
Targeting Store-Operated Calcium Entry Regulates the Inflammation-Induced Proliferation and Migration of Breast Cancer Cells
 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Reagents
2.2. Real-Time Quantitative Reverse Transcription Polymerase Chain Reaction
2.3. Wound Healing Assay
2.4. Cell Proliferation Assay
2.5. Western Blotting
2.6. Enzyme-Linked Immunosorbent Assay
2.7. Statistical Analysis
3. Results
3.1. LPS Treatment Upregulated the Gene Expression of COX-2 and IL-6 in MDA-MB-231 but Not in MCF-7 Cells
3.2. SOCE Inhibition Blocked LPS-Induced Activation of the COX-2/PGE2 Pathway
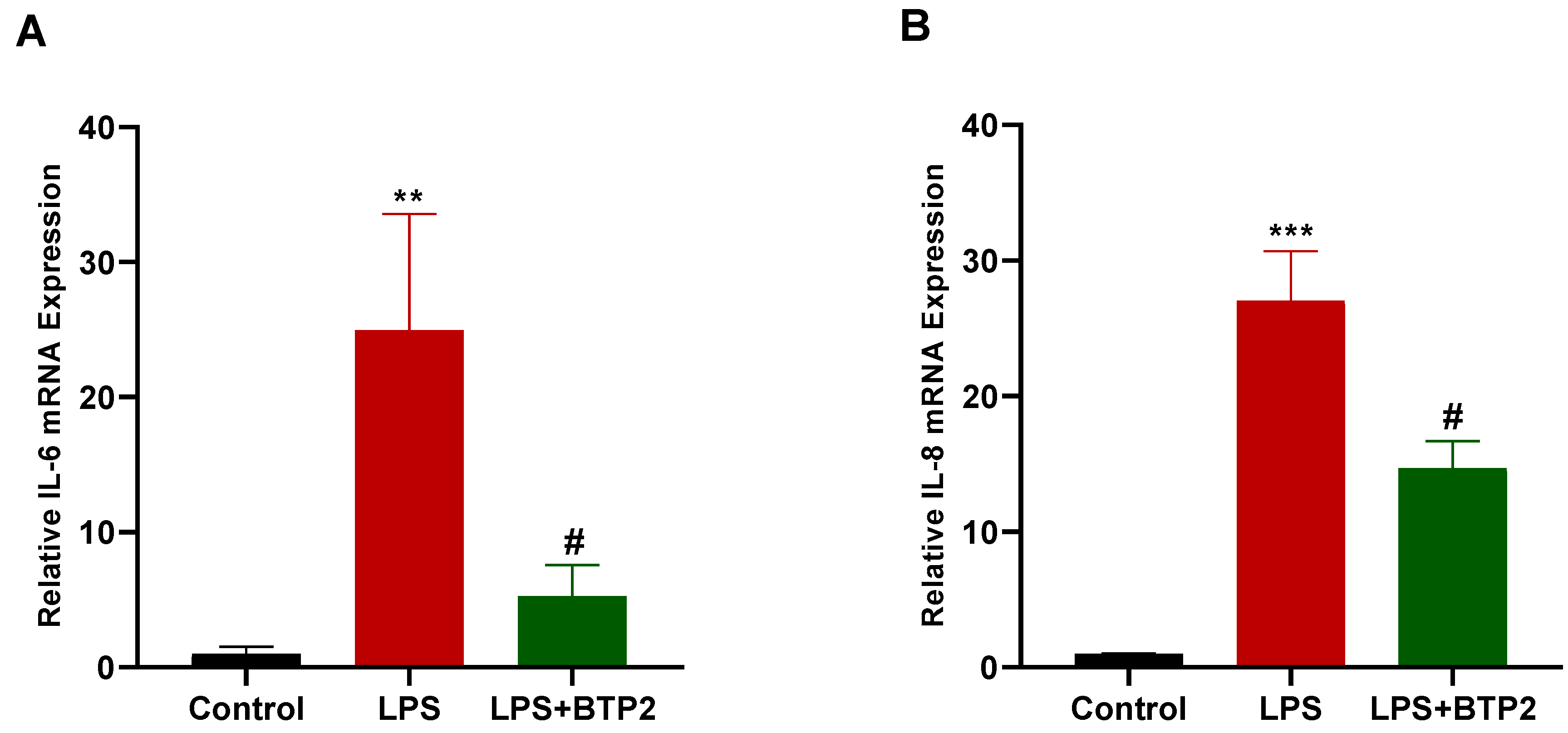
3.3. SOCE Inhibition Suppressed LPS-Induced Inflammatory Gene Production in MDA-MB-231 Cells
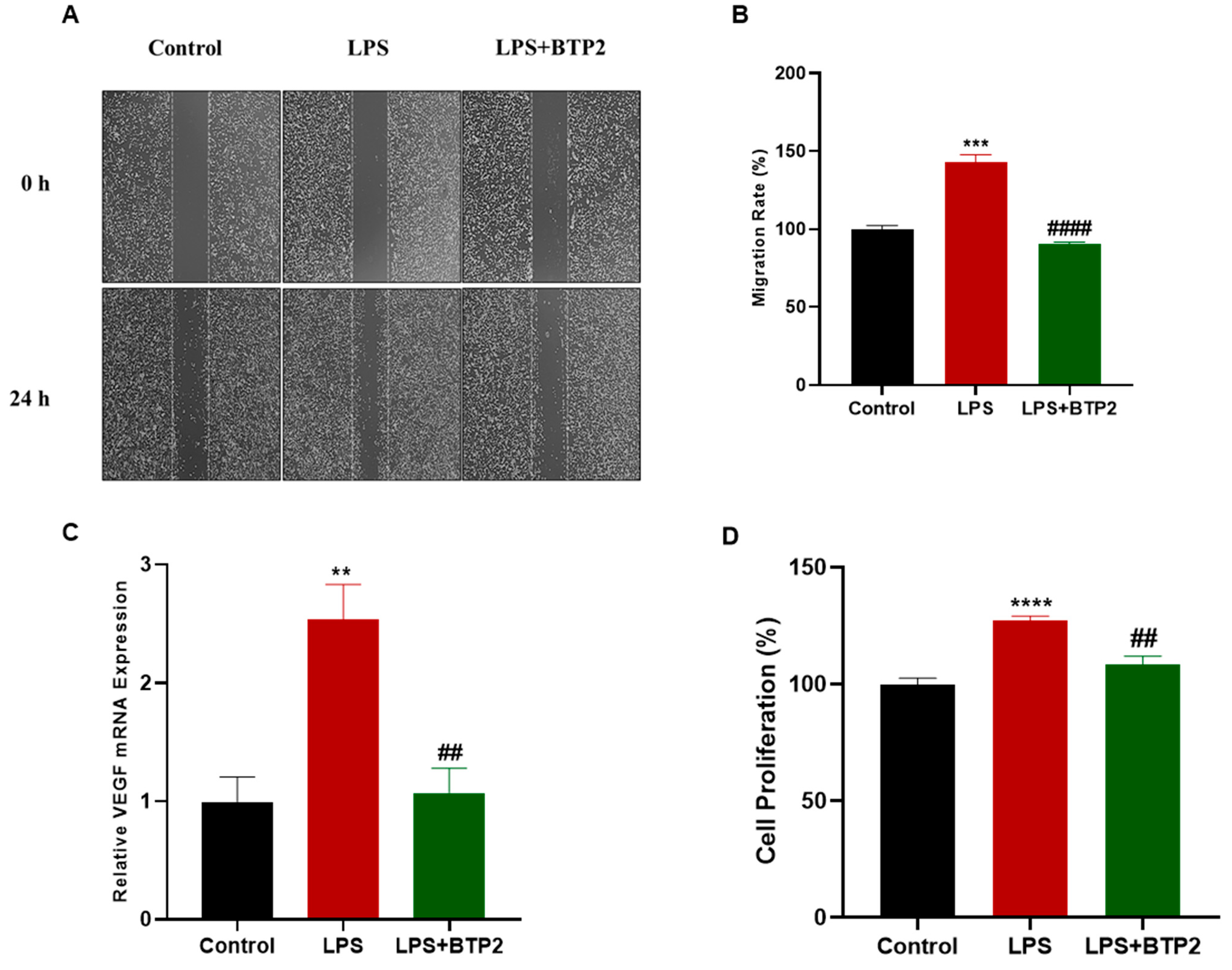
3.4. LPS-Induced Migration and Proliferation of MDA-MB-231 Cells Were Suppressed by SOCE Inhibition
3.5. Thapsigargin Treatment Potentiated LPS-Induced Production of Inflammatory Genes
3.6. Thapsigargin Enhanced the LPS-Induced Migration and Proliferation of MDA-MB-231 Cells
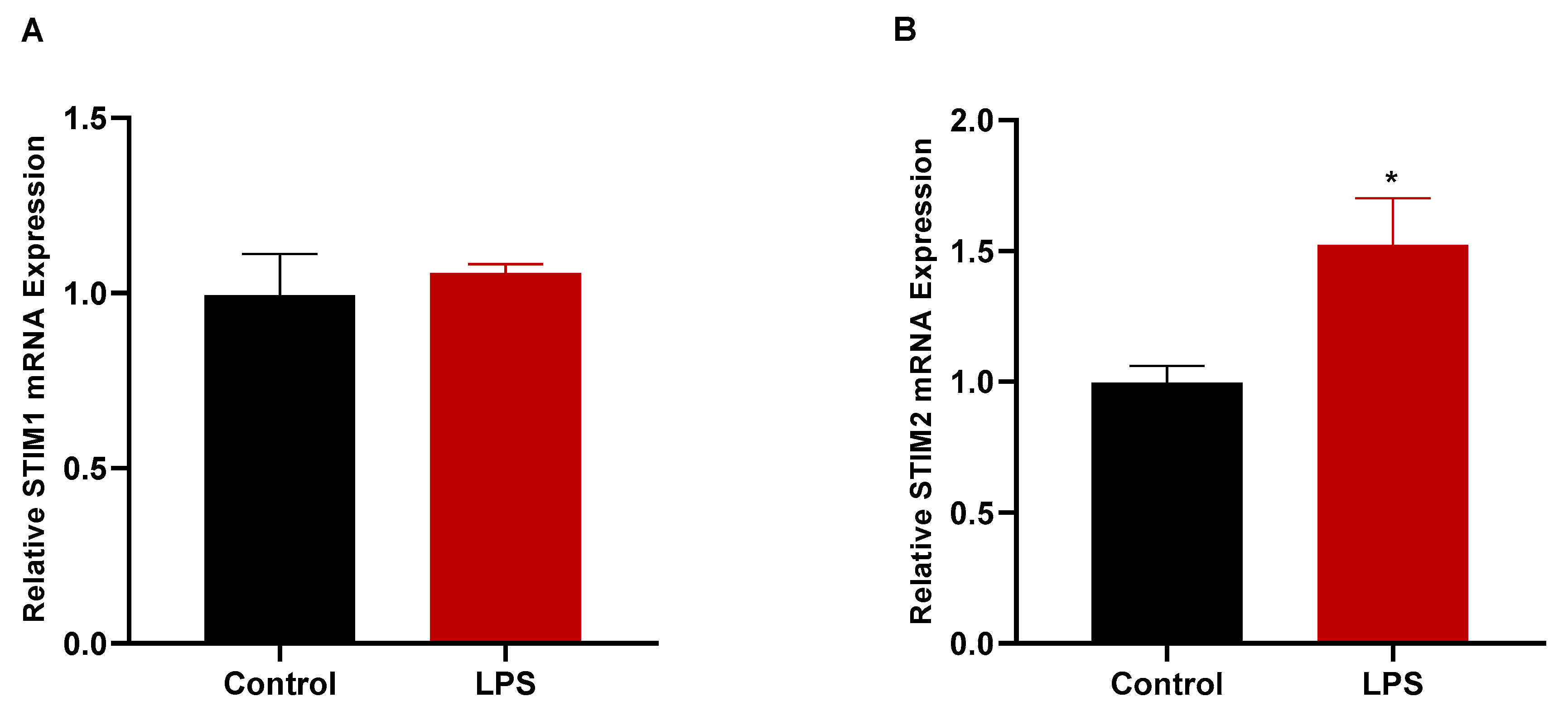
3.7. LPS Treatment Increased the Gene Expression of STIM2
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2022. CA. Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Perrot-Applanat, M.; Di Benedetto, M. Autocrine Functions of VEGF in Breast Tumor Cells. Cell Adh. Migr. 2012, 6, 547–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeuchi, Y.; Gotoh, N. Inflammatory cytokine-enriched microenvironment plays key roles in the development of breast cancers. Cancer Sci. 2023, 114, 1792–1799. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.; Cao, S.; Tang, L. The Tumor Microenvironment and Inflammatory Breast Cancer. J. Cancer 2017, 8, 1884–1891. [Google Scholar] [CrossRef]
