
H3G34-Mutant Gliomas—A Review of Molecular Pathogenesis and Therapeutic Options
 , ,
, ,
Abstract
:1. Introduction
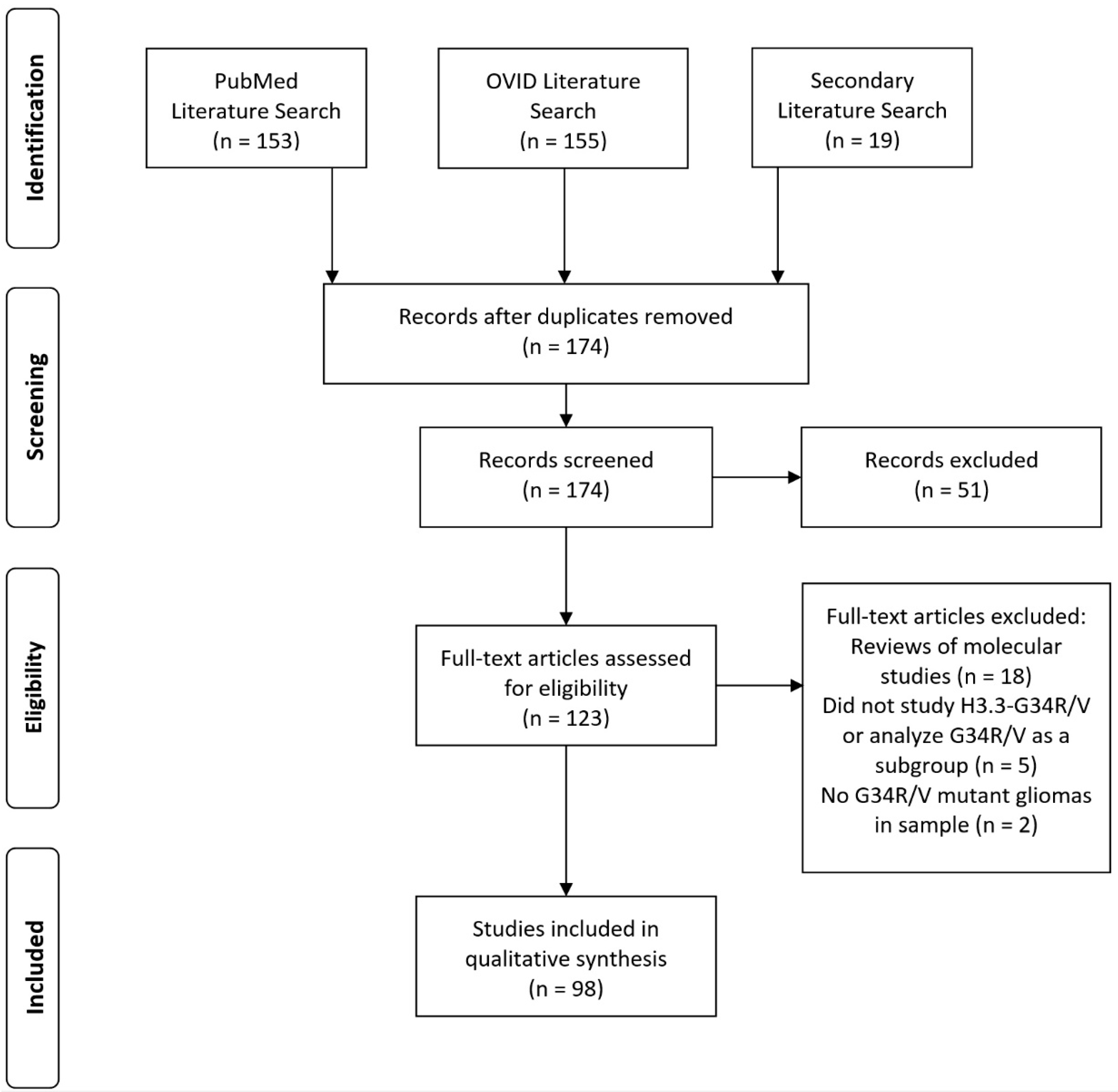
2. Methods
3. Literature Review
3.1. Brief Overview of Epigenetics
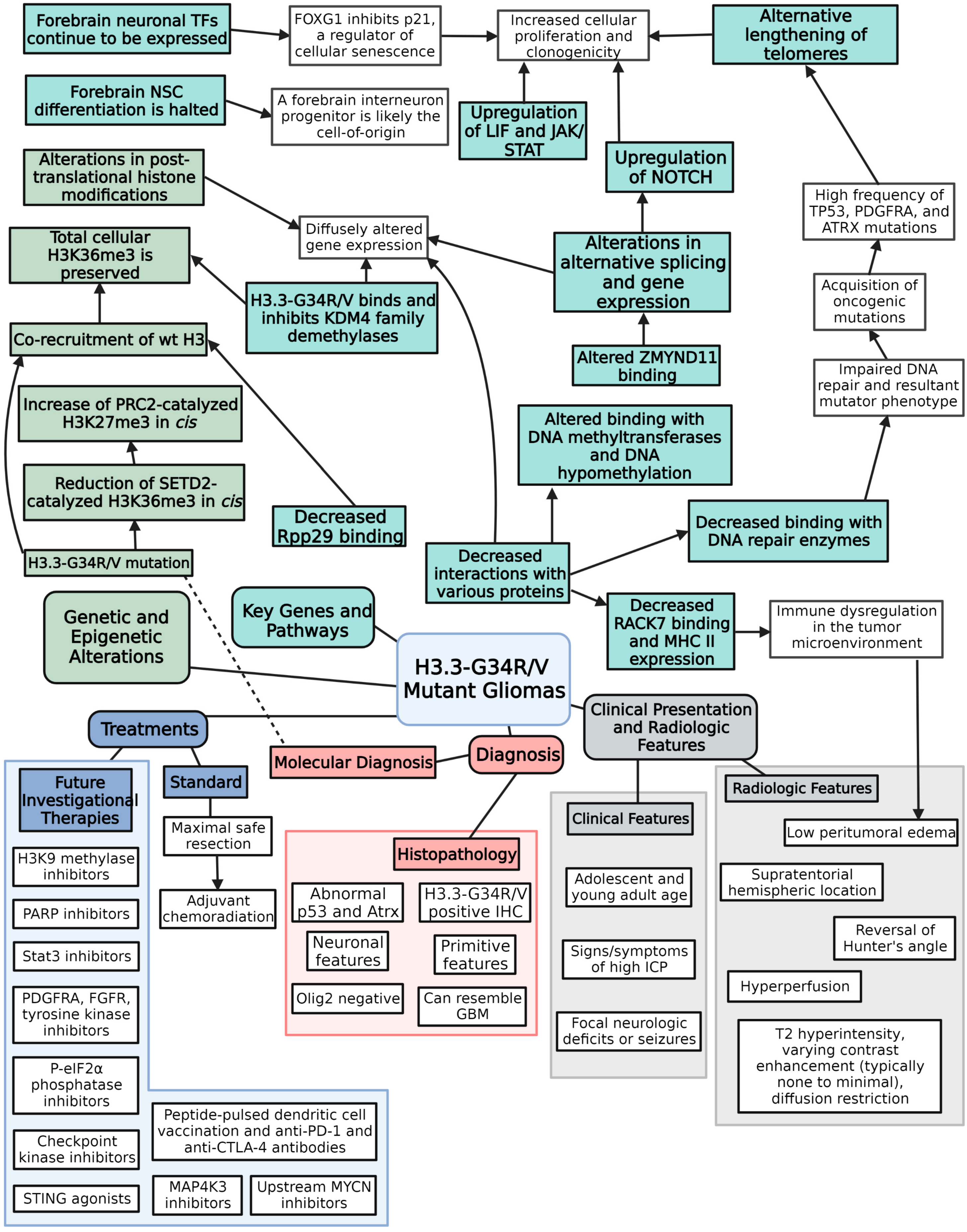
3.2. Histone H3.3-G34R/V Mutations
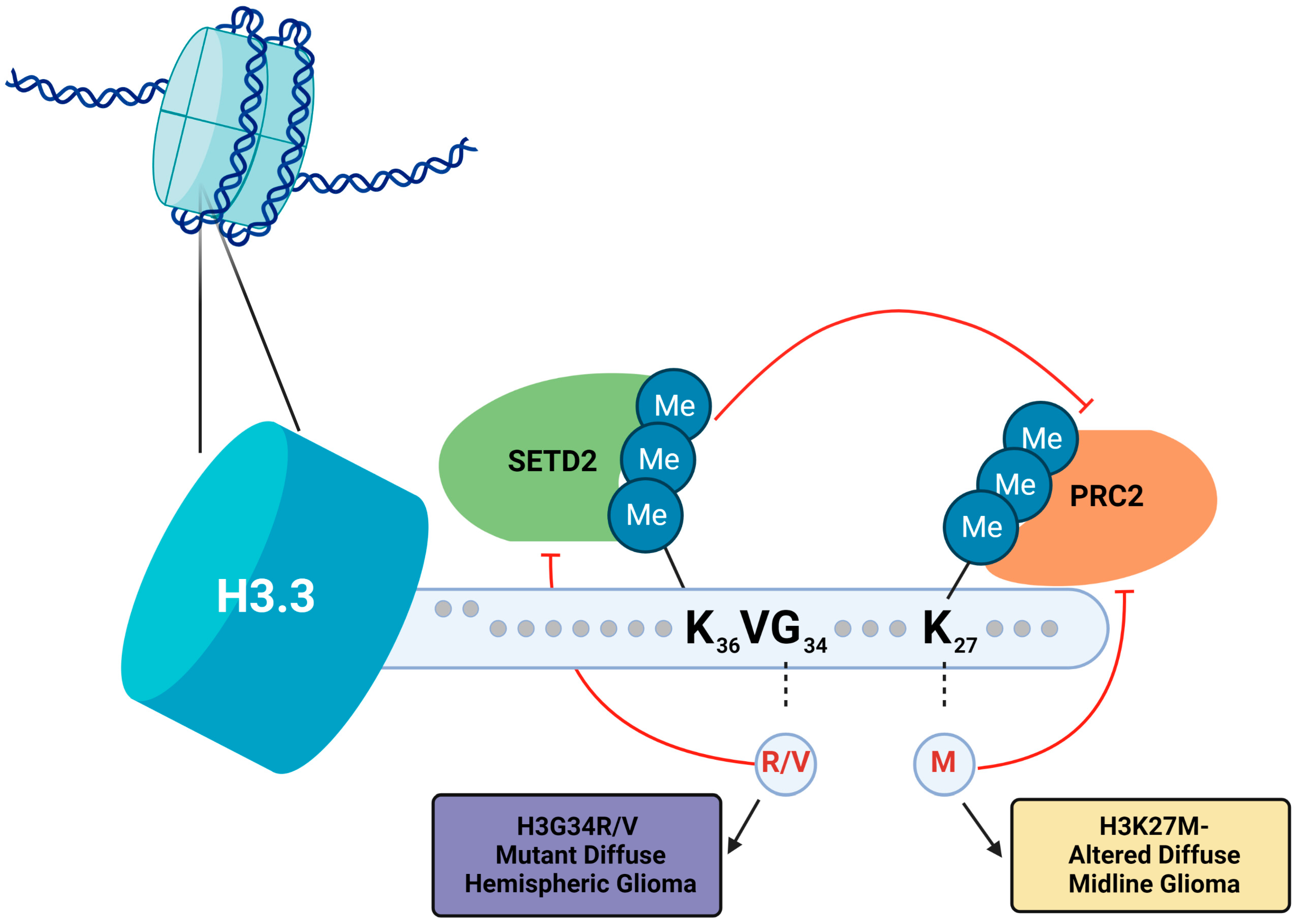
3.3. Histone H3.3-G34R/V Mutations Affect Post-Translational Modification of H3K36 and H3K27
3.4. Histone H3.3-G34R/V Mutations Affect DNA by Altering Methylation Patterns, Repair Mechanisms, and Genetic Stability
3.5. TP53, ATRX, and PDGFRA Are Heavily Implicated in Tumorigenesis of H3.3-G34R/V Mutant Gliomas
3.6. Forebrain Neuronal Identity Is a Critical Component of Tumorigenesis of H3.3-G34R/V Mutant Gliomas
3.7. Other Possible Key Proteins and Pathways Involved in Gliomagenesis
3.8. Baseline Information and Clinical Presentation of Patients with H3.3-G34R/V Mutant Gliomas
3.9. Radiologic Findings Common to H3.3-G34R/V Mutant Gliomas
3.10. Histopathologic Analysis and Subsequent Diagnosis of H3.3-G34R/V Mutant Gliomas
3.11. Treatment, Outcomes, and Factors Associated with Improved Prognosis in Patients with H3.3-G34R/V Mutant Gliomas
3.12. Possible Therapeutics and Clinical Trials (Future Directions)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ostrom, Q.T.; Cioffi, G.; Gittleman, H.; Patil, N.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2012–2016. Neuro-oncology 2019, 21, v1–v100. [Google Scholar] [CrossRef]
- Mackay, A.; Burford, A.; Carvalho, D.; Izquierdo, E.; Fazal-Salom, J.; Taylor, K.R.; Bjerke, L.; Clarke, M.; Vinci, M.; Nandhabalan, M.; et al. Integrated Molecular Meta-Analysis of 1,000 Pediatric High-Grade and Diffuse Intrinsic Pontine Glioma. Cancer Cell 2017, 32, 520–537.e5. [Google Scholar] [CrossRef] [Green Version]
- Korshunov, A.; Capper, D.; Reuss, D.; Schrimpf, D.; Ryzhova, M.; Hovestadt, V.; Sturm, D.; Meyer, J.; Jones, C.; Zheludkova, O.; et al. Histologically distinct neuroepithelial tumors with histone 3 G34 mutation are molecularly similar and comprise a single nosologic entity. Acta Neuropathol. 2016, 131, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Crowell, C.; Mata-Mbemba, D.; Bennett, J.; Matheson, K.; Mackley, M.; Perreault, S.; Erker, C. Systematic review of diffuse hemispheric glioma, H3 G34-mutant: Outcomes and associated clinical factors. Neuro-Oncol. Adv. 2022, 4, vdac133. [Google Scholar] [CrossRef]
- Gianno, F.; Giovannoni, I.; Cafferata, B.; Diomedi-Camassei, F.; Minasi, S.; Barresi, S.; Buttarelli, F.R.; Alesi, V.; Cardoni, A.; Antonelli, M.; et al. Paediatric-type diffuse high-grade gliomas in the 5th CNS WHO Classification. Pathologica 2022, 114, 422–435. [Google Scholar] [CrossRef] [PubMed]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.-Y.; Jones, D.T.W.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.-A.K.; Tönjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Broniscer, A.; McEachron, T.A.; Lu, C.; Paugh, B.S.; Becksfort, J.; Qu, C.; Ding, L.; Huether, R.; Parker, M.; et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet. 2012, 44, 251–253. [Google Scholar] [CrossRef] [Green Version]
- Sturm, D.; Witt, H.; Hovestadt, V.; Khuong-Quang, D.-A.; Jones, D.T.W.; Konermann, C.; Pfaff, E.; Tönjes, M.; Sill, M.; Bender, S.; et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 2012, 22, 425–437. [Google Scholar] [CrossRef] [Green Version]
- Lewis, P.W.; Müller, M.M.; Koletsky, M.S.; Cordero, F.; Lin, S.; Banaszynski, L.A.; Garcia, B.A.; Muir, T.W.; Becher, O.J.; Allis, C.D. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 2013, 340, 857–861. [Google Scholar] [CrossRef] [Green Version]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [Green Version]
- Barritault, M.; Meyronet, D.; Ducray, F. Molecular classification of adult gliomas: Recent advances and future perspectives. Curr. Opin. Oncol. 2018, 30, 375–382. [Google Scholar] [CrossRef]
- Weller, M.; Reifenberger, G. Beyond the World Health Organization classification of central nervous system tumors 2016: What are the new developments for gliomas from a clinician’s perspective? Curr. Opin. Neurol. 2020, 33, 701–706. [Google Scholar] [CrossRef]
- Huchedé, P.; Leblond, P.; Castets, M. The Intricate Epigenetic and Transcriptional Alterations in Pediatric High-Grade Gliomas: Targeting the Crosstalk as the Oncogenic Achilles’ Heel. Biomedicines 2022, 10, 1311. [Google Scholar] [CrossRef] [PubMed]
- Wood, M.D.; Halfpenny, A.M.; Moore, S.R. Applications of molecular neuro-oncology–a review of diffuse glioma integrated diagnosis and emerging molecular entities. Diagn. Pathol. 2019, 14, 29. [Google Scholar] [CrossRef] [Green Version]
- Di Nunno, V.; Franceschi, E.; Gatto, L.; Tosoni, A.; Bartolini, S.; Brandes, A.A. How to treat histone 3 altered gliomas: Molecular landscape and therapeutic developments. Expert Rev. Clin. Pharmacol. 2023, 16, 17–26. [Google Scholar] [CrossRef]
- Jones, C.; Karajannis, M.A.; Jones, D.T.W.; Kieran, M.W.; Monje, M.; Baker, S.J.; Becher, O.J.; Cho, Y.-J.; Gupta, N.; Hawkins, C.; et al. Pediatric high-grade glioma: Biologically and clinically in need of new thinking. Neuro-oncology 2017, 19, 153–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, L.; Han, J. Histone H3G34 Mutation in Brain and Bone Tumors. Adv. Exp. Med. Biol. 2021, 1283, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Crick, F. Central dogma of molecular biology. Nature 1970, 227, 561–563. [Google Scholar] [CrossRef]
- Tachiwana, H.; Osakabe, A.; Shiga, T.; Miya, Y.; Kimura, H.; Kagawa, W.; Kurumizaka, H. Structures of human nucleosomes containing major histone H3 variants. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 578–583. [Google Scholar] [CrossRef]
