
Oxalate Homeostasis in Non-Stone-Forming Chronic Kidney Disease: A Review of Key Findings and Perspectives


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction to Health Oxalate Homeostasis
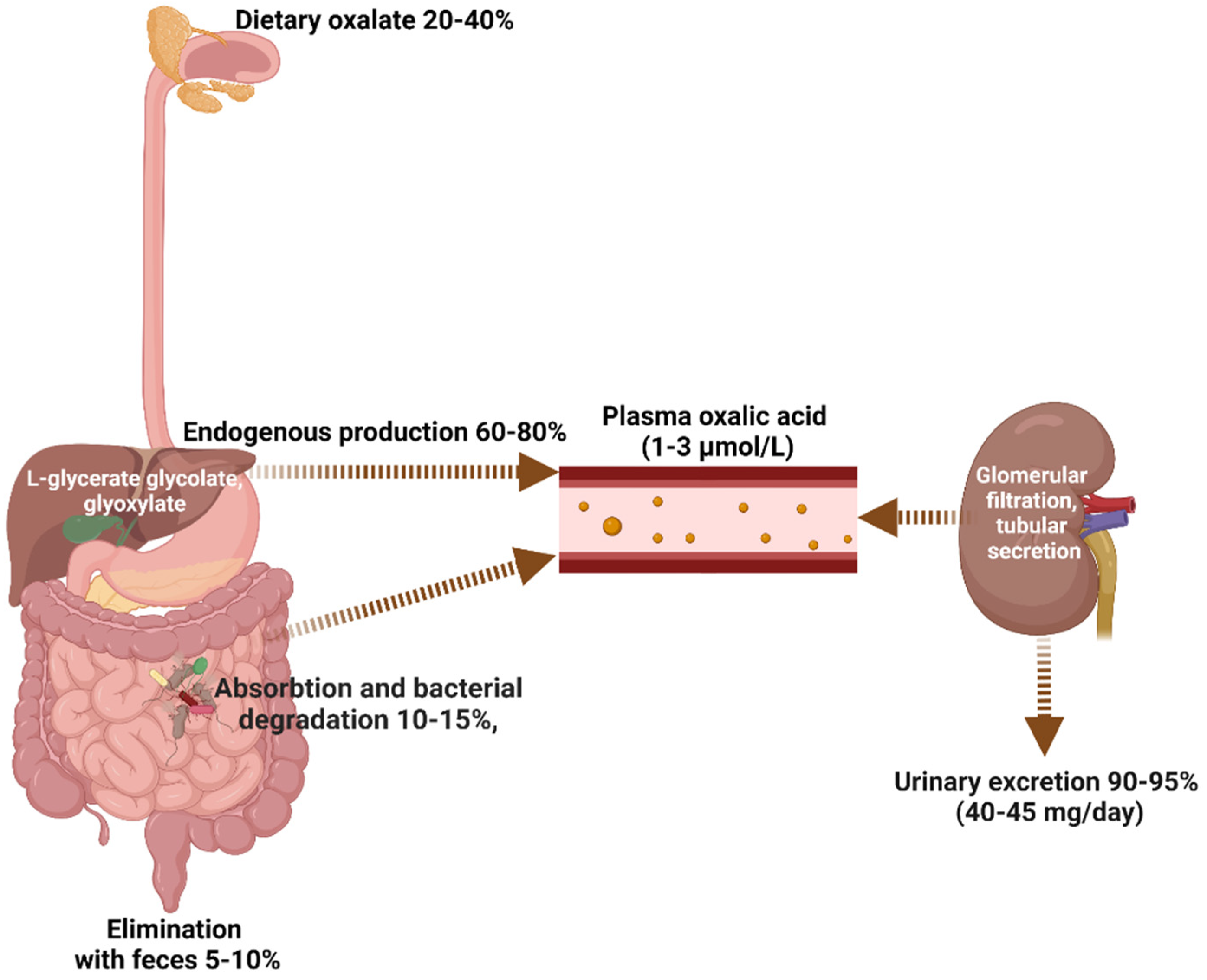
1.1. Dietary Oxalate Intake
1.2. Endogenous Oxalate Metabolism
1.3. Intestinal Oxalate Absorption and Bacterial Degradation
1.4. Oxalate Excretion
2. Oxalate Implications in Health and Disease
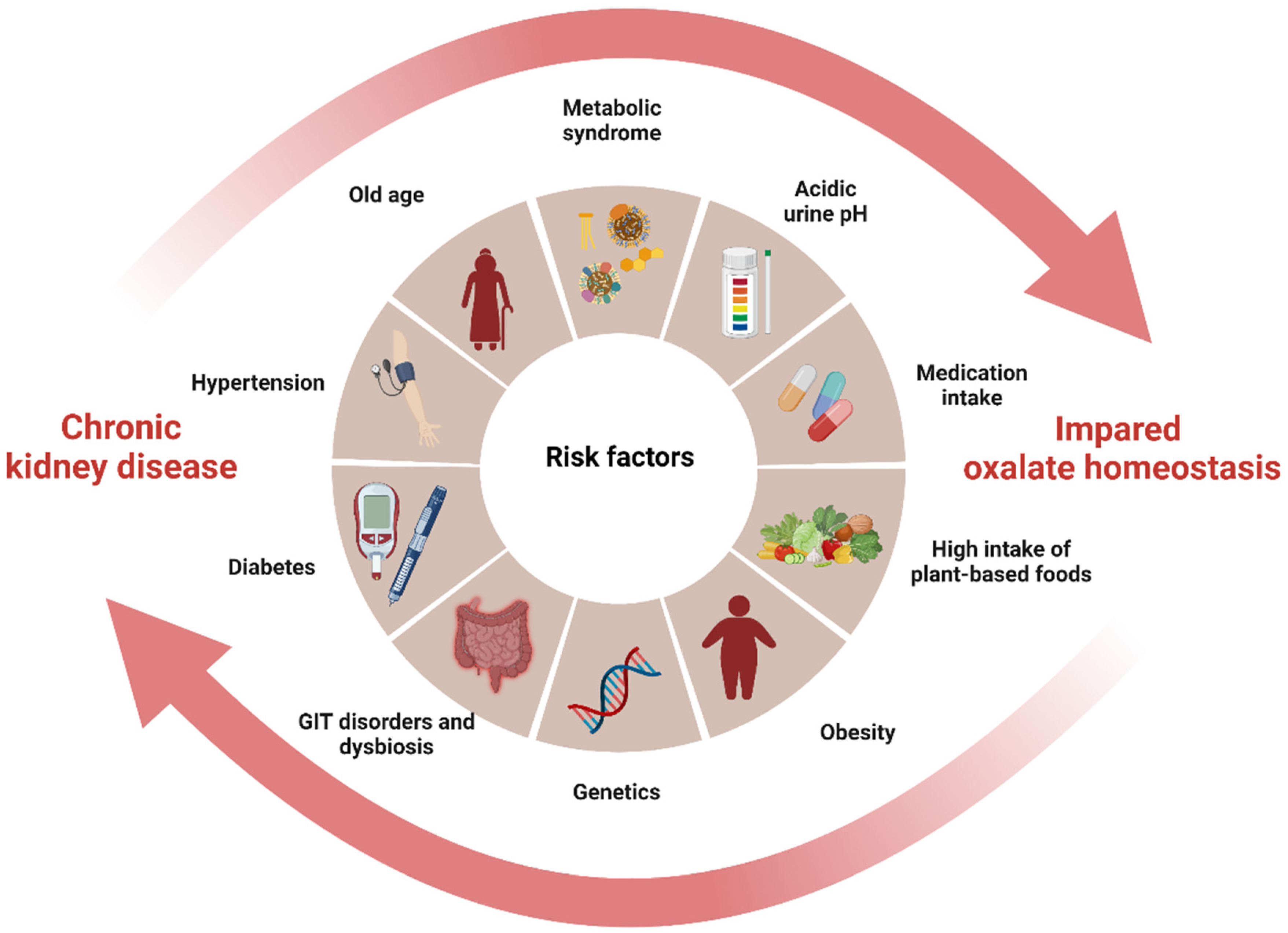
3. The Interplay between Oxalate and CKD: A Vicious Cycle of Shared Risk Factors
4. Oxalate’s Role in the Pathogenesis of CKD: From Silent Culprit to Active Player
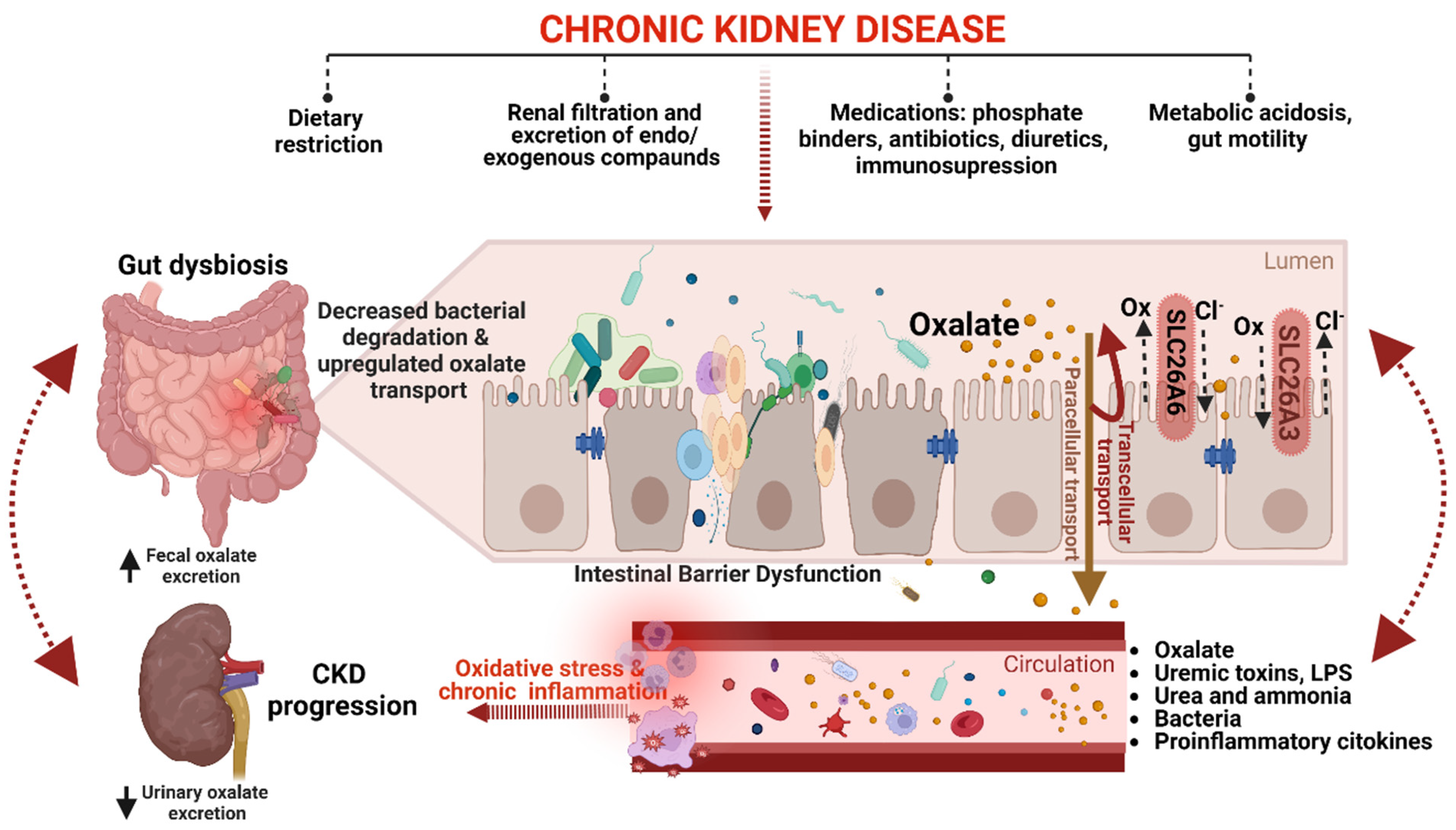
5. Gut-Kidney Axis in CKD Oxalate Homeostasis
6. Oxalate as a Clinical Marker for CKD Progression and Prognosis
7. Targeting Oxalate Homeostasis to Reduce CKD Progression and the Risk of Cardiovascular Events
7.1. Dialysis Treatment for Management Oxalate Burden in Patients with Kidney Failure
7.2. Modifying the Shared Risk Factors to Prevent CKD Progression
7.3. Medication Adjustments
- Calcium-based phosphate binders. Calcium-based phosphate binders, including calcium acetate and calcium carbonate, are commonly used to prevent mineral-bone disorders associated with CKD and are also considered to prevent CaOx stone formation [23,128]. These supplements work by binding with phosphate in the intestine, thus reducing its bioavailability and absorption, resulting in lower urinary oxalate excretion and a decreased risk of hyperoxaluria and CaOx stone formation [23]. To achieve a neutral calcium balance and avoid the adverse effects of either negative or positive calcium balance, a calcium intake of around 1000 mg/day is recommended [128,129]. However, calcium supplementation, especially if administered between meals, can raise urinary calcium excretion without any positive impact on oxalate, thereby elevating the risk of stone formation [23]. Moreover, caution should be exercised when using calcium supplements in patients with advanced CKD because they may cause hypercalcemia and vascular calcification [128,130]. Therefore, the decision to use calcium-based phosphate binders in patients with advanced CKD should be made on a case-by-case basis, balancing the individual needs and risks of each patient against the risk of worsening vascular calcification.
- Noncalcium phosphate binders. Noncalcium phosphate binders, such as lanthanum carbonate, have also been shown to reduce UOx excretion in CKD patients by decreasing gut absorption of dietary oxalate [131,132]. This is thought to be due to the ability of lanthanum to form insoluble complexes with oxalate, thus reducing its bioavailability for absorption [131,133]. However, more extensive studies are needed to establish the efficacy and safety of noncalcium phosphate binders in preventing hyperoxalemia/hyperoxaluria and CKD progression.
- Calcium channel blockers. Verapamil has been shown to increase urinary oxalate excretion and reduce the risk of CaOx stone formation in animal models [134]. However, their efficacy in patients with hyperoxalemia/hyperoxaluria has not been established, and the risk-benefit ratio for the individual patient should determine the treatment option.
