
Therapeutic Targets of Monoclonal Antibodies Used in the Treatment of Cancer: Current and Emerging

 , ,
, ,  ,
,
Abstract
:1. Introduction
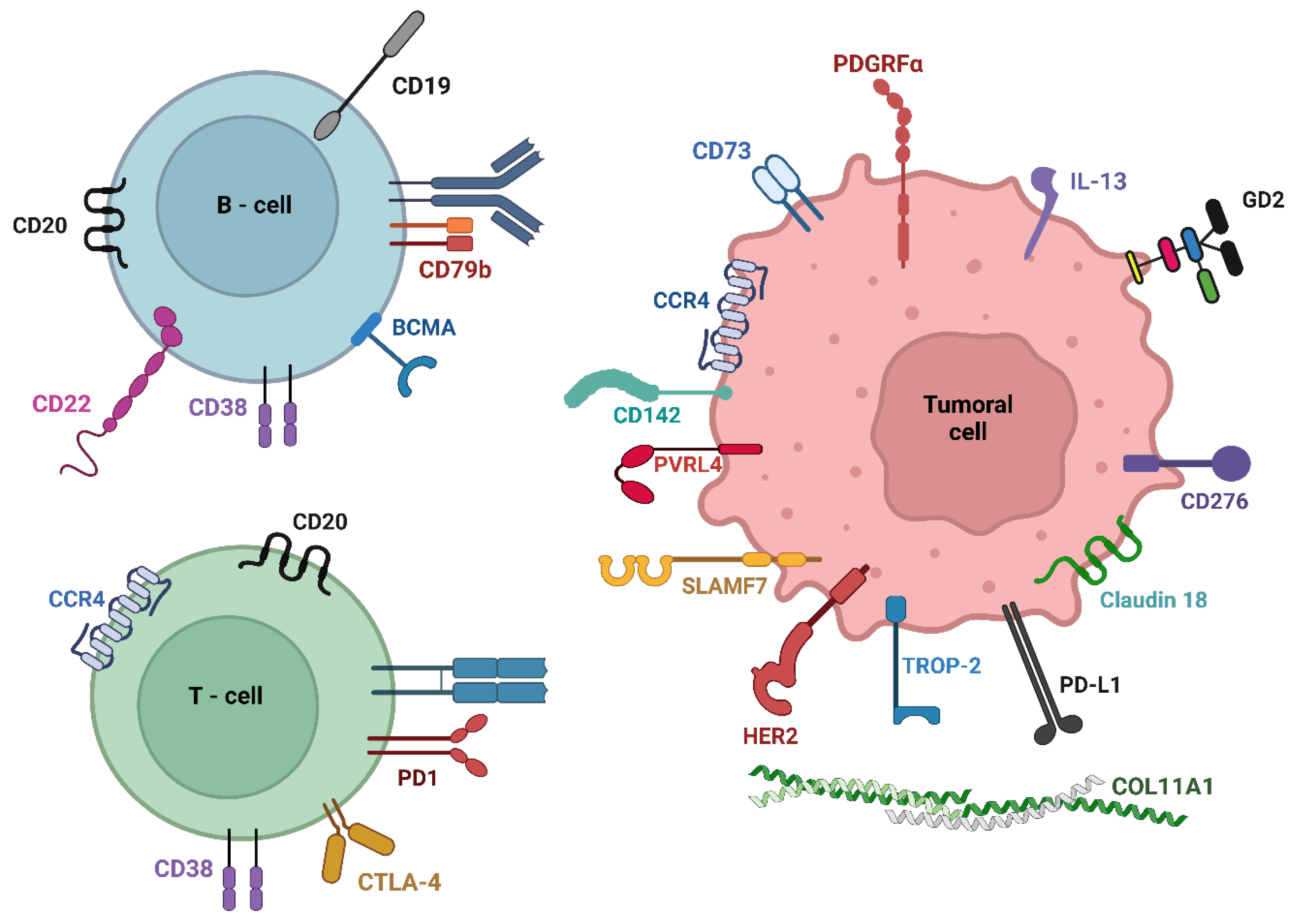
2. Current Immunotherapeutic Targets for Cancer Treatment
2.1. Programmed Cell Death Protein 1 (PD1)/Programmed Cell Death Ligand 1 (PD-L1) Axis
2.2. B-Lymphocyte Antigen CD20 (CD20)
2.3. Human Epidermal Growth Factor Receptor 2 (Receptor Tyrosine Kinase) (HER2)
2.4. B-Lymphocyte Antigen CD19 (CD19)
2.5. Disialoganglioside GD2 (GD2)
2.6. B-Cell Maturation Antigen (BCMA or CD269)
2.7. Trophoblast Cell-Surface Antigen 2 (TROP-2)
2.8. Adp-Ribosyl Cyclase/Cyclic Adp-Ribose Hydrolase 1 (CD38)
2.9. Nectin-4 or Poliovirus Receptor-like 4 (PVRL4)
2.10. Cd79b (ADC) Diffuse Large B-Cell Lymphoma
2.11. CD22
2.12. CC Chemokine Receptor Type 4 (CCR4)
2.13. PDGRFα
2.14. SLAMF7 (CD319)
2.15. Tissue Factor/CD142
2.16. CTLA-4
3. Emerging Immunotherapeutic Targets for Cancer Treatment
3.1. COL11A1
3.2. Claundin 18
3.3. CD73
3.4. B7-H3 (CD276)
3.5. Interleukin-13 (IL13)
4. Mechanisms of Action of the mAbs Currently Used in the Treatment of Cancer
4.1. Blocking Ligand Binding
4.1.1. Nonconjugated mAbs
- Steric hindrance: the antibody binds to the antigen receptor or ligand, occupying the region of interaction. By physically blocking the binding, it prevents the transmission of signals or the initiation of unwanted biological responses [255].