- Lowe, B.R.; Maxham, L.A.; Hamey, J.J.; Wilkins, M.R.; Partridge, J.F. Histone H3 Mutations: An Updated View of Their Role in Chromatin Deregulation and Cancer. Cancers 2019, 11, 660. [Google Scholar] [CrossRef] [Green Version]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Alaskhar Alhamwe, B.; Khalaila, R.; Wolf, J.; von Bülow, V.; Harb, H.; Alhamdan, F.; Hii, C.S.; Prescott, S.L.; Ferrante, A.; Renz, H.; et al. Histone modifications and their role in epigenetics of atopy and allergic diseases. Allergy Asthma Clin. Immunol. 2018, 14, 39. [Google Scholar] [CrossRef] [Green Version]
- Kanwal, R.; Gupta, S. Epigenetic modifications in cancer. Clin. Genet. 2012, 81, 303–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting epigenetic regulators for cancer therapy: Mechanisms and advances in clinical trials. Signal Transduct. Target. Ther. 2019, 4, 62. [Google Scholar] [CrossRef] [Green Version]
- Xiao, C.; Fan, T.; Tian, H.; Zheng, Y.; Zhou, Z.; Li, S.; Li, C.; He, J. H3K36 trimethylation-mediated biological functions in cancer. Clin. Epigenet. 2021, 13, 199. [Google Scholar] [CrossRef]
- Jain, S.U.; Khazaei, S.; Marchione, D.M.; Lundgren, S.M.; Wang, X.; Weinberg, D.N.; Deshmukh, S.; Juretic, N.; Lu, C.; Allis, C.D.; et al. Histone H3.3 G34 mutations promote aberrant PRC2 activity and drive tumor progression. Proc. Natl. Acad. Sci. USA 2020, 117, 27354–27364. [Google Scholar] [CrossRef]
- Zhang, Y.; Shan, C.-M.; Wang, J.; Bao, K.; Tong, L.; Jia, S. Molecular basis for the role of oncogenic histone mutations in modulating H3K36 methylation. Sci. Rep. 2017, 7, 43906. [Google Scholar] [CrossRef] [Green Version]
- Fang, J.; Huang, Y.; Mao, G.; Yang, S.; Rennert, G.; Gu, L.; Li, H.; Li, G.-M. Cancer-driving H3G34V/R/D mutations block H3K36 methylation and H3K36me3-MutSα interaction. Proc. Natl. Acad. Sci. USA 2018, 115, 9598–9603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Zheng, X.; Lu, C.; Li, G.-M.; Allis, C.D.; Li, H. Molecular basis for oncohistone H3 recognition by SETD2 methyltransferase. Genes Dev. 2016, 30, 1611–1616. [Google Scholar] [CrossRef] [Green Version]
- Huang, T.Y.-T.; Piunti, A.; Qi, J.; Morgan, M.; Bartom, E.; Shilatifard, A.; Saratsis, A.M. Effects of H3.3G34V mutation on genomic H3K36 and H3K27 methylation patterns in isogenic pediatric glioma cells. Acta Neuropathol. Commun. 2020, 8, 219. [Google Scholar] [CrossRef]
- Lemon, L.D.; Kannan, S.; Mo, K.W.; Adams, M.; Choi, H.G.; Gulka, A.O.D.; Withers, E.S.; Nurelegne, H.T.; Gomez, V.; Ambrocio, R.E.; et al. A Saccharomyces cerevisiae model and screen to define the functional consequences of oncogenic histone missense mutations. G3 Bethesda Md 2022, 12, jkac120. [Google Scholar] [CrossRef] [PubMed]
- Haase, S.; Banerjee, K.; Mujeeb, A.A.; Hartlage, C.S.; Núñez, F.M.; Núñez, F.J.; Alghamri, M.S.; Kadiyala, P.; Carney, S.; Barissi, M.N.; et al. H3.3-G34 mutations impair DNA repair and promote cGAS/STING-mediated immune responses in pediatric high-grade glioma models. J. Clin. Investig. 2022, 132, e154229. [Google Scholar] [CrossRef]
- Voon, H.P.J.; Udugama, M.; Lin, W.; Hii, L.; Law, R.H.P.; Steer, D.L.; Das, P.P.; Mann, J.R.; Wong, L.H. Inhibition of a K9/K36 demethylase by an H3.3 point mutation found in paediatric glioblastoma. Nat. Commun. 2018, 9, 3142. [Google Scholar] [CrossRef] [Green Version]
- Siddaway, R.; Canty, L.; Pajovic, S.; Milos, S.; Coyaud, E.; Sbergio, S.-G.; Vadivel Anguraj, A.K.; Lubanszky, E.; Yun, H.Y.; Portante, A.; et al. Oncohistone interactome profiling uncovers contrasting oncogenic mechanisms and identifies potential therapeutic targets in high grade glioma. Acta Neuropathol. 2022, 144, 1027–1048. [Google Scholar] [CrossRef] [PubMed]
- Fontebasso, A.M.; Schwartzentruber, J.; Khuong-Quang, D.-A.; Liu, X.-Y.; Sturm, D.; Korshunov, A.; Jones, D.T.W.; Witt, H.; Kool, M.; Albrecht, S.; et al. Mutations in SETD2 and genes affecting histone H3K36 methylation target hemispheric high-grade gliomas. Acta Neuropathol. 2013, 125, 659–669. [Google Scholar] [CrossRef] [Green Version]
- Yadav, R.K.; Jablonowski, C.M.; Fernandez, A.G.; Lowe, B.R.; Henry, R.A.; Finkelstein, D.; Barnum, K.J.; Pidoux, A.L.; Kuo, Y.-M.; Huang, J.; et al. Histone H3G34R mutation causes replication stress, homologous recombination defects and genomic instability in S. pombe. eLife 2017, 6, e27406. [Google Scholar] [CrossRef]
- Shastrula, P.K.; Lund, P.J.; Garcia, B.A.; Janicki, S.M. Rpp29 regulates histone H3.3 chromatin assembly through transcriptional mechanisms. J. Biol. Chem. 2018, 293, 12360–12377. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.; Park, J.H.; Baude, A.; Fellenberg, J.; Zustin, J.; Haller, F.; Krücken, I.; Kang, H.G.; Park, Y.J.; Plass, C.; et al. Transcriptome and protein interaction profiling in cancer cells with mutations in histone H3.3. Sci. Data 2018, 5, 180283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjerke, L.; Mackay, A.; Nandhabalan, M.; Burford, A.; Jury, A.; Popov, S.; Bax, D.A.; Carvalho, D.; Taylor, K.R.; Vinci, M.; et al. Histone H3.3. mutations drive pediatric glioblastoma through upregulation of MYCN. Cancer Discov. 2013, 3, 512–519. [Google Scholar] [CrossRef] [Green Version]
- Chan, K.M.; Han, J.; Fang, D.; Gan, H.; Zhang, Z. A lesson learned from the H3.3K27M mutation found in pediatric glioma: A new approach to the study of the function of histone modifications in vivo? Cell Cycle Georget. Tex 2013, 12, 2546–2552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, T.-G.; Zhou, Q.; Ma, X.-S.; Liu, X.-Y.; Meng, Q.-R.; Huang, X.-J.; Liu, H.-L.; Lei, W.-L.; Zhao, Z.-H.; Ouyang, Y.-C.; et al. PRC2 and EHMT1 regulate H3K27me2 and H3K27me3 establishment across the zygote genome. Nat. Commun. 2020, 11, 6354. [Google Scholar] [CrossRef]
- Bressan, R.B.; Southgate, B.; Ferguson, K.M.; Blin, C.; Grant, V.; Alfazema, N.; Wills, J.C.; Marques-Torrejon, M.A.; Morrison, G.M.; Ashmore, J.; et al. Regional identity of human neural stem cells determines oncogenic responses to histone H3.3 mutants. Cell Stem Cell 2021, 28, 877–893.e9. [Google Scholar] [CrossRef]