- Thiazide and thiazide-like diuretics. Thiazide diuretics have been found to be effective in treating hypertension in patients with ESKD [135] and in reducing the formation of CaOx kidney stones. Thiazide-type diuretics (hydrochlorothiazide, chlorthalidone, and indapamide) act on the distal tubule of the kidney, increasing calcium reabsorption and decreasing the excretion of calcium in the urine, resulting in a decrease in UOx excretion [136,137]. However, the use of thiazide and thiazide-like diuretics in CKD patients is often restricted due to concerns regarding their safety and effectiveness, such as the risk of electrolyte imbalances, volume depletion, and a decline in eGFR [138].
- Magnesium. Magnesium has been recognized as a potent inhibitor of calcium oxalate (CaOx) crystals due to its ability to bind with oxalate, forming a soluble complex. This inhibitory effect is particularly significant when magnesium is combined with citrate and remains effective even in acidic environments [139]. Magnesium also inhibits the absorption of dietary oxalate from the gut lumen, as well as citrate-rich food [140]. Furthermore, magnesium plays a multifaceted role in the management of CKD. It has been shown to suppress the secretion of parathyroid hormones, activate the calcium-sensing receptor, promote osteoblast activity, and reduce intestinal phosphate absorption. These mechanisms contribute to the regulation of mineral metabolism and prevent the development of secondary hyperparathyroidism and vascular calcification in CKD patients. Additionally, magnesium has been associated with a decrease in the incidence of vascular calcification and improvements in cardiac function [141,142,143]. Incorporating magnesium supplementation as a medical adjustment in patients with CKD may not only help address oxalate burden but also maintain mineral balance, reduce the risk of complications, and improve overall cardiac health. However, further studies are needed to determine optimal dosing strategies and assess the long-term effects of magnesium supplementation in CKD populations.
- Vitamins B6 and D. Extensive research has been conducted on the association between deficiencies in vitamins B6 and D and the development of CaOx urolithiasis [23,144,145,146]. However, the effectiveness of vitamin supplementation in preventing hyperoxalemia/hyperoxaluria is still a controversial issue [122,145,146]. Additionally, there is a lack of knowledge regarding the potential of these vitamins to prevent the oxalate burden in CKD patients. More research is required to ascertain the potential benefits of these vitamins in managing disrupted oxalate homeostasis in CKD.
7.4. Enhancement of Intestinal Oxalate Handling with Promising New Pharmacological Targets
- ODB. ODB have been extensively studied as a potential treatment option for patients with hyperoxaluria and urolithiasis [147]. By degrading oxalate in the gut, ODB may prevent the absorption of oxalate into the bloodstream and reduce the burden of oxalate in CKD patients. Studies have shown that probiotics and synbiotics can be considered good sources of naturally occurring oxalate-degrading agents in the human colon. Pro- and/or synbiotics supplements containing O. formigenes, Bifidobacterium lactis, Lactobacillus strains, and others have been found to decrease hyperoxalemia/hyperoxaluria [21,147,148,149,150], but the results of in vitro and experimental studies do not always reflect the ability of bacteria to degrade oxalate in humans [18,147,151]. Specific clinical studies are scarce, and further research is needed to determine the optimal dosages and benefits of ODB supplementation in the management of the oxalate burden in CKD.