- Zhao, H.; Wu, L.; Yan, G.; Chen, Y.; Zhou, M.; Wu, Y.; Li, Y. Inflammation and Tumor Progression: Signaling Pathways and Targeted Intervention. Signal Transduct. Target. Ther. 2021, 6, 263. [Google Scholar] [CrossRef]
- Bell, E. TLR4 Signalling. Nat. Rev. Immunol. 2008, 8, 241. [Google Scholar] [CrossRef]
- Lu, Y.-C.; Yeh, W.-C.; Ohashi, P.S. LPS/TLR4 Signal Transduction Pathway. Cytokine 2008, 42, 145–151. [Google Scholar] [CrossRef]
- Yang, H.; Wang, B.; Wang, T.; Xu, L.; He, C.; Wen, H.; Yan, J.; Su, H.; Zhu, X. Toll-Like Receptor 4 Prompts Human Breast Cancer Cells Invasiveness via Lipopolysaccharide Stimulation and Is Overexpressed in Patients with Lymph Node Metastasis. PLoS ONE 2014, 9, e109980. [Google Scholar] [CrossRef]
- Yang, H.; Zhou, H.; Feng, P.; Zhou, X.; Wen, H.; Xie, X.; Shen, H.; Zhu, X. Reduced Expression of Toll-like Receptor 4 Inhibits Human Breast Cancer Cells Proliferation and Inflammatory Cytokines Secretion. J. Exp. Clin. Cancer Res. 2010, 29, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, A.; Wang, G.; Zhao, H.; Zhang, Y.; Han, Q.; Zhang, C.; Tian, Z.; Zhang, J. TLR4 Signaling Promotes a COX-2/PGE 2 /STAT3 Positive Feedback Loop in Hepatocellular Carcinoma (HCC) Cells. Oncoimmunology 2016, 5, e1074376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; DuBois, R.N. The Role of COX-2 in Intestinal Inflammation and Colorectal Cancer. Oncogene 2010, 29, 781–788. [Google Scholar] [CrossRef] [Green Version]