- Khazaei, S.; Chen, C.C.L.; Andrade, A.F.; Kabir, N.; Azarafshar, P.; Morcos, S.M.; França, J.A.; Lopes, M.; Lund, P.J.; Danieau, G.; et al. Single substitution in H3.3G34 alters DNMT3A recruitment to cause progressive neurodegeneration. Cell 2023, 186, 1162–1178.e20. [Google Scholar] [CrossRef] [PubMed]
- Funato, K.; Smith, R.C.; Saito, Y.; Tabar, V. Dissecting the impact of regional identity and the oncogenic role of human-specific NOTCH2NL in an hESC model of H3.3G34R-mutant glioma. Cell Stem Cell 2021, 28, 894–905.e7. [Google Scholar] [CrossRef]
- Lowe, B.R.; Yadav, R.K.; Henry, R.A.; Schreiner, P.; Matsuda, A.; Fernandez, A.G.; Finkelstein, D.; Campbell, M.; Kallappagoudar, S.; Jablonowski, C.M.; et al. Surprising phenotypic diversity of cancer-associated mutations of Gly 34 in the histone H3 tail. eLife 2021, 10, e65369. [Google Scholar] [CrossRef]
- Mahrez, W.; Arellano, M.S.T.; Moreno-Romero, J.; Nakamura, M.; Shu, H.; Nanni, P.; Köhler, C.; Gruissem, W.; Hennig, L. H3K36ac Is an Evolutionary Conserved Plant Histone Modification That Marks Active Genes. Plant Physiol. 2016, 170, 1566–1577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, S.A.; Rao, B.; Garcia, B.A.; Hake, S.B.; Diaz, R.L.; Shabanowitz, J.; Hunt, D.F.; Allis, C.D.; Lieb, J.D.; Strahl, B.D. Identification of histone H3 lysine 36 acetylation as a highly conserved histone modification. J. Biol. Chem. 2007, 282, 7632–7640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sweha, S.R.; Chung, C.; Natarajan, S.K.; Panwalkar, P.; Pun, M.; Ghali, A.; Bayliss, J.; Pratt, D.; Shankar, A.; Ravikumar, V.; et al. Epigenetically defined therapeutic targeting in H3.3G34R/V high-grade gliomas. Sci. Transl. Med. 2021, 13, eabf7860. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Botero, G.; Giry, M.; Mokhtari, K.; Labussière, M.; Idbaih, A.; Delattre, J.-Y.; Laigle-Donadey, F.; Sanson, M. Molecular analysis of diffuse intrinsic brainstem gliomas in adults. J. Neurooncol. 2014, 116, 405–411. [Google Scholar] [CrossRef]
- Reuss, D.E.; Kratz, A.; Sahm, F.; Capper, D.; Schrimpf, D.; Koelsche, C.; Hovestadt, V.; Bewerunge-Hudler, M.; Jones, D.T.W.; Schittenhelm, J.; et al. Adult IDH wild type astrocytomas biologically and clinically resolve into other tumor entities. Acta Neuropathol. 2015, 130, 407–417. [Google Scholar] [CrossRef] [Green Version]
- Hu, W.; Duan, H.; Zhong, S.; Zeng, J.; Mou, Y. High frequency of PDGFRA and MUC family gene mutations in diffuse hemispheric glioma, H3 G34-mutant: A glimmer of hope? J. Transl. Med. 2022, 20, 64. [Google Scholar] [CrossRef] [PubMed]
- Fontebasso, A.M.; Papillon-Cavanagh, S.; Schwartzentruber, J.; Nikbakht, H.; Gerges, N.; Fiset, P.-O.; Bechet, D.; Faury, D.; De Jay, N.; Ramkissoon, L.A.; et al. Recurrent somatic mutations in ACVR1 in pediatric midline high-grade astrocytoma. Nat. Genet. 2014, 46, 462–466. [Google Scholar] [CrossRef] [Green Version]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 mutations in human cancers: Origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2010, 2, a001008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vuong, H.G.; Le, H.T.; Dunn, I.F. The prognostic significance of further genotyping H3G34 diffuse hemispheric gliomas. Cancer 2022, 128, 1907–1912. [Google Scholar] [CrossRef]
- Udugama, M.; Hii, L.; Garvie, A.; Cervini, M.; Vinod, B.; Chan, F.-L.; Das, P.P.; Mann, J.R.; Collas, P.; Voon, H.P.J.; et al. Mutations inhibiting KDM4B drive ALT activation in ATRX-mutated glioblastomas. Nat. Commun. 2021, 12, 2584. [Google Scholar] [CrossRef]
- Abdallah, A.S.; Cardona, H.J.; Gadd, S.L.; Brat, D.J.; Powla, P.P.; Alruwalli, W.S.; Shen, C.; Picketts, D.J.; Li, X.-N.; Becher, O.J. Novel genetically engineered H3.3G34R model reveals cooperation with ATRX loss in upregulation of Hoxa cluster genes and promotion of neuronal lineage. Neuro-Oncol. Adv. 2023, 5, vdad003. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.L.; Deshmukh, S.; Jessa, S.; Hadjadj, D.; Lisi, V.; Andrade, A.F.; Faury, D.; Jawhar, W.; Dali, R.; Suzuki, H.; et al. Histone H3.3G34-Mutant Interneuron Progenitors Co-opt PDGFRA for Gliomagenesis. Cell 2020, 183, 1617–1633.e22. [Google Scholar] [CrossRef]
- McNicholas, M.; De Cola, A.; Bashardanesh, Z.; Foss, A.; Lloyd, C.B.; Hebert, S.; Faury, D.; Andrade, A.F.; Jabado, N.; Kleinman, C.L.; et al. A Compendium of Syngeneic, Transplantable Pediatric High-Grade Glioma Models Reveals Subtype-Specific Therapeutic Vulnerabilities. Cancer Discov. 2023, 13, 1592–1615. [Google Scholar] [CrossRef]
- Marker, D.F.; Agnihotri, S.; Amankulor, N.; Murdoch, G.H.; Pearce, T.M. The dominant TP53 hotspot mutation in IDH -mutant astrocytoma, R273C, has distinctive pathologic features and sex-specific prognostic implications. Neuro-Oncol. Adv. 2022, 4, vdab182. [Google Scholar] [CrossRef]
- Sasaki, S.; Tomomasa, R.; Nobusawa, S.; Hirato, J.; Uchiyama, T.; Boku, E.; Miyasaka, T.; Hirose, T.; Ohbayashi, C. Anaplastic pleomorphic xanthoastrocytoma associated with an H3G34 mutation: A case report with review of literature. Brain Tumor Pathol. 2019, 36, 169–173. [Google Scholar] [CrossRef]
- Cheng, Y.; Bao, W.; Wu, Q. Cerebral hemispheric glioblastoma with PNET-like morphology and histone H3.3 G34 mutation in younger patients: Report of three rare cases and diagnostic pitfalls. Indian J. Pathol. Microbiol. 2020, 63, 262–266. [Google Scholar] [CrossRef] [PubMed]
- Crotty, E.E.; Leary, S.E.S.; Geyer, J.R.; Olson, J.M.; Millard, N.E.; Sato, A.A.; Ermoian, R.P.; Cole, B.L.; Lockwood, C.M.; Paulson, V.A.; et al. Children with DIPG and high-grade glioma treated with temozolomide, irinotecan, and bevacizumab: The Seattle Children’s Hospital experience. J. Neurooncol. 2020, 148, 607–617. [Google Scholar] [CrossRef]