- Oxalate-degrading enzymes. Oxalate-degrading enzymes represent a new class of enzymes that can effectively degrade oxalate into nontoxic compounds [150,151]. As described above, enzymes, such as OxdC and Oxc, have been found naturally in the gut microbiota [18]. Two promising oxalate-degrading enzymes, reloxaliase and Oxazyme®, are currently under investigation as potential treatments for oxalate-related diseases. Reloxaliase, also known as ALLN-177, is a recombinant OxdC enzyme derived from Bacillus subtilis and expressed in Escherichia coli and developed for the treatment of enteric hyperoxaluria [152]. Clinical trials have demonstrated the safety and efficacy of reloxaliase, showing a significant reduction in UOx levels in patients with enteric hyperoxaluria [152,153]. Notably, reloxaliase has also shown promising results in lowering urine and plasma oxalate in patients with CKD, including those with moderate to severe kidney dysfunction. Specifically, it has led to a ~30% decrease in UOx in two patients with grade 3b CKD and a similar reduction in POx in seven patients with grade 5 CKD [154]. Oxazyme® is a synthetic enzyme engineered to effectively degrade oxalate in the gastrointestinal tract [155]. Although clinical trials for Oxazyme® are still in the early stages, preclinical studies have demonstrated its ability to degrade oxalate in laboratory settings [155]. Further research and clinical trials are needed to establish their efficacy, safety, and optimal dosing regimens for the CKD patient population.
- Small-molecule inhibitor. A small-molecule inhibitor of the intestinal anion exchanger SLC26A3 has been identified for the treatment of hyperoxaluria [151]. The small-molecule SLC26A3 inhibitor (DRAinh-A270) selectively inhibits SLC26A3-mediated chloride/bicarbonate exchange and oxalate/chloride exchange [13,17]. In colonic closed loops in mice, luminal DRAinh-A270 inhibited oxalate absorption by 70% [17]. By selectively inhibiting SLC26A3-mediated oxalate absorption, this inhibitor has the potential to alleviate the burden of oxalate-related complications and improve the management of hyperoxaluria. Continued research and clinical investigations are necessary to fully explore the therapeutic potential of this small-molecule inhibitor and advance its translation into clinical practice.
8. Conclusions and Future Directions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ermer, T.; Eckardt, K.U.; Aronson, P.S.; Knauf, F. Oxalate, Inflammasome, and Progression of Kidney Disease. Curr. Opin. Nephrol. Hypertens. 2016, 25, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zhang, Y.H.; Chi, Z.P.; Huang, R.; Huang, H.; Liu, G.Y.; Zhang, Y.F.; Yang, H.S.; Lin, J.H.; Yang, T.H.; et al. The Handling of Oxalate in the Body and the Origin of Oxalate in Calcium Oxalate Stones. Urol. Int. 2020, 104, 167–176. [Google Scholar] [CrossRef]
- Robertson, D.S. The Function of Oxalic Acid in the Human Metabolism. Clin. Chem. Lab. Med. 2011, 49, 1405–1412. [Google Scholar] [CrossRef]
- Brzica, H.; Breljak, D.; Burckhardt, B.C.; Burckhardt, G.; Sabolić, I. Oxalate: From the Environment to Kidney Stones. Arh. Hig. Rada Toksikol. 2013, 64, 609–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ermer, T.; Nazzal, L.; Tio, M.C.; Waikar, S.; Aronson, P.S.; Knauf, F. Oxalate Homeostasis. Nat. Rev. Nephrol. 2023, 19, 123–138. [Google Scholar] [CrossRef]
- Mitchell, T.; Kumar, P.; Reddy, T.; Wood, K.D.; Knight, J.; Assimos, D.G.; Holmes, R.P. Dietary Oxalate and Kidney Stone Formation. Am. J. Physiol. Ren. Physiol. 2019, 316, F409–F413. [Google Scholar] [CrossRef] [PubMed]
- Crivelli, J.J.; Mitchell, T.; Knight, J.; Wood, K.D.; Assimos, D.G.; Holmes, R.P.; Fargue, S. Contribution of Dietary Oxalate and Oxalate Precursors to Urinary Oxalate Excretion. Nutrients 2021, 13, 62. [Google Scholar] [CrossRef]
- Zimmermann, D.J.; Hesse, A.; von Unruh, G.E. Influence of a High-Oxalate Diet on Intestinal Oxalate Absorption. World J. Urol. 2005, 23, 324–329. [Google Scholar] [CrossRef]
- Holmes, R.P.; Ambrosius, W.T.; Assimos, D.G. Dietary Oxalate Loads and Renal Oxalate Handling. J. Urol. 2005, 174, 943–947. [Google Scholar] [CrossRef]
- Lange, J.N.; Wood, K.D.; Knight, J.; Assimos, D.G.; Holmes, R.P. Glyoxal Formation and Its Role in Endogenous Oxalate Synthesis. Adv. Urol. 2012, 2012, 819202. [Google Scholar] [CrossRef] [Green Version]
- Groothoff, J.W.; Metry, E.; Deesker, L.; Garrelfs, S.; Acquaviva, C.; Almardini, R.; Beck, B.B.; Boyer, O.; Cerkauskiene, R.; Ferraro, P.M.; et al. Clinical Practice Recommendations for Primary Hyperoxaluria: An Expert Consensus Statement from ERKNet and OxalEurope. Nat. Rev. Nephrol. 2023, 19, 194–211. [Google Scholar] [CrossRef]
- Knight, J.; Madduma-Liyanage, K.; Mobley, J.A.; Assimos, D.G.; Holmes, R.P. Ascorbic Acid Intake and Oxalate Synthesis. Urolithiasis 2016, 44, 289–297. [Google Scholar] [CrossRef] [Green Version]
- Whittamore, J.M.; Hatch, M. The Role of Intestinal Oxalate Transport in Hyperoxaluria and the Formation of Kidney Stones in Animals and Man. Urolithiasis 2017, 45, 89–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Wang, W.; Wang, H.; Tuo, B. Physiological and Pathological Functions of SLC26A6. Front. Med. 2021, 7, 1156. [Google Scholar] [CrossRef] [PubMed]
- Neumeier, L.I.; Thomson, R.B.; Reichel, M.; Eckardt, K.U.; Aronson, P.S.; Knauf, F. Enteric Oxalate Secretion Mediated by Slc26a6 Defends against Hyperoxalemia in Murine Models of Chronic Kidney Disease. J. Am. Soc. Nephrol. 2020, 31, 1987–1995. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q. Slc26a3 (DRA) in the Gut: Expression, Function, Regulation, Role in Infectious Diarrhea and Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2021, 27, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Cil, O.; Chu, T.; Lee, S.; Haggie, P.M.; Verkman, A.S. Small-Molecule Inhibitor of Intestinal Anion Exchanger SLC26A3 for Treatment of Hyperoxaluria and Nephrolithiasis. JCI Insight 2022, 7, e153359. [Google Scholar] [CrossRef]
- Karamad, D.; Khosravi-Darani, K.; Khaneghah, A.M.; Miller, A.W. Probiotic Oxalate-Degrading Bacteria: New Insight of Environmental Variables and Expression of the Oxc and Frc Genes on Oxalate Degradation Activity. Foods 2022, 11, 2876. [Google Scholar] [CrossRef]
- Liu, M.; Devlin, J.C.; Hu, J.; Volkova, A.; Battaglia, T.W.; Ho, M.; Asplin, J.R.; Byrd, A.; Loke, P.; Li, H.; et al. Microbial Genetic and Transcriptional Contributions to Oxalate Degradation by the Gut Microbiota in Health and Disease. Elife 2021, 10, e63642. [Google Scholar] [CrossRef]