- Petkova, D.K.; Clelland, C.; Ronan, J.; Pang, L.; Coulson, J.M.; Lewis, S.; Knox, A.J. Overexpression of Cyclooxygenase-2 in Non-Small Cell Lung Cancer. Respir. Med. 2004, 98, 164–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shim, J.Y.; An, H.J.; Lee, Y.H.; Kim, S.K.; Lee, K.P.; Lee, K.S. Overexpression of Cyclooxygenase-2 Is Associated with Breast Carcinoma and Its Poor Prognostic Factors. Mod. Pathol. 2003, 16, 1199–1204. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.-J.; Kim, H.-S.; Lee, S.-H.; Yang, Y.; Lee, M.-S.; Lim, J.-S. NDRG2 Controls COX-2/PGE2-Mediated Breast Cancer Cell Migration and Invasion. Mol. Cells 2014, 37, 759–765. [Google Scholar] [CrossRef] [Green Version]
- Zheng, S.; Wang, X.; Zhao, D.; Liu, H.; Hu, Y. Calcium Homeostasis and Cancer: Insights from Endoplasmic Reticulum-Centered Organelle Communications. Trends Cell Biol. 2023, 33, 312–323. [Google Scholar] [CrossRef]
- Putney, J.W.; Tomita, T. Phospholipase C Signaling and Calcium Influx. Adv. Biol. Regul. 2012, 52, 152–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, N.T.; Han, W.; Cao, W.; Wang, Y.; Wen, S.; Huang, Y.; Li, M.; Du, L.; Zhou, Y. Store-Operated Calcium Entry Mediated by ORAI and STIM. In Comprehensive Physiology; Wiley: Hoboken, NJ, USA, 2018; pp. 981–1002. [Google Scholar]
- Shaw, P.J.; Feske, S. Regulation of Lymphocyte Function by ORAI and STIM Proteins in Infection and Autoimmunity. J. Physiol. 2012, 590, 4157–4167. [Google Scholar] [CrossRef]
- Wendimu, M.Y.; Alqinyah, M.; Vella, S.; Dean, P.; Almutairi, F.; Davila-Rivera, R.; Rayatpisheh, S.; Wohlschlegel, J.; Moreno, S.; Hooks, S.B. RGS10 Physically and Functionally Interacts with STIM2 and Requires Store-Operated Calcium Entry to Regulate pro-Inflammatory Gene Expression in Microglia. Cell. Signal. 2021, 83, 109974. [Google Scholar] [CrossRef]