- Morris, M.; Driscoll, M.; Henson, J.W.; Cobbs, C.; Jiang, L.; Gocke, C.D.; Chen, L.; Rodriguez, F.J. Low-Grade Gemistocytic Morphology in H3 G34R-Mutant Gliomas and Concurrent K27M Mutation: Clinicopathologic Findings. J. Neuropathol. Exp. Neurol. 2020, 79, 1038–1043. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.Y.; Won, J.K.; Park, C.-K.; Kim, S.-K.; Choi, S.H.; Kim, T.; Yun, H.; Park, S.-H. H3 G34-mutant high-grade glioma. Brain Tumor Pathol. 2021, 38, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Gianno, F.; Antonelli, M.; Di Dio, T.; Minasi, S.; Donofrio, V.; Buccoliero, A.M.; Gardiman, M.P.; Pollo, B.; Diomedi Camassei, F.; Rossi, S.; et al. Correlation Between Immunohistochemistry and Sequencing in H3G34-Mutant Gliomas. Am. J. Surg. Pathol. 2021, 45, 200–204. [Google Scholar] [CrossRef]
- Wang, L.; Shao, L.; Li, H.; Yao, K.; Duan, Z.; Zhi, C.; Song, S.; Cheng, Y.; Wang, F.; Wang, W.; et al. Histone H3.3 G34-mutant Diffuse Gliomas in Adults. Am. J. Surg. Pathol. 2022, 46, 249–257. [Google Scholar] [CrossRef]
- Lucas, C.-H.G.; Mueller, S.; Reddy, A.; Taylor, J.W.; Oberheim Bush, N.A.; Clarke, J.L.; Chang, S.M.; Gupta, N.; Berger, M.S.; Perry, A.; et al. Diffuse hemispheric glioma, H3 G34-mutant: Genomic landscape of a new tumor entity and prospects for targeted therapy. Neuro-oncology 2021, 23, 1974–1976. [Google Scholar] [CrossRef]
- Trejo-Lopez, J.A.; Praska, C.E.; Zepeda Mendoza, C.; Kollmeyer, T.M.; Raghunathan, A.; Giannini, C.; Vaubel, R.A.; Nguyen, A.; Jentoft, M.E.; Donev, K.; et al. H3 G34 mutation assessment for diffuse gliomas in adults: When would testing be most diagnostically useful? J. Neuropathol. Exp. Neurol. 2022, 82, 93–95. [Google Scholar] [CrossRef]
- Yu, N.; Lee, H.S.; Raslan, O.A.; Jin, L.-W.; Aboud, O. H3G34-mutant diffuse hemispheric glioma with osseous metastases: A case report and literature review. CNS Oncol. 2023, 12, CNS95. [Google Scholar] [CrossRef]
- Bedics, G.; Kotmayer, L.; Zajta, E.; Hegyi, L.L.; Brückner, E.Á.; Rajnai, H.; Reiniger, L.; Bödör, C.; Garami, M.; Scheich, B. Germline MUTYH mutations and high-grade gliomas: Novel evidence for a potential association. Genes. Chromosomes Cancer 2022, 61, 622–628. [Google Scholar] [CrossRef]
- Hwang, E.I.; Kool, M.; Burger, P.C.; Capper, D.; Chavez, L.; Brabetz, S.; Williams-Hughes, C.; Billups, C.; Heier, L.; Jaju, A.; et al. Extensive Molecular and Clinical Heterogeneity in Patients with Histologically Diagnosed CNS-PNET Treated as a Single Entity: A Report From the Children’s Oncology Group Randomized ACNS0332 Trial. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2018, 36, JCO2017764720. [Google Scholar] [CrossRef]
- Korshunov, A.; Okonechnikov, K.; Schmitt-Hoffner, F.; Ryzhova, M.; Sahm, F.; Stichel, D.; Schrimpf, D.; Reuss, D.E.; Sievers, P.; Suwala, A.K.; et al. Molecular analysis of pediatric CNS-PNET revealed nosologic heterogeneity and potent diagnostic markers for CNS neuroblastoma with FOXR2-activation. Acta Neuropathol. Commun. 2021, 9, 20. [Google Scholar] [CrossRef] [PubMed]
- Onishi, S.; Amatya, V.J.; Karlowee, V.; Takeshima, Y.; Sugiyama, K.; Kurisu, K.; Yamasaki, F. Radiological and Immunostaining Characteristics of H3.3 G34R-Mutant Glioma: A Report of 3 Cases and Review of the Literature. Pediatr. Neurosurg. 2020, 55, 319–325. [Google Scholar] [CrossRef]
- Picart, T.; Barritault, M.; Poncet, D.; Berner, L.-P.; Izquierdo, C.; Tabouret, E.; Figarella-Branger, D.; Idbaïh, A.; Bielle, F.; Bourg, V.; et al. Characteristics of diffuse hemispheric gliomas, H3 G34-mutant in adults. Neuro-Oncol. Adv. 2021, 3, vdab061. [Google Scholar] [CrossRef] [PubMed]
- Lavrador, J.P.; Reisz, Z.; Sibtain, N.; Rajwani, K.; Baig Mirza, A.; Vergani, F.; Gullan, R.; Bhangoo, R.; Ashkan, K.; Bleil, C.; et al. H3 G34-mutant high-grade gliomas: Integrated clinical, imaging and pathological characterisation of a single-centre case series. Acta Neurochir. 2023, 165, 1615–1633. [Google Scholar] [CrossRef]
- Yoshimoto, K.; Hatae, R.; Sangatsuda, Y.; Suzuki, S.O.; Hata, N.; Akagi, Y.; Kuga, D.; Hideki, M.; Yamashita, K.; Togao, O.; et al. Prevalence and clinicopathological features of H3.3 G34-mutant high-grade gliomas: A retrospective study of 411 consecutive glioma cases in a single institution. Brain Tumor Pathol. 2017, 34, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Neumann, J.E.; Dorostkar, M.M.; Korshunov, A.; Mawrin, C.; Koch, A.; Giese, A.; Schüller, U. Distinct Histomorphology in Molecular Subgroups of Glioblastomas in Young Patients. J. Neuropathol. Exp. Neurol. 2016, 75, 408–414. [Google Scholar] [CrossRef] [Green Version]
- Ebrahimi, A.; Skardelly, M.; Bonzheim, I.; Ott, I.; Mühleisen, H.; Eckert, F.; Tabatabai, G.; Schittenhelm, J. ATRX immunostaining predicts IDH and H3F3A status in gliomas. Acta Neuropathol. Commun. 2016, 4, 60. [Google Scholar] [CrossRef] [Green Version]
- Salloum, R.; McConechy, M.K.; Mikael, L.G.; Fuller, C.; Drissi, R.; DeWire, M.; Nikbakht, H.; De Jay, N.; Yang, X.; Boue, D.; et al. Characterizing temporal genomic heterogeneity in pediatric high-grade gliomas. Acta Neuropathol. Commun. 2017, 5, 78. [Google Scholar] [CrossRef]
- Minasi, S.; Baldi, C.; Gianno, F.; Antonelli, M.; Buccoliero, A.M.; Pietsch, T.; Massimino, M.; Buttarelli, F.R. Alternative lengthening of telomeres in molecular subgroups of paediatric high-grade glioma. Childs Nerv. Syst. ChNS Off. J. Int. Soc. Pediatr. Neurosurg. 2021, 37, 809–818. [Google Scholar] [CrossRef]
- Cooley, L.D.; Lansdon, L.A.; Laurence, K.; Herriges, J.C.; Zhang, L.; Repnikova, E.A.; Joyce, J.; Thakor, P.; Warren, L.; Smith, S.C.; et al. Integrated genetic profiling of archival pediatric high-grade glial tumors and reassessment with 2021 WHO classification of paediatric CNS tumours. Cancer Genet. 2023, 274–275, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Pathak, P.; Jha, P.; Purkait, S.; Sharma, V.; Suri, V.; Sharma, M.C.; Faruq, M.; Suri, A.; Sarkar, C. Altered global histone-trimethylation code and H3F3A-ATRX mutation in pediatric GBM. J. Neurooncol. 2015, 121, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Diaz, A.K.; Paugh, B.S.; Rankin, S.L.; Ju, B.; Li, Y.; Zhu, X.; Qu, C.; Chen, X.; Zhang, J.; et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat. Genet. 2014, 46, 444–450. [Google Scholar] [CrossRef] [Green Version]