- Jiang, T.; Chen, W.; Cao, L.; He, Y.; Zhou, H.; Mao, H. Abundance, Functional, and Evolutionary Analysis of Oxalyl-Coenzyme A Decarboxylase in Human Microbiota. Front. Microbiol. 2020, 11, 672. [Google Scholar] [CrossRef]
- Stepanova, N.; Akulenko, I.; Serhiichuk, T.; Dovbynchuk, T.; Savchenko, S.; Tolstanova, G. Synbiotic Supplementation and Oxalate Homeostasis in Rats: Focus on Microbiota Oxalate-Degrading Activity. Urolithiasis 2022, 50, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Hatch, M. Gut Microbiota and Oxalate Homeostasis. Ann. Transl. Med. 2017, 5, 36. [Google Scholar] [CrossRef] [PubMed]
- Bargagli, M.; Ferraro, P.M.; Vittori, M.; Lombardi, G.; Gambaro, G.; Somani, B. Calcium and Vitamin D Supplementation and Their Association with Kidney Stone Disease: A Narrative Review. Nutrients 2021, 13, 4363. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, M.D.; Hsi, R.S.; Chi, T.; Shara, N.; Wactawski-Wende, J.; Kahn, A.J.; Wang, H.; Hou, L.; Stoller, M.L. Dietary Intake of Fiber, Fruit, and Vegetables Decrease the Risk of Incident Kidney Stones in Women: A Women’s Health Initiative (WHI) Report. J. Urol. 2014, 192, 1694. [Google Scholar] [CrossRef] [Green Version]
- Miller, A.W.; Dearing, D. The Metabolic and Ecological Interactions of Oxalate-Degrading Bacteria in the Mammalian Gut. Pathogens 2013, 2, 636–652. [Google Scholar] [CrossRef] [Green Version]
- Nazzal, L.; Francois, F.; Henderson, N.; Liu, M.; Li, H.; Koh, H.; Wang, C.; Gao, Z.; Perez, G.P.; Asplin, J.R.; et al. Effect of Antibiotic Treatment on Oxalobacter Formigenes Colonization of the Gut Microbiome and Urinary Oxalate Excretion. Sci. Rep. 2021, 11, 16428. [Google Scholar] [CrossRef]
- Ramos, C.I.; Armani, R.G.; Canziani, M.E.; Ribeiro Dolenga, C.J.; Nakao, L.S.; Campbell, K.L.; Cuppari, L. Bowel Habits and the Association with Uremic Toxins in Non-Dialysis-Dependent Chronic Kidney Disease Patients. J. Ren. Nutr. 2020, 30, 31–35. [Google Scholar] [CrossRef]
- Demoulin, N.; Aydin, S.; Gillion, V.; Morelle, J.; Jadoul, M. Pathophysiology and Management of Hyperoxaluria and Oxalate Nephropathy: A Review. Am. J. Kidney Dis. 2022, 79, 717–727. [Google Scholar] [CrossRef]
- Jiang, H.; Pokhrel, G.; Chen, Y.; Wang, T.; Yin, C.; Liu, J.; Wang, S.; Liu, Z. High Expression of SLC26A6 in the Kidney May Contribute to Renal Calcification via an SLC26A6-Dependent Mechanism. PeerJ 2018, 6, e5192. [Google Scholar] [CrossRef] [Green Version]
- Bergsland, K.J.; Zisman, A.L.; Asplin, J.R.; Worcester, E.M.; Coe, F.L. Evidence for Net Renal Tubule Oxalate Secretion in Patients with Calcium Kidney Stones. Am. J. Physiol. Ren. Physiol. 2011, 300, F311–F318. [Google Scholar] [CrossRef]
- Whittamore, J.M.; Hatch, M. Oxalate Flux Across the Intestine: Contributions from Membrane Transporters. Compr. Physiol. 2022, 12, 2835–2875. [Google Scholar] [CrossRef]
- Verhulst, A.; De Broe, M.E. Oxalate. In Clinical Nephrotoxins; De Broe, M.E., Porter, G.A., Bennett, W.M., Deray, G., Eds.; Springer: Boston, MA, USA, 2008; pp. 749–756. [Google Scholar] [CrossRef]
- Penniston, K.L. Dietary Oxalate and Calcium Oxalate Stones: A Theoretical or Real Concern. In Practical Controversies in Medical Management of Stone Disease; Pearle, M., Nakada, S., Eds.; Springer: New York, NY, USA, 2014; pp. 7–28. [Google Scholar] [CrossRef]
- Solubility Product Constant (Ksp) Values at 25 °C. Available online: https://users.stlcc.edu/gkrishnan/ksptable.html (accessed on 31 May 2023).
- López-Moreno, M.; Garcés-Rimón, M.; Miguel, M. Antinutrients: Lectins, Goitrogens, Phytates and Oxalates, Friends or Foe? J. Funct. Foods 2022, 89, 104938. [Google Scholar] [CrossRef]
- Castellaro, A.M.; Tonda, A.; Cejas, H.H.; Ferreyra, H.; Caputto, B.L.; Pucci, O.A.; Gil, G.A. Oxalate Induces Breast Cancer. BMC Cancer 2015, 15, 761. [Google Scholar] [CrossRef] [Green Version]
- Markovich, D.; Aronson, P.S. Specificity and Regulation of Renal Sulfate Transporters. Annu. Rev. Physiol. 2007, 69, 361–375. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, S.; Brandl, H.; Schönfels, C. Human Oxalate—Really Just an End-Product of Metabolism? Angew. Chem. Int. 1994, 33, 1780–1781. [Google Scholar] [CrossRef]
- Rosenstock, J.L.; Joab, T.M.J.; Devita, M.V.; Yang, Y.; Sharma, P.D.; Bijol, V. Oxalate Nephropathy: A Review. Clin. Kidney J. 2022, 15, 194–204. [Google Scholar] [CrossRef]
- Demoulin, N.; Issa, Z.; Crott, R.; Morelle, J.; Danse, E.; Wallemacq, P.; Jadoul, M.; Deprez, P.H. Enteric Hyperoxaluria in Chronic Pancreatitis. Medicine 2017, 96, e6758. [Google Scholar] [CrossRef] [PubMed]
- Efe, O.; Verma, A.; Waikar, S.S. Urinary Oxalate as a Potential Mediator of Kidney Disease in Diabetes Mellitus and Obesity. Curr. Opin. Nephrol. Hypertens. 2019, 28, 316–320. [Google Scholar] [CrossRef]
- Konstantynowicz, J.; Porowski, T.; Zoch-Zwierz, W.; Wasilewska, J.; Kadziela-Olech, H.; Kulak, W.; Owens, S.C.; Piotrowska-Jastrzebska, J.; Kaczmarski, M. A Potential Pathogenic Role of Oxalate in Autism. Eur. J. Paediatr. Neurol. 2012, 16, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhao, Y.; Shukha, Y.; Lu, H.; Wang, L.; Liu, Z.; Liu, C.; Zhao, Y.; Wang, H.; Zhao, G.; et al. Dysregulated Oxalate Metabolism Is a Driver and Therapeutic Target in Atherosclerosis. Cell Rep. 2021, 36, 109420. [Google Scholar] [CrossRef]
- Bahadoran, Z.; Mirmiran, P.; Azizi, F. Dietary Oxalate to Calcium Ratio and Incident Cardiovascular Events: A 10-Year Follow-up among an Asian Population. Nutr. J. 2022, 21, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Bhasin, B.; Ürekli, H.M.; Atta, M.G. Primary and secondary hyperoxaluria: Understanding the enigma. World J. Nephrol. 2015, 4, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Haghayeghi, K.; Najibi, M.; Wang, H.; Donegan, L.; Wang, Y. Clinicopathologic Update of Calcium Oxalate in Breast: A 15-Year Retrospective Review. Breast J. 2020, 26, 1736–1741. [Google Scholar] [CrossRef] [PubMed]
- Oda, M.; Saraya, T.; Wakayama, M.; Shibuya, K.; Ogawa, Y.; Inui, T.; Yokoyama, E.; Inoue, M.; Shimoyamada, H.; Fujiwara, M.; et al. Calcium Oxalate Crystal Deposition in a Patient with Aspergilloma Due to Aspergillus Niger. J. Thorac. Dis. 2013, 5, E174–E178. [Google Scholar] [CrossRef] [PubMed]
- Wahl, R.; Fuchs, R.; Kailee, E. Oxalate in the Human Thyroid Gland. Eur. J. Clin. Chem. Clin. Biochem. 1993, 31, 559–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dessombz, A.; Méria, P.; Bazin, D.; Daudon, M. Prostatic Stones: Evidence of a Specific Chemistry Related to Infection and Presence of Bacterial Imprints. PLoS ONE 2012, 7, e51691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yavorskyy, A.; Hernandez-Santana, A.; McCarthy, G.; McMahon, G. Detection of Calcium Phosphate Crystals in the Joint Fluid of Patients with Osteoarthritis—Analytical Approaches and Challenges. Analyst 2008, 133, 302–318. [Google Scholar] [CrossRef] [Green Version]