- Birla, H.; Xia, J.; Gao, X.; Zhao, H.; Wang, F.; Patel, S.; Amponsah, A.; Bekker, A.; Tao, Y.-X.; Hu, H. Toll-like Receptor 4 Activation Enhances Orai1-Mediated Calcium Signal Promoting Cytokine Production in Spinal Astrocytes. Cell Calcium 2022, 105, 102619. [Google Scholar] [CrossRef]
- Bird, G.S.; DeHaven, W.I.; Smyth, J.T.; Putney, J.W. Methods for Studying Store-Operated Calcium Entry. Methods 2008, 46, 204–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Rashed, F.; Thomas, R.; Al-Roub, A.; Al-Mulla, F.; Ahmad, R. LPS Induces GM-CSF Production by Breast Cancer MDA-MB-231 Cells via Long-Chain Acyl-CoA Synthetase 1. Molecules 2020, 25, 4709. [Google Scholar] [CrossRef]
- Go, J.; Wei, J.; Park, J.; Ahn, K.; Kim, J. Wogonin suppresses the LPS-enhanced invasiveness of MDA-MB-231 breast cancer cells by inhibiting the 5-LO/BLT2 cascade. Int. J. Mol. Med. 2018, 42, 1899–1908. [Google Scholar] [CrossRef] [Green Version]
- Seol, M.-A.; Park, J.-H.; Jeong, J.H.; Lyu, J.; Han, S.Y.; Oh, S.-M. Role of TOPK in Lipopolysaccharide-Induced Breast Cancer Cell Migration and Invasion. Oncotarget 2017, 8, 40190–40203. [Google Scholar] [CrossRef] [Green Version]
- Mehmeti, M.; Allaoui, R.; Bergenfelz, C.; Saal, L.H.; Ethier, S.P.; Johansson, M.E.; Jirström, K.; Leandersson, K. Expression of Functional Toll like Receptor 4 in Estrogen Receptor/Progesterone Receptor-Negative Breast Cancer. Breast Cancer Res. 2015, 17, 130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faggioli, L.; Costanzo, C.; Merola, M.; Bianchini, E.; Furia, A.; Carsana, A.; Palmieri, M. Nuclear Factor k B (NF-k B), Nuclear Factor Interleukin-6 (NFIL-6 or C/EBPbeta) and Nuclear Factor Interleukin-6beta (NFIL6-Beta or C/EBPdelta) Are Not Sufficient to Activate the Endogenous Interleukin-6 Gene in the Human Breast Carcinoma Cell Line MCF-7. Eur. J. Biochem. 