- Andreiuolo, F.; Lisner, T.; Zlocha, J.; Kramm, C.; Koch, A.; Bison, B.; Gareton, A.; Zanello, M.; Waha, A.; Varlet, P.; et al. H3F3A-G34R mutant high grade neuroepithelial neoplasms with glial and dysplastic ganglion cell components. Acta Neuropathol. Commun. 2019, 7, 78. [Google Scholar] [CrossRef] [Green Version]
- Recurrent MET fusion genes represent a drug target in pediatric glioblastoma. Nat. Med. 2016, 22, 1314–1320. [CrossRef] [PubMed]
- Paugh, B.S.; Zhu, X.; Qu, C.; Endersby, R.; Diaz, A.K.; Zhang, J.; Bax, D.A.; Carvalho, D.; Reis, R.M.; Onar-Thomas, A.; et al. Novel oncogenic PDGFRA mutations in pediatric high-grade gliomas. Cancer Res. 2013, 73, 6219–6229. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, D.; Mackay, A.; Bjerke, L.; Grundy, R.G.; Lopes, C.; Reis, R.M.; Jones, C. The prognostic role of intragenic copy number breakpoints and identification of novel fusion genes in paediatric high grade glioma. Acta Neuropathol. Commun. 2014, 2, 23. [Google Scholar] [CrossRef] [Green Version]
- Puget, S.; Philippe, C.; Bax, D.A.; Job, B.; Varlet, P.; Junier, M.-P.; Andreiuolo, F.; Carvalho, D.; Reis, R.; Guerrini-Rousseau, L.; et al. Mesenchymal transition and PDGFRA amplification/mutation are key distinct oncogenic events in pediatric diffuse intrinsic pontine gliomas. PLoS ONE 2012, 7, e30313. [Google Scholar] [CrossRef] [Green Version]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Nambirajan, A.; Gurung, N.; Suri, V.; Sarkar, C.; Kumar, A.; Singh, M.; Sharma, M.C. C19MC amplification and expression of Lin28A and Olig2 in the classification of embryonal tumors of the central nervous system: A 14-year retrospective study from a tertiary care center. Childs Nerv. Syst. ChNS Off. J. Int. Soc. Pediatr. Neurosurg. 2021, 37, 1067–1075. [Google Scholar] [CrossRef]
- Schäfer, S.; Behling, F.; Skardelly, M.; Koch, M.; Ott, I.; Paulsen, F.; Tabatabai, G.; Schittenhelm, J. Low FoxG1 and high Olig-2 labelling indices define a prognostically favourable subset in isocitrate dehydrogenase (IDH)-mutant gliomas. Neuropathol. Appl. Neurobiol. 2018, 44, 207–223. [Google Scholar] [CrossRef]
- Kumari, R.; Jat, P. Mechanisms of Cellular Senescence: Cell Cycle Arrest and Senescence Associated Secretory Phenotype. Front. Cell Dev. Biol. 2021, 9, 645593. [Google Scholar] [CrossRef]
- Wichterle, H.; Garcia-Verdugo, J.M.; Herrera, D.G.; Alvarez-Buylla, A. Young neurons from medial ganglionic eminence disperse in adult and embryonic brain. Nat. Neurosci. 1999, 2, 461–466. [Google Scholar] [CrossRef]
- Pan, P.; Bale, T.; Miller, A.; Ladanyi, M.; Rosenblum, M.; Haggiagi, A. PATH-17. NOVEL DUAL HISTONE 3 K27M AND G34R POINT MUTATIONS IN A MIDLINE GLIOMA. Neuro-oncology 2020, 22, ii167. [Google Scholar] [CrossRef]
- Nery, S.; Fishell, G.; Corbin, J.G. The caudal ganglionic eminence is a source of distinct cortical and subcortical cell populations. Nat. Neurosci. 2002, 5, 1279–1287. [Google Scholar] [CrossRef]
- Trabelsi, S.; Chabchoub, I.; Ksira, I.; Karmeni, N.; Mama, N.; Kanoun, S.; Burford, A.; Jury, A.; Mackay, A.; Popov, S.; et al. Molecular Diagnostic and Prognostic Subtyping of Gliomas in Tunisian Population. Mol. Neurobiol. 2017, 54, 2381–2394. [Google Scholar] [CrossRef]
- Mefford, H.C.; Sharp, A.J.; Baker, C.; Itsara, A.; Jiang, Z.; Buysse, K.; Huang, S.; Maloney, V.K.; Crolla, J.A.; Baralle, D.; et al. Recurrent rearrangements of chromosome 1q21.1 and variable pediatric phenotypes. N. Engl. J. Med. 2008, 359, 1685–1699. [Google Scholar] [CrossRef] [Green Version]
- van Bever, Y.; Rooms, L.; Laridon, A.; Reyniers, E.; van Luijk, R.; Scheers, S.; Wauters, J.; Kooy, R.F. Clinical report of a pure subtelomeric 1qter deletion in a boy with mental retardation and multiple anomalies adds further evidence for a specific phenotype. Am. J. Med. Genet. A 2005, 135, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Aaroe, A.; Liang, J.; Puduvalli, V.K. Tumor microenvironment in glioblastoma: Current and emerging concepts. Neuro-Oncol. Adv. 2023, 5, vdad009. [Google Scholar] [CrossRef] [PubMed]
- Jiao, F.; Li, Z.; He, C.; Xu, W.; Yang, G.; Liu, T.; Shen, H.; Cai, J.; Anastas, J.N.; Mao, Y.; et al. RACK7 recognizes H3.3G34R mutation to suppress expression of MHC class II complex components and their delivery pathway in pediatric glioblastoma. Sci. Adv. 2020, 6, eaba2113. [Google Scholar] [CrossRef] [PubMed]
- Takamura, Y.; Ikeda, H.; Kanaseki, T.; Toyota, M.; Tokino, T.; Imai, K.; Houkin, K.; Sato, N. Regulation of MHC class II expression in glioma cells by class II transactivator (CIITA). Glia 2004, 45, 392–405. [Google Scholar] [CrossRef]
- Chih, Y.; Sahm, K.; Sadik, A.; Bunse, T.; Trautwein, N.; Pusch, S.; Stevanovic, S.; Opitz, C.; Deimling, A.; Wick, W.; et al. KS01.3.A Tumoral MHC class II expression in gliomas drives T cell exhaustion. Neuro Oncol 2021, 23, ii3. [Google Scholar] [CrossRef]
- Zagzag, D.; Salnikow, K.; Chiriboga, L.; Yee, H.; Lan, L.; Ali, M.A.; Garcia, R.; Demaria, S.; Newcomb, E.W. Downregulation of major histocompatibility complex antigens in invading glioma cells: Stealth invasion of the brain. Lab. Investig. J. Tech. Methods Pathol. 2005, 85, 328–341. [Google Scholar] [CrossRef] [Green Version]
- Graf, U.; Casanova, E.A.; Cinelli, P. The Role of the Leukemia Inhibitory Factor (LIF)–Pathway in Derivation and Maintenance of Murine Pluripotent Stem Cells. Genes 2011, 2, 280–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lutsik, P.; Baude, A.; Mancarella, D.; Öz, S.; Kühn, A.; Toth, R.; Hey, J.; Toprak, U.H.; Lim, J.; Nguyen, V.H.; et al. Globally altered epigenetic landscape and delayed osteogenic differentiation in H3.3-G34W-mutant giant cell tumor of bone. Nat. Commun. 2020, 11, 5414. [Google Scholar] [CrossRef] [PubMed]