- Froberg, K.; Dorion, R.P.; McMartin, K.E. The Role of Calcium Oxalate Crystal Deposition in Cerebral Vessels During Ethylene Glycol Poisoning. Clin. Toxicol. 2008, 44, 315–318. [Google Scholar] [CrossRef]
- Zeng, Z.; Xu, S.; Wang, F.; Peng, X.; Zhang, W.; Zhan, Y.; Ding, Y.; Liu, Z.; Liang, L. HAO1-Mediated Oxalate Metabolism Promotes Lung Pre-Metastatic Niche Formation by Inducing Neutrophil Extracellular Traps. Oncogene 2022, 41, 3719–3731. [Google Scholar] [CrossRef]
- Sun, X.; Kour, B.D.; Wang, X.; Hu, X.; Wang, Y.; Vuree, S. Insights into Association between Urolithiasis and Prostate Cancer. J Men’s Health 2021, 17, 52–61. [Google Scholar] [CrossRef]
- Kovesdy, C.P. Epidemiology of Chronic Kidney Disease: An Update. Kidney Int. Suppl. 2022, 12, 7–11. [Google Scholar] [CrossRef]
- Carney, E.F. The Impact of Chronic Kidney Disease on Global Health. Nat. Rev. Nephrol. 2020, 16, 251. [Google Scholar] [CrossRef] [Green Version]
- Dhondup, T.; Kittanamongkolchai, W.; Vaughan, L.E.; Mehta, R.A.; Chhina, J.K.; Enders, F.T.; Hickson, L.T.J.; Lieske, J.C.; Rule, A.D. Risk of ESRD and Mortality in Kidney and Bladder Stone Formers. Am. J. Kidney Dis. 2018, 72, 790–797. [Google Scholar] [CrossRef] [Green Version]
- Pfau, A.; Wytopil, M.; Chauhan, K.; Reichel, M.; Coca, S.G.; Aronson, P.S.; Eckardt, K.U.; Knauf, F. Assessment of Plasma Oxalate Concentration in Patients with CKD. Kidney Int. Rep. 2020, 5, 2013–2020. [Google Scholar] [CrossRef]
- Stepanova, N.; Driianska, V.; Korol, L.; Snisar, L.; Lebed, L. Plasma Oxalic Acid and Cardiovascular Risk in End-Stage Renal Disease Patients: A Prospective, Observational Cohort Pilot Study. Korean J. Intern. Med. 2021, 37, 167–178. [Google Scholar] [CrossRef]
- Pfau, A.; Ermer, T.; Coca, S.G.; Tio, M.C.; Genser, B.; Reichel, M.; Finkelstein, F.O.; März, W.; Wanner, C.; Waikar, S.S.; et al. High Oxalate Concentrations Correlate with Increased Risk for Sudden Cardiac Death in Dialysis Patients. J. Am. Soc. Nephrol. 2021, 32, 2375–2385. [Google Scholar] [CrossRef] [PubMed]
- Mulay, S.R.; Eberhard, J.N.; Pfann, V.; Marschner, J.A.; Darisipudi, M.N.; Daniel, C.; Romoli, S.; Desai, J.; Grigorescu, M.; Kumar, S.V.; et al. Oxalate-Induced Chronic Kidney Disease with Its Uremic and Cardiovascular Complications in C57BL/6 Mice. Am. J. Physiol. Ren. Physiol. 2016, 310, F785–F795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres, J.A.; Rezaei, M.; Broderick, C.; Lin, L.; Wang, X.; Hoppe, B.; Cowley, B.D.; Savica, V.; Torres, V.E.; Khan, S.; et al. Crystal Deposition Triggers Tubule Dilation That Accelerates Cystogenesis in Polycystic Kidney Disease. J. Clin. Investig. 2019, 129, 4506–4522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snijders, M.L.H.; Hesselink, D.A.; Clahsen-Van Groningen, M.C.; Roodnat, J.I. Oxalate Deposition in Renal Allograft Biopsies within 3 Months after Transplantation Is Associated with Allograft Dysfunction. PLoS ONE 2019, 14, e0214940. [Google Scholar] [CrossRef] [PubMed]
- Krogstad, V.; Elgstøen, K.B.P.; Johnsen, L.F.; Hartmann, A.; Mørkrid, L.; Åsberg, A. High Plasma Oxalate Levels Early After Kidney Transplantation Are Associated with Impaired Long-Term Outcomes. Transpl. Int. 2022, 35, 10240. [Google Scholar] [CrossRef]
- Alshaikh, A.E.; Hassan, H.A. Gut-Kidney Axis in Oxalate Homeostasis. Curr. Opin. Nephrol. Hypertens. 2021, 30, 264–274. [Google Scholar] [CrossRef]
- Mirmiran, P.; Bahadoran, Z.; Azizi, F. Dietary Oxalate-Calcium Balance and the Incidence of Hypertension and Chronic Kidney Disease: A Prospective Study among an Asian Population. Nutr. Metab. 2022, 19, 74. [Google Scholar] [CrossRef]
- Xiang, H.; Chen, H.; Liu, Y.; Dodd, D.; Pao, A.C. Role of Insulin Resistance and the Gut Microbiome on Urine Oxalate Excretion in Ob/Ob Mice. Physiol. Rep. 2022, 10, e15357. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.W.; Ke, H.L.; Lee, J.I.; Lee, Y.C.; Jhan, J.H.; Wang, H.S.; Shen, J.T.; Tsao, Y.H.; Huang, S.P.; Geng, J.H. Metabolic Syndrome Increases the Risk of Kidney Stone Disease: A Cross-Sectional and Longitudinal Cohort Study. J. Pers. Med. 2021, 11, 1154. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Shao, X.; Gao, P.; Zou, H.; Zhang, X. Metabolic Syndrome Components and Chronic Kidney Disease in a Community Population Aged 40 Years and Older in Southern China: A Cross-Sectional Study. Diabetes Metab. Syndr. Obes. 2022, 15, 839–848. [Google Scholar] [CrossRef] [PubMed]
- Cornière, N.; Thomson, R.B.; Thauvin, S.; Villoutreix, B.O.; Karp, S.; Dynia, D.W.; Burlein, S.; Brinkmann, L.; Badreddine, A.; Dechaume, A.; et al. Dominant Negative Mutation in Oxalate Transporter SLC26A6 Associated with Enteric Hyperoxaluria and Nephrolithiasis. J. Med. Genet. 2022, 59, 1035–1043. [Google Scholar] [CrossRef]
- Soleimani, M. SLC26 Cl-/HCO3- Exchangers in the Kidney: Roles in Health and Disease. Kidney Int. 2013, 84, 657–666. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Xue, X.; Terkeltaub, R.; Dalbeth, N.; Merriman, T.R.; Mount, D.B.; Feng, Z.; Li, X.; Cui, L.; Liu, Z.; et al. Association of Acidic Urine PH with Impaired Renal Function in Primary Gout Patients: A Chinese Population-Based Cross-Sectional Study. Arthritis Res. Ther. 2022, 24, 32. [Google Scholar] [CrossRef]
- Nakanishi, N.; Fukui, M.; Tanaka, M.; Toda, H.; Imai, S.; Yamazaki, M.; Hasegawa, G.; Oda, Y.; Nakamura, N. Low Urine PH Is a Predictor of Chronic Kidney Disease. Kidney Blood Press. Res. 2012, 35, 77–81. [Google Scholar] [CrossRef]
- Manissorn, J.; Fong-Ngern, K.; Peerapen, P.; Thongboonkerd, V. Systematic evaluation for effects of urine pH on calcium oxalate crystallization, crystal-cell adhesion and internalization into renal tubular cells. Sci. Rep. 2017, 7, 1798. [Google Scholar] [CrossRef]
- Goraya, N.; Simoni, J.; Sager, L.N.; Madias, N.E.; Wesson, D.E. Urine citrate excretion as a marker of acid retention in patients with chronic kidney disease without overt metabolic acidosis. Kidney Int. 2019, 95, 1190–1196. [Google Scholar] [CrossRef] [PubMed]
- Saranya, G.R.; Viswanathan, P. Gut Microbiota Dysbiosis in AKI to CKD Transition. Biomed. Pharmacother. 2023, 161, 114447. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.C.; Liao, M.H.; Kung, W.M.; Wang, Y.C. Proton Pump Inhibitors and Risk of Chronic Kidney Disease: Evidence from Observational Studies. J. Clin. Med. 2023, 12, 2262. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Yoo, D.M.; Bang, W.J.; Choi, H.G. Association between Urolithiasis and History Proton Pump Inhibitor Medication: A Nested Case-Control Study. J. Clin. Med. 2022, 11, 5693. [Google Scholar] [CrossRef]