1996, 239, 624–631. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yao, J.J.; Chen, Z.; Lei, W.; Duan, R.; Yao, Z. Lipopolysaccharide Sensitizes the Therapeutic Response of Breast Cancer to IAP Antagonist. Front. Immunol. 2022, 13, 906357. [Google Scholar] [CrossRef]
- Wu, K.; Zhang, H.; Fu, Y.; Zhu, Y.; Kong, L.; Chen, L.; Zhao, F.; Yu, L.; Chen, X. TLR4/MyD88 Signaling Determines the Metastatic Potential of Breast Cancer Cells. Mol. Med. Rep. 2018, 18, 3411–3420. [Google Scholar] [CrossRef] [Green Version]
- Sogkas, G.; Stegner, D.; Syed, S.N.; Vögtle, T.; Rau, E.; Gewecke, B.; Schmidt, R.E.; Nieswandt, B.; Gessner, J.E. Cooperative and Alternate Functions for STIM1 and STIM2 in Macrophage Activation and in the Context of Inflammation. Immun. Inflamm. Dis. 2015, 3, 154–170. [Google Scholar] [CrossRef] [Green Version]
- Wong, J.-H.; Ho, K.-H.; Nam, S.; Hsu, W.-L.; Lin, C.-H.; Chang, C.-M.; Wang, J.-Y.; Chang, W.-C. Store-Operated Ca2+ Entry Facilitates the Lipopolysaccharide-Induced Cyclooxygenase-2 Expression in Gastric Cancer Cells. Sci. Rep. 2017, 7, 12813. [Google Scholar] [CrossRef] [Green Version]
- Gu, C.; Zhang, W.; Yang, E.; Gu, C.; Zhang, Z.; Ke, J.; Wang, X.; Wu, S.; Li, S.; Wu, F. Blockage of Orai1-Nucleolin Interaction Meditated Calcium Influx Attenuates Breast Cancer Cells Growth. Oncogenesis 2022, 11, 55. [Google Scholar] [CrossRef]