- Colli, S.L.; Cardoso, N.; Massone, C.A.; Cores, M.; García Lombardi, M.; De Matteo, E.N.; Lorenzetti, M.A.; Preciado, M.V. Molecular alterations in the integrated diagnosis of pediatric glial and glioneuronal tumors: A single center experience. PLoS ONE 2022, 17, e0266466. [Google Scholar] [CrossRef]
- Gessi, M.; Gielen, G.H.; Hammes, J.; Dörner, E.; Mühlen, A.Z.; Waha, A.; Pietsch, T. H3.3 G34R mutations in pediatric primitive neuroectodermal tumors of central nervous system (CNS-PNET) and pediatric glioblastomas: Possible diagnostic and therapeutic implications? J. Neurooncol. 2013, 112, 67–72. [Google Scholar] [CrossRef]
- Zhang, R.-Q.; Shi, Z.; Chen, H.; Chung, N.Y.-F.; Yin, Z.; Li, K.K.-W.; Chan, D.T.-M.; Poon, W.S.; Wu, J.; Zhou, L.; et al. Biomarker-based prognostic stratification of young adult glioblastoma. Oncotarget 2016, 7, 5030–5041. [Google Scholar] [CrossRef] [Green Version]
- Jethanandani, A.; Gule-Monroe, M.K.; Chen, M.; Johnson, J.M. Extraneural Metastases From a High-Grade Glioma (HGG) with an H3F3A G34R Mutation. Front. Oncol. 2019, 9, 373. [Google Scholar] [CrossRef]
- Kay, M.D.; Pariury, H.E.; Perry, A.; Winegar, B.A.; Kuo, P.H. Extracranial Metastases From Glioblastoma with Primitive Neuronal Components on FDG PET/CT. Clin. Nucl. Med. 2020, 45, e162–e164. [Google Scholar] [CrossRef]
- Roux, A.; Pallud, J.; Saffroy, R.; Edjlali-Goujon, M.; Debily, M.-A.; Boddaert, N.; Sanson, M.; Puget, S.; Knafo, S.; Adam, C.; et al. High-grade gliomas in adolescents and young adults highlight histomolecular differences from their adult and pediatric counterparts. Neuro-oncology 2020, 22, 1190–1202. [Google Scholar] [CrossRef] [PubMed]
- Gilani, A.; Kleinschmidt-DeMasters, B.K. Morphological diversity in H3G34-mutant high-grade gliomas: Ganglionic and epithelioid features. Clin. Neuropathol. 2021, 40, 4–10. [Google Scholar] [CrossRef]
- Oliveira, V.F.; De Sousa, G.R.; Dos Santos, A.C.; Saggioro, F.P.; Machado, H.R.; de Oliveira, R.S.; Tone, L.G.; Valera, E.T. Evaluating H3F3A K27M and G34R/V somatic mutations in a cohort of pediatric brain tumors of different and rare histologies. Childs Nerv. Syst. ChNS Off. J. Int. Soc. Pediatr. Neurosurg. 2021, 37, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Mohiuddin, S.; Maraka, S.; Usman Baig, M.; Gupta, S.; Muzzafar, T.; Valyi-Nagy, T.; Lindsay, H.; Moody, K.; Razvi, S.; Paulino, A.; et al. Case series of diffuse extraneural metastasis in H3F3A mutant high-grade gliomas: Clinical, molecular phenotype and literature review. J. Clin. Neurosci. Off. J. Neurosurg. Soc. Australas. 2021, 89, 405–411. [Google Scholar] [CrossRef]
- Schoof, M.; Kordes, U.; Volk, A.E.; Al-Kershi, S.; Kresbach, C.; Schüller, U. Malignant gliomas with H3F3A G34R mutation or MYCN amplification in pediatric patients with Li Fraumeni syndrome. Acta Neuropathol. 2021, 142, 591–593. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Chen, Y.; Wang, L.; Gao, Y.; Xu, J. The clinicopathological features and prognosis of multifocal high-grade gliomas in adults with H3F3A mutation. Neurosci. Riyadh Saudi Arab. 2023, 28, 42–47. [Google Scholar] [CrossRef]
- Wood, M.D.; Neff, T.; Nickerson, J.P.; Sayama, C.; Raslan, A.M.; Ambady, P.; Corless, C.L.; Nazemi, K.J. Post-treatment hypermutation in a recurrent diffuse glioma with H3.3 p.G34 Mutation. Neuropathol. Appl. Neurobiol. 2021, 47, 460–463. [Google Scholar] [CrossRef]
- Hodgson, J.M.; Douch, C.; Hartley, L.; Merve, A.; Devadass, A.; Chatterjee, F. Problem solving in clinical practice: An unusual cause of multifocal brain lesions. Arch. Dis. Child. Educ. Pract. Ed. 2021, 106, 299–303. [Google Scholar] [CrossRef]
- Korshunov, A.; Ryzhova, M.; Hovestadt, V.; Bender, S.; Sturm, D.; Capper, D.; Meyer, J.; Schrimpf, D.; Kool, M.; Northcott, P.A.; et al. Integrated analysis of pediatric glioblastoma reveals a subset of biologically favorable tumors with associated molecular prognostic markers. Acta Neuropathol. 2015, 129, 669–678. [Google Scholar] [CrossRef]
- Kurokawa, R.; Baba, A.; Kurokawa, M.; Pinarbasi, E.S.; Makise, N.; Ota, Y.; Kim, J.; Srinivasan, A.; Moritani, T. Neuroimaging features of diffuse hemispheric glioma, H3 G34-mutant: A case series and systematic review. J. Neuroimaging Off. J. Am. Soc. Neuroimaging 2022, 32, 17–27. [Google Scholar] [CrossRef]
- Kalelioglu, T.; Emerson, D.; Luk, A.; Lopes, B.; Patel, S.H. Imaging features of diffuse hemispheric glioma, H3 G34-mutant: Report of 4 cases. J. Neuroradiol. J. Neuroradiol. 2022, 50, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Grill, J.; Massimino, M.; Bouffet, E.; Azizi, A.A.; McCowage, G.; Cañete, A.; Saran, F.; Le Deley, M.-C.; Varlet, P.; Morgan, P.S.; et al. Phase II, Open-Label, Randomized, Multicenter Trial (HERBY) of Bevacizumab in Pediatric Patients with Newly Diagnosed High-Grade Glioma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2018, 36, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Hummel, T.R.; Salloum, R.; Drissi, R.; Kumar, S.; Sobo, M.; Goldman, S.; Pai, A.; Leach, J.; Lane, A.; Pruitt, D.; et al. A pilot study of bevacizumab-based therapy in patients with newly diagnosed high-grade gliomas and diffuse intrinsic pontine gliomas. J. Neurooncol. 2016, 127, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez Gutierrez, D.; Jones, C.; Varlet, P.; Mackay, A.; Warren, D.; Warmuth-Metz, M.; Aliaga, E.S.; Calmon, R.; Hargrave, D.R.; Cañete, A.; et al. Radiological Evaluation of Newly Diagnosed Non-Brainstem Pediatric High-Grade Glioma in the HERBY Phase II Trial. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2020, 26, 1856–1865. [Google Scholar] [CrossRef] [Green Version]