- Cantorna, M.T.; Snyder, L.; Arora, J. Vitamin A and Vitamin D Regulate the Microbial Complexity, Barrier Function and the Mucosal Immune Responses to Insure Intestinal Homeostasis. Crit. Rev. Biochem. Mol. Biol. 2019, 54, 184–192. [Google Scholar] [CrossRef]
- Chmiel, J.A.; Stuivenberg, G.A.; Al, K.F.; Akouris, P.P.; Razvi, H.; Burton, J.P.; Bjazevic, J. Vitamins as Regulators of Calcium-Containing Kidney Stones—New Perspectives on the Role of the Gut Microbiome. Nat. Rev. Urol. 2023, 1–23. [Google Scholar] [CrossRef]
- Rule, A.D.; Krambeck, A.E.; Lieske, J.C. Chronic Kidney Disease in Kidney Stone Formers. Clin. J. Am. Soc. Nephrol. 2011, 6, 2069. [Google Scholar] [CrossRef] [Green Version]
- Sigurjonsdottir, V.K.; Runolfsdottir, H.L.; Indridason, O.S.; Palsson, R.; Edvardsson, V.O. Impact of Nephrolithiasis on Kidney Function. BMC Nephrol. 2015, 16, 149. [Google Scholar] [CrossRef] [Green Version]
- Cao, L.C.; Honeyman, T.W.; Cooney, R.; Kennington, L.; Scheid, C.R.; Jonassen, J.A. Mitochondrial Dysfunction Is a Primary Event in Renal Cell Oxalate Toxicity. Kidney Int. 2004, 66, 1890–1900. [Google Scholar] [CrossRef] [Green Version]
- Takemura, K.; Nishi, H.; Inagi, R. Mitochondrial Dysfunction in Kidney Disease and Uremic Sarcopenia. Front. Physiol. 2020, 11, 565023. [Google Scholar] [CrossRef]
- Braga, P.C.; Alves, M.G.; Rodrigues, A.S.; Oliveira, P.F. Mitochondrial Pathophysiology on Chronic Kidney Disease. Int. J. Mol. Sci. 2022, 23, 1776. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.; Yarlagadda, V.; Adedoyin, O.; Saini, V.; Assimos, D.G.; Holmes, R.P.; Mitchell, T. Oxalate Induces Mitochondrial Dysfunction and Disrupts Redox Homeostasis in a Human Monocyte Derived Cell Line. Redox Biol. 2018, 15, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Barbero, N.M.; Oller, J.; Sanz, A.B.; Ramos, A.M.; Ortiz, A.; Ruiz-Ortega, M.; Rayego-Mateos, S. Mitochondrial Dysfunction in the Cardio-Renal Axis. Int. J. Mol. Sci. 2023, 24, 8209. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Tang, X.; Song, S.; Gao, Y.; Yu, H.; Sun, N.; Wen, B.; Mei, C. Hyperoxalemia Leads to Oxidative Stress in Endothelial Cells and Mice with Chronic Kidney Disease. Kidney Blood Press. Res. 2021, 46, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Recht, P.A.; Tepedino, G.J.; Siecke, N.W.; Buckley, M.T.; Mandeville, J.T.; Maxfield, F.R.; Levin, R.I. Oxalic Acid Alters Intracellular Calcium in Endothelial Cells. Atherosclerosis 2004, 173, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Saini, K.; Saini, V.; Mitchell, T. Oxalate Alters Cellular Bioenergetics, Redox Homeostasis, Antibacterial Response, and Immune Response in Macrophages. Front. Immunol. 2021, 12, 4432. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, T.; Yang, J.; Wang, S.; Yang, W.; Liu, J.; Ye, Z. Calcium Oxalate Monohydrate Crystals Stimulate Monocyte Chemoattractant Protein-1 and Transforming Growth Factor Β1 Expression in Human Renal Epithelial Cells. Mol. Med. Rep. 2012, 5, 1241–1244. [Google Scholar] [CrossRef]
- Convento, M.; Pessoa, E.; Aragão, A.; Schor, N.; Borges, F.; Convento, M.; Pessoa, E.; Aragão, A.; Schor, N.; Borges, F. Oxalate Induces Type II Epithelial to Mesenchymal Transition (EMT) in Inner Medullary Collecting Duct Cells (IMCD) in Vitro and Stimulate the Expression of Osteogenic and Fibrotic Markers in Kidney Medulla in Vivo. Oncotarget 2019, 10, 1102–1118. [Google Scholar] [CrossRef] [Green Version]
- Ming, S.; Tian, J.; Ma, K.; Pei, C.; Li, L.; Wang, Z.; Fang, Z.; Liu, M.; Dong, H.; Li, W.; et al. Oxalate-Induced Apoptosis through ERS-ROS–NF-ΚB Signalling Pathway in Renal Tubular Epithelial Cell. Mol. Med. 2022, 28, 88. [Google Scholar] [CrossRef]
- Dominguez-Gutierrez, P.R.; Kusmartsev, S.; Canales, B.K.; Khan, S.R. Calcium Oxalate Differentiates Human Monocytes Into Inflammatory M1 Macrophages. Front. Immunol. 2018, 9, 1863. [Google Scholar] [CrossRef]
- Mazur, T.; Demikhova, N.; Rudenko, T.; Yurchenko, A.; Yezhova, O.; Bokova, S.; Demikhov, A. Chronic Inflammation and Progression of Chronic Kidney Disease in Patients with Type 2 Diabetes. Ukr. J. Nephrol. Dial. 2021, 4, 36–43. [Google Scholar] [CrossRef]
- Lau, W.L.; Kalantar-Zadeh, K.; Vaziri, N.D. The Gut as a Source of Inflammation in Chronic Kidney Disease. Nephron 2015, 130, 92–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cobo, G.; Lindholm, B.; Stenvinkel, P. Chronic Inflammation in End-Stage Renal Disease and Dialysis. Nephrol. Dial. Transplant. 2018, 33 (Suppl. 3), iii35–iii40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wehedy, E.; Shatat, I.F.; Al Khodor, S. The Human Microbiome in Chronic Kidney Disease: A Double-Edged Sword. Front. Med. 2022, 8, 790783. [Google Scholar] [CrossRef]
- Bargagli, M.; Tio, M.C.; Waikar, S.S.; Ferraro, P.M. Dietary Oxalate Intake and Kidney Outcomes. Nutrients 2020, 12, 2673. [Google Scholar] [CrossRef]
- Ramezani, A.; Massy, Z.A.; Meijers, B.; Evenepoel, P.; Vanholder, R.; Raj, D.S. Role of the Gut Microbiome in Uremia: A Potential Therapeutic Target. Am. J. Kidney Dis. 2016, 67, 483–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stepanova, N.; Tolstanova, G.; Akulenko, I.; Nepomnyashchyi, V.; Savchenko, S.; Zholos, A.; Kolesnyk, M. Pilot Testing for Long-Term Impact of Glycerol-Induced Acute Kidney Injury on Oxalate Homeostasis in Rats. Ukr. J. Nephrol. Dial. 2022, 2, 15–24. [Google Scholar] [CrossRef]
- Stepanova, N.; Korol, L.; Tolstanova, G.; Akulenko, I. Low Oxalate-Degrading Activity in Fecal Microbiota Is Associated with High Serum Indoxyl Sulfate and Plasma Oxalic Acid Concentrations in Dialysis Patients. Am. J. Kidney Dis. 2022, 79, S13. [Google Scholar] [CrossRef]
- Stepanova, N.; Tolstanova, G.; Korol, L.; Akulenko, I.; Savchenko, O.; Kolesnyk, M. A Potential Role of Fecal Oxalate-Degrading Activity in Oxalate Homeostasis in End-Stage Renal Disease Patients; a Descriptive Pilot Study. J. Ren. Inj. Prev. 2021, 10, e19. [Google Scholar] [CrossRef]
- Zupcic, A.; Slezak, P.; Radloff, J. The Gastrointestinal Microbiota as a Potential Cause and Target in Chronic Kidney Disease Accentuating Treatment and Intervention Strategies. Appl. Sci. 2023, 13, 3212. [Google Scholar] [CrossRef]
- Noce, A.; Marchetti, M.; Marrone, G.; Di Renzo, L.; Di Lauro, M.; Di Daniele, F.; Albanese, M.; Di Daniele, N.; De Lorenzo, A. Link between Gut Microbiota Dysbiosis and Chronic Kidney Disease. Eur. Rev. Med. Pharmacol. Sci. 2022, 26, 2057–2074. [Google Scholar] [CrossRef] [PubMed]
