021, 28, 378–393. [Google Scholar] [CrossRef]
- Dhani, N.; Fyles, A.; Hedley, D.; Milosevic, M. The Clinical Significance of Hypoxia in Human Cancers. Semin. Nucl. Med. 2015, 45, 110–121. [Google Scholar] [CrossRef]
- Ashton, T.M.; McKenna, W.G.; Kunz-Schughart, L.A.; Higgins, G.S. Oxidative Phosphorylation as an Emerging Target in Cancer Therapy. Clin. Cancer Res. 2018, 24, 2482–2490. [Google Scholar] [CrossRef]
- Ekstrand, M.I.; Falkenberg, M.; Rantanen, A.; Park, C.B.; Gaspari, M.; Hultenby, K.; Rustin, P.; Gustafsson, C.M.; Larsson, N.-G. Mitochondrial Transcription Factor A Regulates MtDNA Copy Number in Mammals. Hum. Mol. Genet. 2004, 13, 935–944. [Google Scholar] [CrossRef]
- Rong, Z.; Tu, P.; Xu, P.; Sun, Y.; Yu, F.; Tu, N.; Guo, L.; Yang, Y. The Mitochondrial Response to DNA Damage. Front. Cell Dev. Biol. 2021, 9, 669379. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S.; Schumacker, P.T. Cells Depleted of Mitochondrial DNA (Ρ0) Yield Insight into Physiological Mechanisms. FEBS Lett. 1999, 454, 173–176. [Google Scholar] [CrossRef]
- Stepanenko, A.A.; Dmitrenko, V.V. HEK293 in Cell Biology and Cancer Research: Phenotype, Karyotype, Tumorigenicity, and Stress-Induced Genome-Phenotype Evolution. Gene 2015, 569, 182–190. [Google Scholar] [CrossRef]
- Shen, C.; Gu, M.; Song, C.; Miao, L.; Hu, L.; Liang, D.; Zheng, C. The Tumorigenicity Diversification in Human Embryonic Kidney 293 Cell Line Cultured in Vitro. Biologicals 2008, 36, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.; Kline, C.L.; Ralff, M.D.; Avital, L.; Amriti, L.; Lanlan, Z.; Olson, G.L.; Nallaganchu, B.R.; Benes, C.H.; Allen, J.E.; et al. Preclinical Evaluation of the Imipridone Family, Analogs of Clinical Stage Anti-Cancer Small Molecule ONC201, Reveals Potent Anti-Cancer Effects of ONC212. Cell Cycle 2017, 16, 1790–1799. [Google Scholar] [CrossRef]
- Mennuni, M.; Filograna, R.; Felser, A.; Bonekamp, N.A.; Giavalisco, P.; Lytovchenko, O.; Larsson, N.-G. Metabolic Resistance to the Inhibition of Mitochondrial Transcription Revealed by CRISPR-Cas9 Screen. EMBO Rep. 2022, 23, e53054. [Google Scholar] [CrossRef] [PubMed]
- Jacques, S.; van der Sloot, A.M.; Huard, C.C.; Coulombe-Huntington, J.; Tsao, S.; Tollis, S.; Bertomeu, T.; Culp, E.J.; Pallant, D.; Cook, M.A.; et al. Imipridone Anticancer Compounds Ectopically Activate the ClpP Protease and Represent a New Scaffold for Antibiotic Development. Genetics 2020, 214, 1103–1120. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Dang, C.V. Time to Hit Pause on Mitochondria-Targeting Cancer Therapies. Nat. Med. 2023, 29, 29–30. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tissue | Cell Line | IMT1 IC50 (µM) * | ClpP Agonist | ClpP Agonist IC50 (µM) | Source |
|---|---|---|---|---|---|
| Bladder | J82 | >30 | ONC201 | 2.45 | [70] |
| Breast | MDAMB436 | 0.56 | ONC201 | 3.94 | [70] |
| MDAMB468 | 0.13 | ONC201 | 1–6.3 | [39,41] | |
| MDAMB231 | >30 | ONC201 | 3.0–7.0 | [41,70,71] | |
| IMP075 | 10.1 | [71] | |||
| ONC212 | <0.5 | [103] | |||
| ONC206 | <1 | [103] | |||
| TR-57 | 0.017–0.0193 | [35,78] | |||
| TR-107 | 0.023–0.0294 | [34,35] | |||
| Colon | CACO2 | 0.15 | ONC201 | 15.5 | [71] |
| IMP075 | 2.5 | [71] | |||
| HT29 | 1.22 | ONC201 | 5.07 | [70] | |
| SW620 | 0.22 | ONC201 | 9.8 | [71] | |
| IMP075 | 0.6 | [71] | |||
| Kidney | HEK293T | 0.185 | TR-57 | 0.0147 | Daglish, et al. |
| Liver | HEPG2 | 0.12 | ONC201 | 6.2–12.4 | [39,71] |
| ONC212 | <1 | [103] | |||
| ONC206 | <1 | [103] | |||
| IMP075 | 3.1 | [71] | |||
| Lung | A549 | 0.64 | ONC201 | 9 | [71] |
| IMP075 | 1.6 | [71] | |||
| Ovary | IGROV1 | >30 | ONC201 | 5.55 | [70] |
| OVCAR3 | 2.23 | ONC201 | 1.98 | [70] | |
| SKOV3 | >30 | ONC201 | 2.3–20.5 | [70,71] | |
| IMP075 | 4.1 | [71] | |||
| Pancreas | BXPC3 | 0.27 | ONC201 | 12.5 | [72] |
| ONC212 | 0.25 | [72] | |||
| PANC1 | 0.79 | ONC201 | 3.13 | [72] | |
| ONC212 | 0.17 | [72] | |||
| Prostate | 22RV1 | 0.82 | ONC201 | 1.0–2.5 | [70,72] |
| DU145 | >30 | ONC201 | >10 | [72] | |
| PC3 | >30 | ONC201 | 2.2–20.1 | [39,70,72] | |
| Skin | A375 | >30 | ONC201 | 12.2 | [71] |
| IMP075 | 5 | [71] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Daglish, S.C.D.; Fennell, E.M.J.; Graves, L.M. Targeting Mitochondrial DNA Transcription by POLRMT Inhibition or Depletion as a Potential Strategy for Cancer Treatment. Biomedicines 2023, 11, 1598. https://doi.org/10.3390/biomedicines11061598
Daglish SCD, Fennell EMJ, Graves LM. Targeting Mitochondrial DNA Transcription by POLRMT Inhibition or Depletion as a Potential Strategy for Cancer Treatment. Biomedicines. 2023; 11(6):1598. https://doi.org/10.3390/biomedicines11061598
Chicago/Turabian StyleDaglish, Sabrina C. D., Emily M. J. Fennell, and Lee M. Graves. 2023. "Targeting Mitochondrial DNA Transcription by POLRMT Inhibition or Depletion as a Potential Strategy for Cancer Treatment" Biomedicines 11, no. 6: 1598. https://doi.org/10.3390/biomedicines11061598