- Kitakami, K.; Beppu, T.; Sato, Y.; Kurose, A.; Ogasawara, K. Utility of arterial spin labeling for objective assessment of intratumoral microvessels in diffuse hemispheric glioma, H3 G34R-mutant: A case report and literature review. Radiol. Case Rep. 2023, 18, 856–861. [Google Scholar] [CrossRef]
- Vettermann, F.J.; Felsberg, J.; Reifenberger, G.; Hasselblatt, M.; Forbrig, R.; Berding, G.; la Fougère, C.; Galldiks, N.; Schittenhelm, J.; Weis, J.; et al. Characterization of Diffuse Gliomas with Histone H3-G34 Mutation by MRI and Dynamic 18F-FET PET. Clin. Nucl. Med. 2018, 43, 895–898. [Google Scholar] [CrossRef] [PubMed]
- Puntonet, J.; Dangouloff-Ros, V.; Saffroy, R.; Pagès, M.; Andreiuolo, F.; Grill, J.; Puget, S.; Boddaert, N.; Varlet, P. Historadiological correlations in high-grade glioma with the histone 3.3 G34R mutation. J. Neuroradiol. J. Neuroradiol. 2018, 45, 316–322. [Google Scholar] [CrossRef]
- Lasocki, A.; Abdalla, G.; Chow, G.; Thust, S.C. Imaging features associated with H3 K27-altered and H3 G34-mutant gliomas: A narrative systematic review. Cancer Imaging Off. Publ. Int. Cancer Imaging Soc. 2022, 22, 63. [Google Scholar] [CrossRef]
- Gonçalves, F.G.; Viaene, A.N.; Vossough, A. Advanced Magnetic Resonance Imaging in Pediatric Glioblastomas. Front. Neurol. 2021, 12, 733323. [Google Scholar] [CrossRef]
- Rao, S.; Sahay, A.; Epari, S. Paediatric type diffuse high grade gliomas in the WHO CNS5 classification: What the pathologist needs to know? Indian J. Pathol. Microbiol. 2022, 65, S50–S58. [Google Scholar] [CrossRef]
- Haque, F.; Varlet, P.; Puntonet, J.; Storer, L.; Bountali, A.; Rahman, R.; Grill, J.; Carcaboso, A.M.; Jones, C.; Layfield, R.; et al. Evaluation of a novel antibody to define histone 3.3 G34R mutant brain tumours. Acta Neuropathol. Commun. 2017, 5, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, T.Y.; Piunti, A.; Lulla, R.R.; Qi, J.; Horbinski, C.M.; Tomita, T.; James, C.D.; Shilatifard, A.; Saratsis, A.M. Detection of Histone H3 mutations in cerebrospinal fluid-derived tumor DNA from children with diffuse midline glioma. Acta Neuropathol. Commun. 2017, 5, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pohl, S.; Dimitrova, L.; Grassow-Narlik, M.; Jöhrens, K.; Acker, T.; Dohmen, H.; Herms, J.; Dorostkar, M.; Hartmann, C.; Hasselblatt, M.; et al. Update on quality assurance in neuropathology: Summary of the round robin trials on TERT promoter mutation, H3-3A mutation, 1p/19q codeletion, and KIAA1549::BRAF fusion testing in Germany in 2020 and 2021. Clin. Neuropathol. 2023, 42, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Broniscer, A.; Chamdine, O.; Hwang, S.; Lin, T.; Pounds, S.; Onar-Thomas, A.; Shurtleff, S.; Allen, S.; Gajjar, A.; Northcott, P.; et al. Gliomatosis cerebri in children shares molecular characteristics with other pediatric gliomas. Acta Neuropathol. 2016, 131, 299–307. [Google Scholar] [CrossRef] [Green Version]
- Hatoum, R.; Chen, J.-S.; Lavergne, P.; Shlobin, N.A.; Wang, A.; Elkaim, L.M.; Dodin, P.; Couturier, C.P.; Ibrahim, G.M.; Fallah, A.; et al. Extent of Tumor Resection and Survival in Pediatric Patients with High-Grade Gliomas: A Systematic Review and Meta-analysis. JAMA Netw. Open 2022, 5, e2226551. [Google Scholar] [CrossRef]
- Herrlinger, U.; Jones, D.T.W.; Glas, M.; Hattingen, E.; Gramatzki, D.; Stuplich, M.; Felsberg, J.; Bähr, O.; Gielen, G.H.; Simon, M.; et al. Gliomatosis cerebri: No evidence for a separate brain tumor entity. Acta Neuropathol. 2016, 131, 309–319. [Google Scholar] [CrossRef] [Green Version]
- Eytan, K.; Versano, Z.; Oren, R.; Jacob-Hirsch, J.; Leitner, M.; Harmelin, A.; Rechavi, G.; Toren, A.; Paglin, S.; Yalon, M. Pediatric glioblastoma cells are sensitive to drugs that inhibit eIF2α dephosphorylation and its phosphomimetic S51D variant. Front. Oncol. 2022, 12, 959133. [Google Scholar] [CrossRef]
- Karajannis, M.; Thomas, A.O.; Baxter, P.; Butingan, N.; Fuller, C.; Gajjar, A.; Haque, S.; Jabado, N.; Lin, T.; Lucas, J.; et al. CTNI-31. Cog Acns1721: Phase 2 Study of Veliparib and Local Irradiation, Followed by Maintenance Veliparib and Temozolomide, in Patients with Newly Diagnosed High-Grade Glioma Without H3 K27m Or Braf Mutations. Neuro-oncology 2022, 24, vii78. [Google Scholar] [CrossRef]
- Vanan, M.I.; Underhill, D.A.; Eisenstat, D.D. Targeting Epigenetic Pathways in the Treatment of Pediatric Diffuse (High Grade) Gliomas. Neurother. J. Am. Soc. Exp. Neurother. 2017, 14, 274–283. [Google Scholar] [CrossRef] [Green Version]
- Yoda, H.; Inoue, T.; Shinozaki, Y.; Lin, J.; Watanabe, T.; Koshikawa, N.; Takatori, A.; Nagase, H. Direct Targeting of MYCN Gene Amplification by Site-Specific DNA Alkylation in Neuroblastoma. Cancer Res. 2019, 79, 830–840. [Google Scholar] [CrossRef] [Green Version]
- Mackay, A.; Burford, A.; Molinari, V.; Jones, D.T.W.; Izquierdo, E.; Brouwer-Visser, J.; Giangaspero, F.; Haberler, C.; Pietsch, T.; Jacques, T.S.; et al. Molecular, Pathological, Radiological, and Immune Profiling of Non-brainstem Pediatric High-Grade Glioma from the HERBY Phase II Randomized Trial. Cancer Cell 2018, 33, 829–842.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
| Study | Inclusion Age | Total Patients | Median Age (Years) a | Number of Males/Females a | Number of H3.3-G34R/V Mutants a | Median EFS or PFS a,b,c,d,e (Months) | Median OS c,d,e (Months) | Treatments |
|---|---|---|---|---|---|---|---|---|
| Korshunov A et al. (2015) [119] | 1–18 years | 24 | 14 | 17/7 | 24/0 | N/A | ~30 | 24: Surgery (GTR or STR) followed by RT + TMZ. Recurrent tumors treated with polychemotherapy (not specified) |
| Hummel TR et al. (2015) [123] | 3–30 years | 1 | 22 | 0/1 | 1/0 | 19.6 | 22.9 | 1: GTR followed by RT + TMZ + BEV |
| Broniscer A et al. (2016) [134] | <22 years | 4 | 12.2 | 3/1 | 4/0 | N/A | 11.4 | 4: Biopsy followed by RT + chemotherapy (not specified) |