- Stuivenberg, G.A.; Chmiel, J.; Akouris, P.P.; Al, K.; Bjazevic, J.; Burton, J. MPBS2-14 Gut Microbiota Derived Uremic Toxins Enhance Calcium Oxalate Stone Formation in Vitro and in Vivo. J. Endourol. 2022, 36 (Suppl. 1), A1–A315. [Google Scholar] [CrossRef]
- Liu, Y.; Jin, X.; Ma, Y.; Jian, Z.; Wei, Z.; Xiang, L.; Sun, Q.; Qi, S.; Wang, K.; Li, H. Short-Chain Fatty Acids Reduced Renal Calcium Oxalate Stones by Regulating the Expression of Intestinal Oxalate Transporter SLC26A6. mSystems 2021, 6, e0104521. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Priyamvada, S.; Ge, Y.; Jayawardena, D.; Singhal, M.; Anbazhagan, A.N.; Chatterjee, I.; Dayal, A.; Patel, M.; Zadeh, K.; et al. A Novel Role of SLC26A3 in the Maintenance of Intestinal Epithelial Barrier Integrity. Gastroenterology 2021, 160, 1240–1255.E3. [Google Scholar] [CrossRef] [PubMed]
- Freel, R.W.; Whittamore, J.M.; Hatch, M. Transcellular Oxalate and Cl− Absorption in Mouse Intestine Is Mediated by the DRA Anion Exchanger Slc26a3, and DRA Deletion Decreases Urinary Oxalate. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G520–G527. [Google Scholar] [CrossRef]
- Nazzal, L.; Puri, S.; Goldfarb, D.S. Enteric Hyperoxaluria: An Important Cause of End-Stage Kidney Disease. Nephrol. Dial. Transplant. 2016, 31, 375–382. [Google Scholar] [CrossRef]
- Witting, C.; Langman, C.B.; Assimos, D.; Baum, M.A.; Kausz, A.; Milliner, D.; Tasian, G.; Worcester, E.; Allain, M.; West, M.; et al. Pathophysiology and Treatment of Enteric Hyperoxaluria. Clin. J. Am. Soc. Nephrol. 2021, 16, 487–495. [Google Scholar] [CrossRef]
- Selistre, L.D.S.; Cochat, P.; Rech, D.L.; Parant, F.; Souza, V.C.D.; Dubourg, L. Association between Glomerular Filtration Rate (Measured by High-Performance Liquid Chromatography with Iohexol) and Plasma Oxalate. J. Bras. Nephrol. 2018, 40, 73–76. [Google Scholar] [CrossRef] [Green Version]
- Perinpam, M.; Enders, F.T.; Mara, K.C.; Vaughan, L.E.; Mehta, R.A.; Voskoboev, N.; Milliner, D.S.; Lieske, J.C. Plasma Oxalate in Relation to EGFR in Patients with Primary Hyperoxaluria, Enteric Hyperoxaluria and Urinary Stone Disease. Clin. Biochem. 2017, 50, 1014–1019. [Google Scholar] [CrossRef]
- Metry, E.L.; Garrelfs, S.F.; Peters-Sengers, H.; Vaz, F.M.; Bijlsma, J.A.; Neradova, A.; Oosterveld, M.J.S.; Groothoff, J.W. Plasma Oxalate and Glycolate Concentrations in Dialysis Patients with and without Primary Hyperoxaluria Type 1. Nephrol. Dial. Transplant. 2023, gfad049. [Google Scholar] [CrossRef]
- Ermer, T.; Kopp, C.; Asplin, J.R.; Granja, I.; Perazella, M.A.; Reichel, M.; Nolin, T.D.; Eckardt, K.U.; Aronson, P.S.; Finkelstein, F.O.; et al. Impact of Regular or Extended Hemodialysis and Hemodialfiltration on Plasma Oxalate Concentrations in Patients with End-Stage Renal Disease. Kidney Int. Rep. 2017, 2, 1050–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waikar, S.S.; Srivastava, A.; Palsson, R.; Shafi, T.; Hsu, C.Y.; Sharma, K.; Lash, J.P.; Chen, J.; He, J.; Lieske, J.; et al. Association of Urinary Oxalate Excretion with the Risk of Chronic Kidney Disease Progression. JAMA Intern. Med. 2019, 179, 542–551. [Google Scholar] [CrossRef] [PubMed]
- Shifris, I.; Korol, L.; Krasiuk, E.; Dudar, S. Activation of Oxidative Stress, Comorbidity and Survival of End-Stage Renal Disease Patients Treated with Hemodialysis. Ukr. J. Nephrol. Dial. 2021, 4, 67–77. [Google Scholar] [CrossRef]
- Steven, S.; Frenis, K.; Oelze, M.; Kalinovic, S.; Kuntic, M.; Jimenez, M.T.B.; Vujacic-Mirski, K.; Helmstädter, J.; Kröller-Schön, S.; Münzel, T.; et al. Vascular Inflammation and Oxidative Stress: Major Triggers for Cardiovascular Disease. Oxid. Med. Cell. Longev. 2019, 7092151. [Google Scholar] [CrossRef] [Green Version]
- Nishizawa, Y.; Higuchi, C.; Nakaoka, T.; Omori, H.; Ogawa, T.; Sakura, H.; Nitta, K. Compositional Analysis of Coronary Artery Calcification in Dialysis Patients in Vivo by Dual-Energy Computed Tomography Angiography. Ther. Apher. Dial. 2018, 22, 365–370. [Google Scholar] [CrossRef] [Green Version]
- Falconi, C.A.; Junho, C.V.D.C.; Fogaça-Ruiz, F.; Vernier, I.C.S.; da Cunha, R.S.; Stinghen, A.E.M.; Carneiro-Ramos, M.S. Uremic Toxins: An Alarming Danger Concerning the Cardiovascular System. Front. Physiol. 2021, 12, 686249. [Google Scholar] [CrossRef]
- Glassock, R.J.; Massry, S.G. Uremic Toxins: An Integrated Overview of Classification and Pathobiology. In Nutritional Management of Renal Disease, 4th ed.; Kopple, J.D., Shaul, G.M., Kalantar-Zadeh, K., Fouque, D., Eds.; Academic Press: Cambridge, MA, USA, 2022; pp. 77–89. [Google Scholar] [CrossRef]
- Franssen, C.F.M. Oxalate Clearance by Haemodialysis—A Comparison of Seven Dialysers. Nephrol. Dial. Transplant. 2005, 20, 1916–1921. [Google Scholar] [CrossRef] [Green Version]
- Marangella, M.; Bagnis, C.; Bruno, M.; Petrarulo, M.; Gabella, P.; Linari, F. Determinants of Oxalate Balance in Patients on Chronic Peritoneal Dialysis. Am. J. Kidney Dis. 1993, 21, 419–426. [Google Scholar] [CrossRef]
- Stepanova, N.; Korol, L.; Lebid, L.; Snisar, L.; Savchenko, S. Oxalate Balance in Peritoneal Dialysis Patients: A Potential Role of Dialysis-Related Peritonitis. In Vivo 2022, 36, 925–933. [Google Scholar] [CrossRef]
- Türk, C.; Petřík, A.; Sarica, K.; Seitz, C.; Skolarikos, A.; Straub, M.; Knoll, T. EAU Guidelines on Diagnosis and Conservative Management of Urolithiasis. Eur. Urol. 2016, 69, 468–474. [Google Scholar] [CrossRef]
- Gamage, K.N.; Jamnadass, E.; Sulaiman, S.K.; Pietropaolo, A.; Aboumarzouk, O.; Somani, B.K. The Role of Fluid Intake in the Prevention of Kidney Stone Disease: A Systematic Review over the Last Two Decades. Turk. J. Urol. 2020, 46 (Suppl. 1), S92–S103. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, Y.; Xiong, L.; Luo, Y.; Huang, Z.; Yi, B. Exercise Therapy Improves EGFR, and Reduces Blood Pressure and BMI in Non-Dialysis CKD Patients: Evidence from a Meta-Analysis. BMC Nephrol. 2019, 20, 398. [Google Scholar] [CrossRef] [Green Version]
- Mao, W.; Zhang, L.; Sun, S.; Wu, J.; Zou, X.; Zhang, G.; Chen, M. Physical Activity Reduces the Effect of High Body Mass Index on Kidney Stones in Diabetes Participants from the 2007–2018 NHANES Cycles: A Cross-Sectional Study. Front. Public Health 2022, 10, 936552. [Google Scholar] [CrossRef]
- Hill Gallant, K.M.; Spiegel, D.M. Calcium Balance in Chronic Kidney Disease. Curr. Osteoporos. Rep. 2017, 15, 214–221. [Google Scholar] [CrossRef]
- Taksande, S.R.; Worcester, E.M. Calcium Supplementation in Chronic Kidney Disease. Expert Opin. Drug Saf. 2014, 13, 1175–1185. [Google Scholar] [CrossRef]