- Asghar, M.Y.; Lassila, T.; Paatero, I.; Nguyen, V.D.; Kronqvist, P.; Zhang, J.; Slita, A.; Löf, C.; Zhou, Y.; Rosenholm, J.; et al. Stromal Interaction Molecule 1 (STIM1) Knock down Attenuates Invasion and Proliferation and Enhances the Expression of Thyroid-Specific Proteins in Human Follicular Thyroid Cancer Cells. Cell. Mol. Life Sci. 2021, 78, 5827–5846. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-Y.; Sun, J.; Huang, M.-Y.; Wang, Y.-S.; Hou, M.-F.; Sun, Y.; He, H.; Krishna, N.; Chiu, S.-J.; Lin, S.; et al. STIM1 Overexpression Promotes Colorectal Cancer Progression, Cell Motility and COX-2 Expression. Oncogene 2015, 34, 4358–4367. [Google Scholar] [CrossRef] [Green Version]
- Jaskulska, A.; Janecka, A.E.; Gach-Janczak, K. Thapsigargin—From Traditional Medicine to Anticancer Drug. Int. J. Mol. Sci. 2020, 22, 4. [Google Scholar] [CrossRef] [PubMed]
- DebRoy, A.; Vogel, S.M.; Soni, D.; Sundivakkam, P.C.; Malik, A.B.; Tiruppathi, C. Cooperative Signaling via Transcription Factors NF-ΚB and AP1/c-Fos Mediates Endothelial Cell STIM1 Expression and Hyperpermeability in Response to Endotoxin. J. Biol. Chem. 2014, 289, 24188–24201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vyas, D.; Laput, G.; Vyas, A. Chemotherapy-Enhanced Inflammation May Lead to the Failure of Therapy and Metastasis. Onco. Targets. Ther. 2014, 7, 1015–1023. [Google Scholar] [CrossRef] [Green Version]
- Alhamed, A.S.; Alqinyah, M.; Alsufayan, M.A.; Alhaydan, I.A.; Alassmrry, Y.A.; Alnefaie, H.O.; Algahtani, M.M.; Alghaith, A.F.; Alhamami, H.N.; Albogami, A.M.; et al. Blockade of Store-Operated Calcium Entry Sensitizes Breast Cancer Cells to Cisplatin Therapy via Modulating Inflammatory Response. Saudi Pharm. J. 2023, 31, 245–254. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Forward Sequence | Reverse Sequence |