| Korshunov A et al. (2016) [3] | All | 81 | 19 | 42/32 | 77/4 | 9 | 22 | 48: Surgery (not specified) followed by RT + TMZ 12: Surgery (not specified) followed by combinations of craniospinal RT + polychemotherapy (various combinations of vincristine, lomustine, and cisplatin) |
| Salloum R et al. (2017) [79] | N/A | 1 | 12 | 1/0 | 0/1 | 7.1 | N/A | 1: GTR followed by RT + TMZ and TMZ + BEV |
| Yoshimoto K et al. (2017) [76] | All | 4 | 10.5 | 2/2 | 4/0 | N/A | 15.5 | 2: STR followed by RT + TMZ 1: STR followed by RT + cisplastin + vincristine 1: Biopsy followed by RT + nimustine |
| Hwang EI et al. (2018) [71] | 3–22 years | 8 | 14.6 | 5/3 | N/A | 14.7 | 23.4 | 1: Biopsy followed by RT + vincristine + carboplatin 1: STR followed by RT + vincristine + carboplatin 1: GTR followed by RT + vincristine + carboplatin 1: GTR followed by RT + vincristine + isotretinoin 4: GTR followed by RT + vincristine + isotretinoin |
| Andreiuolo F et al. (2019) [84] | N/A | 2 | 15 | 2/0 | 2/0 | N/A | At least 18 | 1: STR followed by RT + TMZ 1: GTR followed by RT + TMZ |
| Sasaki S et al. (2019) [60] | N/A | 1 | 12 | 1/0 | 1/0 | 3 | At least 12 | 1: STR x2. After progression, STR followed by TMZ + RT. After progression again, STR followed by RT + TMZ + BEV |
| Morris M et al. (2020) [63] | N/A | 1 | 21 | 1/0 | 1/0 | N/A | At least 10 | 1: Biopsy followed by RT + TMZ followed by lomustine + procarbazine + BEV |
| Rodriguez Gutierrez D et al. (2020) [124] | 3–18 years | 7 | 12.6 | 3/4 | N/A | N/A | 12 | 1: Biopsy followed by RT + TMZ 4: Surgery followed by RT + TMZ 1: Biopsy followed by RT + TMZ + BEV 1: Surgery followed by RT + TMZ + BEV |
| Onishi S et al. (2020) [73] | N/A | 3 | 15 | 1/2 | 3/0 | N/A | At least 18 | 1: GTR followed by chemotherapy (not specified) 1: GTR followed by RT + chemotherapy (not specified) 1: STR followed by RT + chemotherapy (not specified) |
| Crotty EE et al. (2020) [62] | <21 years | 2 | 13.5 | N/A | 2/0 | N/A | N/A | 1: GTR followed by RT + TMZ followed by TMZ + irinotecan + BEV 1: STR followed by RT + TMZ followed by TMZ + irinotecan + BEV |
| Cheng Y et al. (2020) [61] | N/A | 3 | 15 | 2/1 | 1/2 | 5 | 11 | 2: GTR followed by RT + TMZ 1: STR followed by RT + TMZ |
| Mohiuddin S et al. (2021) [114] | N/A | 1 | 17 | 0/1 | 1/0 | N/A | 16 | 1: Biopsy followed by craniospinal RT + TMZ |
| Lim KY et al. (2021) [64] | N/A | 4 | 28 | 1/3 | 3/1 | N/A | 24.5 | 1: Biopsy only 1: GTR followed by RT + TMZ 1: GTR followed by RT 1: GTR followed by vincristine + cyclophosphamide + prednisolone |
| Korshunov A et al. (2021) [72] | 3–18 years | 22 | 14 | 15/7 | 20/2 | ~12 | ~25 | 13: GTR followed by HIT-based protocol of RT (craniospinal RT + tumor bed boost) and cyclophosphamide + vincristine + methotrexate + carboplatin + etoposide (non-metastatic cases) or carboplatin + etoposide + intraventricular etoposide + cyclophosphamide + thiotepa + mesna 9: STR followed by HIT-based protocol as above |
| Picart T et al. (2021) [74] | >18 years | 17 | 25 | 11/6 | 17/0 | 8.8 | 12.5 | Combinations of treatments were not specified. 4: GTR 1: STR 12: Biopsy 13: RT + TMZ 1: RT only 1: RT + procarbazine + lomustine + vincristine 1: RT + TMZ |
| Wood MD et al. (2021) [117] | N/A | 1 | 16 | 0/1 | 1/0 | 15 | At least 15 | 1: GTR followed by RT + TMZ |
| Kalelioglu T et al. (2022) [121] | N/A | 4 | 18 | 2/2 | 4/0 | 5 | 25 | 1: Biopsy followed by RT + clinical trial (not specified) 1: Surgery (not specified) followed by RT + TMZ 1: STR followed by RT + TMZ 1: GTR followed by RT + TMZ |
| Kitakami K et al. (2022) [125] | N/A | 1 | 34 | 0/1 | 1/0 | 15 | At least 20 | 1: Biopsy followed by RT + TMZ + BEV |
| Hu W et al. (2022) [51] | 0–35 years | 10 | 20.5 | 4/6 | 9/1 | N/A | 17.5 | 1: Biopsy followed by RT + TMZ 1: GTR followed by dianhydrodulcitol 4: GTR followed by RT + TMZ 3: STR followed by RT + chemotherapy (not specified) 1: STR followed by RT + TMZ + cisplatin |
| Kurokawa R et al. (2022) [120] | N/A | 3 | 19 | 1/2 | 3/0 | At least 10 | At least 10 | 1: GTR followed by RT + TMZ + lomustine 1 GTR followed by RT + TMZ 1: GTR followed by RT + chemotherapy pending |
| Wang L et al. (2022) [66] | >18 years | 30 | 24 | 11/19 | 26/4 | 10 | 14 | 2: STR followed by TMZ 3: Biopsy followed by RT + TMZ 13: STR followed by RT + TM 12: GTR followed by RT + TMZ |
| Yu N et al. (2023) [69] | N/A | 1 | 19 | 0/1 | 1/0 | N/A | N/A | 1: Biopsy followed by RT + vincristine + cyclophosphamide + doxorubicin + mesna followed by ifosfamide + etoposide + mesna (initially diagnosed with sarcoma) followed by TMZ (after glioma diagnosis) |
| Lavrador JP et al. (2023) [75] | All | 12 | 17.5 | 7/5 | 11/1 | N/A | 12 | Combinations of treatments were not specified. 1: Biopsy 10: STR 1: GTR 1: Supratotal resection 9: RT + TMZ 1: RT only 1: TMZ only |
| Zhao Y et al. (2023) [116] | >18 years | 1 | 27 | 0/1 | 1/0 | 13 | 16 | 1: STR followed by RT + TMZ |
| Cooley LD et al. (2023) [81] | 0–17 years | 2 | 12.5 | 1/1 | 2/0 | N/A | 16.5 | 2: STR followed by RT + chemotherapy (not specified) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, A.V.; Soto, J.M.; Gonzalez, S.-M.; Murillo, J.; Trumble, E.R.; Shan, F.Y.; Huang, J.H. H3G34-Mutant Gliomas—A Review of Molecular Pathogenesis and Therapeutic Options. Biomedicines 2023, 11, 2002. https://doi.org/10.3390/biomedicines11072002
Nguyen AV, Soto JM, Gonzalez S-M, Murillo J, Trumble ER, Shan FY, Huang JH. H3G34-Mutant Gliomas—A Review of Molecular Pathogenesis and Therapeutic Options. Biomedicines. 2023; 11(7):2002. https://doi.org/10.3390/biomedicines11072002
Chicago/Turabian StyleNguyen, Anthony V., Jose M. Soto, Sarah-Marie Gonzalez, Jennifer Murillo, Eric R. Trumble, Frank Y. Shan, and Jason H. Huang. 2023. "H3G34-Mutant Gliomas—A Review of Molecular Pathogenesis and Therapeutic Options" Biomedicines 11, no. 7: 2002. https://doi.org/10.3390/biomedicines11072002