- Kim, J.S.; Hwang, H.S. Vascular Calcification in Chronic Kidney Disease: Distinct Features of Pathogenesis and Clinical Implication. Korean Circ. J. 2021, 51, 961–982. [Google Scholar] [CrossRef] [PubMed]
- Robijn, S.; Vervaet, B.A.; Hoppe, B.; D’Haese, P.C.; Verhulst, A. Lanthanum Carbonate Inhibits Intestinal Oxalate Absorption and Prevents Nephrocalcinosis after Oxalate Loading in Rats. J. Urol. 2013, 189, 1960–1966. [Google Scholar] [CrossRef] [PubMed]
- Pozdzik, A.; David, C.; Vekeman, J.; Tielens, F.; Daudon, M. Lanthanum Carbonate to Control Plasma and Urinary Oxalate Level in Type 1 Primary Hyperoxaluria? IJU Case Rep. 2021, 4, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Biruete, A.; Hill Gallant, K.M.; Lindemann, S.R.; Wiese, G.N.; Chen, N.X.; Moe, S.M. Phosphate Binders and Non-Phosphate Effects in the Gastrointestinal Tract. J. Ren. Nutr. 2020, 30, 4–10. [Google Scholar] [CrossRef]
- Sarica, K.; Erturhan, S.; Altay, B. Effect of Verapamil on Urinary Stone-Forming Risk Factors. Urol. Res. 2007, 35, 23–27. [Google Scholar] [CrossRef]
- Teles, F.; Peçanha de Miranda Coelho, J.A.; Albino, R.M.; Verçosa Pacheco, F.C.; Rodrigues de Oliveira, E.; Silveira, M.A.D.; Diógenes, M.; Feitosa, A.; Bezerra, R. Effectiveness of Thiazide and Thiazide-like Diuretics in Advanced Chronic Kidney Disease: A Systematic Review and Meta-Analysis. Ren. Fail. 2023, 45, 2163903. [Google Scholar] [CrossRef]
- Li, D.F.; Gao, Y.L.; Liu, H.C.; Huang, X.C.; Zhu, R.F.; Zhu, C.T. Use of Thiazide Diuretics for the Prevention of Recurrent Kidney Calculi: A Systematic Review and Meta-Analysis. J. Transl. Med. 2020, 18, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todd Alexander, R.; McArthur, E.; Jandoc, R.; Welk, B.; Hayward, J.S.; Jain, A.K.; Braam, B.; Flockerzi, V.; Garg, A.X.; Quinn, R.R. Antihypertensive Medications and the Risk of Kidney Stones in Older Adults: A Retrospective Cohort Study. Hypertens. Res. 2017, 40, 837–842. [Google Scholar] [CrossRef]
- De la Espriella, R.; Cobo, M.; Núñez, J. Thiazides in Chronic Kidney Disease: “Back to the Future”. Clin. Kidney J. 2023, 16, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Riley, J.M.; Kim, H.; Averch, T.D.; Kim, H.J. Effect of magnesium on calcium and oxalate ion binding. J. Endourol. 2013, 27, 1487–1492. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, D.J.; Voss, S.; von Unruh, G.E.; Hesse, A. Importance of magnesium in absorption and excretion of oxalate. Urol. Int. 2005, 74, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Quiñones, H.; Hamdi, T.; Sakhaee, K.; Pasch, A.; Moe, O.W.; Pak, C.Y.C. Control of metabolic predisposition to cardiovascular complications of chronic kidney disease by effervescent calcium magnesium citrate: A feasibility study. J. Nephrol. 2019, 32, 93–100. [Google Scholar] [CrossRef] [Green Version]
- Vermeulen, E.A.; Vervloet, M.G. Magnesium Administration in Chronic Kidney Disease. Nutrients 2023, 15, 547. [Google Scholar] [CrossRef]
- Bressendorff, I.; Hansen, D.; Schou, M.; Silver, B.; Pasch, A.; Bouchelouche, P.; Pedersen, L.; Rasmussen, L.M.; Brandi, L. Oral Magnesium Supplementation in Chronic Kidney Disease Stages 3 and 4: Efficacy, Safety, and Effect on Serum Calcification Propensity-A Prospective Randomized Double-Blinded Placebo-Controlled Clinical Trial. Kidney Int. Rep. 2016, 2, 380–389. [Google Scholar] [CrossRef] [Green Version]
- Mydlík, M.; Derzsiová, K. Vitamin B6 and Oxalic Acid in Clinical Nephrology. J. Ren. Nutr. 2010, 20 (Suppl. 5), S95–S102. [Google Scholar] [CrossRef]
- Tavasoli, S.; Taheri, M. Vitamin D and Calcium Kidney Stones: A Review and a Proposal. Int. Urol. Nephrol. 2018, 51, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Ferraro, P.M.; Taylor, E.N.; Gambaro, G.; Curhan, G.C. Vitamin B6 Intake and the Risk of Incident Kidney Stones. Urolithiasis 2018, 46, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Wigner, P.; Bijak, M.; Saluk-Bijak, J. Probiotics in the Prevention of the Calcium Oxalate Urolithiasis. Cells 2022, 11, 284. [Google Scholar] [CrossRef]
- Klimesova, K.; Whittamore, J.M.; Hatch, M. Bifidobacterium Animalis Subsp. Lactis Decreases Urinary Oxalate Excretion in a Mouse Model of Primary Hyperoxaluria. Urolithiasis 2015, 43, 107–117. [Google Scholar] [CrossRef] [Green Version]
- Hiremath, S.; Viswanathan, P. Oxalobacter Formigenes: A New Hope as a Live Biotherapeutic Agent in the Management of Calcium Oxalate Renal Stones. Anaerobe 2022, 75, 102572. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Yang, H.; Zhu, X.; Li, Y.; Wang, N.; Han, S.; Xu, H.; Chen, Z.; Ye, Z. Oxalate-Degrading Enzyme Recombined Lactic Acid Bacteria Strains Reduce Hyperoxaluria. Urology 2018, 113, 253.e1–253.e7. [Google Scholar] [CrossRef] [PubMed]
- Burns, Z.; Knight, J.; Fargue, S.; Holmes, R.; Assimos, D.; Wood, K. Future Treatments for Hyperoxaluria. Curr. Opin. Urol. 2020, 30, 171–176. [Google Scholar] [CrossRef]
- Lieske, J.C.; Lingeman, J.E.; Ferraro, P.M.; Wyatt, C.M.; Tosone, C.; Kausz, A.T.; Knauf, F. Randomized Placebo-Controlled Trial of Reloxaliase in Enteric Hyperoxaluria. NEJM Evid. 2022, 1, 1–11. [Google Scholar] [CrossRef]
- Lingeman, J.E.; Pareek, G.; Easter, L.; Pease, R.; Grujic, D.; Brettman, L.; Langman, C.B. ALLN-177, Oral Enzyme Therapy for Hyperoxaluria. Int. Urol. Nephrol. 2019, 51, 601–608. [Google Scholar] [CrossRef] [Green Version]
- Pfau, A.; Grujic, D.; Keddis, M.T.; Kausz, A.T.; Lieske, J.C.; Knauf, F. Pilot Study of Reloxaliase in Patients with Severe Enteric Hyperoxaluria and Hyperoxalemia. Nephrol. Dial. Transplant. 2021, 36, 945–948. [Google Scholar] [CrossRef]
- Mufarrij, P.W.; Lange, J.N.; Knight, J.; Assimos, D.G.; Holmes, R.P. The Effects of Oxazyme on Oxalate Degradation: Results and Implications of in Vitro Experiments. J. Endourol. 2013, 27, 284–287. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stepanova, N. Oxalate Homeostasis in Non-Stone-Forming Chronic Kidney Disease: A Review of Key Findings and Perspectives. Biomedicines 2023, 11, 1654. https://doi.org/10.3390/biomedicines11061654
Stepanova N. Oxalate Homeostasis in Non-Stone-Forming Chronic Kidney Disease: A Review of Key Findings and Perspectives. Biomedicines. 2023; 11(6):1654. https://doi.org/10.3390/biomedicines11061654
Chicago/Turabian StyleStepanova, Natalia. 2023. "Oxalate Homeostasis in Non-Stone-Forming Chronic Kidney Disease: A Review of Key Findings and Perspectives" Biomedicines 11, no. 6: 1654. https://doi.org/10.3390/biomedicines11061654
APA StyleStepanova, N. (2023). Oxalate Homeostasis in Non-Stone-Forming Chronic Kidney Disease: A Review of Key Findings and Perspectives. Biomedicines, 11(6), 1654. https://doi.org/10.3390/biomedicines11061654