- Conformational changes: the antibody binds the antigen and induces conformational changes in the target cell. This alteration in structure prevents efficient interaction with ligand [255].
- Internalization of the complex: in some cases, once the antibody has bound to the antigen, the antibody–antigen complex undergoes internationalization. The cell captures the complex through endocytosis, removing the receptor and ligand from contact with each other. This blocks the signaling or biological activity mediated by them [256].
4.1.2. Conjugated mAbs
Action of the Therapeutic Agent
4.2. Blocking Signaling Pathway
4.3. Depletion of Target by Fc Interaction
5. Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
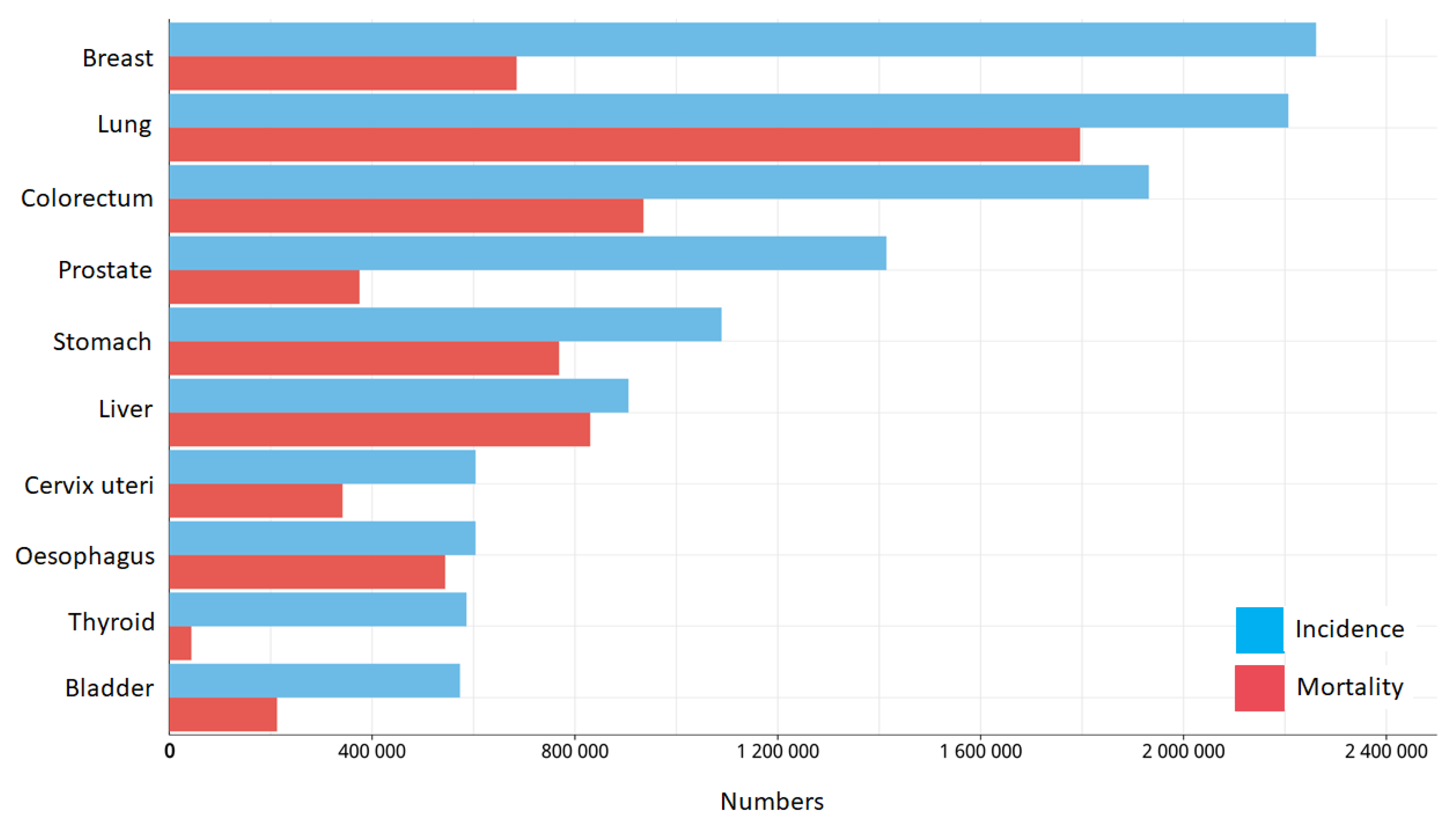
- Ferlay, J.; Ervik, M.; Lam, F.; Colombet, M.; Mery, L.; Piñeros, M.; Znaor, A.; Soerjomataram, I.; Bray, F. Global Cancer Observatory: Cancer Today; International Agency for Research on Cancer: Lyon, Franc. Available online: https://gco.iarc.fr/today (accessed on 20 March 2021).
- Cancer Statistics. Available online: https://www.cancer.gov/about-cancer/understanding/statistics (accessed on 20 March 2021).
- WHO. Cáncer. Available online: https://www.who.int/es/news-room/fact-sheets/detail/cancer (accessed on 20 March 2021).
- Botelho, M.C.; Richter, J. Editorial: Parasites and Cancer. Front. Med. 2019, 6, 55. [Google Scholar] [CrossRef] [PubMed]
- Cao, G.; He, X.; Sun, Q.; Chen, S.; Wan, K.; Xu, X.; Feng, X.; Li, P.; Chen, B.; Xiong, M. The Oncolytic Virus in Cancer Diagnosis and Treatment. Front. Oncol. 2020, 10, 1786. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, S.; Levi-Schaffer, F.; Sela, M.; Yarden, Y. Immunotherapy of Cancer: From Monoclonal to Oligoclonal Cocktails of Anti-Cancer Antibodies: IUPHAR Review 18. Br. J. Pharmacol. 2016, 173, 1407–1424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merino, G. Monoclonal Antibodies. Basic Features. Neurología 2011, 26, 301–306. [Google Scholar] [CrossRef]
- Newsome, B.W.; Ernstoff, M.S. The Clinical Pharmacology of Therapeutic Monoclonal Antibodies in the Treatment of Malignancy; Have the Magic Bullets Arrived? Br. J. Clin. Pharmacol. 2008, 66, 6–19. [Google Scholar] [CrossRef] [Green Version]
- Marrocco, I.; Romaniello, D.; Yarden, Y. Cancer Immunotherapy: The Dawn of Antibody Cocktails. In Human Monoclonal Antibodies: Methods and Protocols; Steinitz, M., Ed.; Springer: Berlin/Heidelberg, Germany, 2019; pp. 11–51. [Google Scholar]
- Mullard, A. FDA Approves 100th Monoclonal Antibody Product. Nat. Rev. Drug Discov. 2021, 20, 491–495. [Google Scholar] [CrossRef]
- Goulet, D.R.; Atkins, W.M. Considerations for the Design of Antibody-Based Therapeutics. J. Pharm. Sci. 2020, 109, 74–103. [Google Scholar] [CrossRef] [Green Version]
- Ramaswami, R.; Longo, D. Monoclonal Antibodies. In Cancer Chemotherapy, Immunotherapy and Biotherapy: Principles and Practice; Chabner, B., Longo, D., Eds.; Wolters Kluwer: Alphen aan den Rijn, The Netherlands, 2019; pp. 782–812. [Google Scholar]
- Karpuz, M.; Silindir-Gunay, M.; Ozer, A.Y. Current and Future Approaches for Effective Cancer Imaging and Treatment. Cancer Biother. Radiopharm. 2018, 33, 39–51. [Google Scholar] [CrossRef]
- Therapeutic Monoclonal Antibodies Approved or in Review in the EU or US. Available online: www.antibodysociety.org (accessed on 12 December 2022).