|---|---|---|
| COX-2 | 5′-CCCTTGGGTGTCAAAGGTAA-3′ | 5′-GCCCTCGCTTATGATCTGTC-3′ |
| IL-6 | 5′-CCAGCTATGAACTCCTTCTC-3′ | 5′-GCTTGTTCCTCACATCTCTC-3′ |
| IL-8 | 5′-AGCCTTCCTGATTTCTGCAG-3′ | 5′-GTCCACTCTCAATCACTCTCAG-3′ |
| VEGF | 5′-GAGGAGCAGTTACGGTCTGT-3′ | 5′-GTAGCTCGTGCTGGTGTT CA-3′ |
| STIM1 | 5′-GCGGGAGGGTACTGAG-3′ | 5′-TCCATGTCATCCACGTCGTCA-3′ |
| STIM2 | 5′-CCCTCACCACCCGCAACA-3′ | 5′-GATGTGTGGCGAGGTTAAGGC-3′ |
| GAPDH | 5′-GCCAAGGTCATCCATGACAACT-3′ | 5′-GAGGGGCCATCCACAGTCTT-3′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alqinyah, M.; Alhamed, A.S.; Alnefaie, H.O.; Algahtani, M.M.; Badr, A.M.; Albogami, A.M.; Mohany, M.; Alassmrry, Y.A.; Alghaith, A.F.; Alhamami, H.N.; et al. Targeting Store-Operated Calcium Entry Regulates the Inflammation-Induced Proliferation and Migration of Breast Cancer Cells. Biomedicines 2023, 11, 1637. https://doi.org/10.3390/biomedicines11061637
Alqinyah M, Alhamed AS, Alnefaie HO, Algahtani MM, Badr AM, Albogami AM, Mohany M, Alassmrry YA, Alghaith AF, Alhamami HN, et al. Targeting Store-Operated Calcium Entry Regulates the Inflammation-Induced Proliferation and Migration of Breast Cancer Cells. Biomedicines. 2023; 11(6):1637. https://doi.org/10.3390/biomedicines11061637
Chicago/Turabian StyleAlqinyah, Mohammed, Abdullah S. Alhamed, Hajar O. Alnefaie, Mohammad M. Algahtani, Amira M. Badr, Abdullah M. Albogami, Mohamed Mohany, Yasseen A. Alassmrry, Adel F. Alghaith, Hussain N. Alhamami, and et al. 2023. "Targeting Store-Operated Calcium Entry Regulates the Inflammation-Induced Proliferation and Migration of Breast Cancer Cells" Biomedicines 11, no. 6: 1637. https://doi.org/10.3390/biomedicines11061637