- Espinoza, J.A.; Riquelme, I.; Sagredo, E.A.; Rosa, L.; García, P.; Bizama, C.; Apud-Bell, M.; Leal, P.; Weber, H.; Benavente, F.; et al. Mucin 5B, Carbonic Anhydrase 9 and Claudin 18 Are Potential Theranostic Markers of Gallbladder Carcinoma. Histopathology 2019, 74, 597–607. [Google Scholar] [CrossRef]
- Han, Y.; Liu, D.; Li, L. PD-1/PD-L1 Pathway: Current Researches in Cancer. Am. J. Cancer Res. 2020, 10, 727–742. [Google Scholar]
- Ahmadzadeh, M.; Johnson, L.A.; Heemskerk, B.; Wunderlich, J.R.; Dudley, M.E.; White, D.E.; Rosenberg, S.A. Tumor Antigen-Specific CD8 T Cells Infiltrating the Tumor Express High Levels of PD-1 and Are Functionally Impaired. Blood 2009, 114, 1537–1544. [Google Scholar] [CrossRef]
- Goodsell, D.S. PD-1 (Programmed Cell Death Protein 1). RCSB Protein Data Bank 2016. [Google Scholar] [CrossRef]
- Ai, L.; Xu, A.; Xu, J. Roles of PD-1/PD-L1 Pathway: Signaling, Cancer, and Beyond. In Advances in Experimental Medicine and Biology 1248—Regulation of Cancer Immune Checkpoints—Molecular and Cellular Mechanisms and Therapy; Xu, J., Ed.; Springer: Berlin/Heidelberg, Germany, 2020; pp. 33–59. [Google Scholar]
- UniProtKB UniProtKB-Q9NZQ7 (PD1L1_HUMAN). Available online: https://www.uniprot.org/uniprot/Q9NZQ7 (accessed on 29 May 2021).
- Sharpe, A.H.; Wherry, E.J.; Ahmed, R.; Freeman, G.J. The Function of Programmed Cell Death 1 and Its Ligands in Regulating Autoimmunity and Infection. Nat. Immunol. 2007, 8, 239–245. [Google Scholar] [CrossRef]
- Zou, W.; Chen, L. Inhibitory B7-Family Molecules in the Tumour Microenvironment. Nat. Rev. Immunol. 2008, 8, 467–477. [Google Scholar] [CrossRef]
- Nunes-Xavier, C.E.; Angulo, J.C.; Pulido, R.; López, J.I. A Critical Insight into the Clinical Translation of PD-1/PD-L1 Blockade Therapy in Clear Cell Renal Cell Carcinoma. Curr. Urol. Rep. 2019, 20, 1. [Google Scholar] [CrossRef]
- The Nobel Assembly at Karolinska Institutet. The Nobel Assembly at Karolinska Institutet Has Today Decided to Award the 2018 Nobel Prize in Physiology or Medicine Jointly to James P. Allison and Tasuku Honjo; 2018; pp. 1–5. [Google Scholar]
- Topalian, S.L.; Sznol, M.; McDermott, D.F.; Kluger, H.M.; Carvajal, R.D.; Sharfman, W.H.; Brahmer, J.R.; Lawrence, D.P.; Atkins, M.B.; Powderly, J.D.; et al. Survival, Durable Tumor Remission, and Long-Term Safety in Patients with Advanced Melanoma Receiving Nivolumab. J. Clin. Oncol. 2014, 32, 1020–1030. [Google Scholar] [CrossRef]
- Sundar, R.; Cho, B.C.; Brahmer, J.R.; Soo, R.A. Nivolumab in NSCLC: Latest Evidence and Clinical Potential. Ther. Adv. Med. Oncol. 2015, 7, 85–96. [Google Scholar] [CrossRef] [Green Version]
- Choueiri, T.K.; Fishman, M.N.; Escudier, B.; McDermott, D.F.; Drake, C.G.; Kluger, H.; Stadler, W.M.; Perez-Gracia, J.L.; McNeel, D.G.; Curti, B.; et al. Immunomodulatory Activity of Nivolumab in Metastatic Renal Cell Carcinoma. Clin. Cancer Res. 2016, 22, 5461–5471. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.; Keam, S.J. Tislelizumab: First Approval. Drugs 2020, 80, 617–624. [Google Scholar] [CrossRef]
- Dhillon, S. Penpulimab: First Approval. Drugs 2021, 81, 2159–2166. [Google Scholar] [CrossRef]
- FDA Www.Fda.Gov. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-retifanlimab-dlwr-metastatic-or-recurrent-locally-advanced-merkel#:~:text=OnMarch22%2C2023%2CtheFoodandDrug,carcinoma%28MCC%29 (accessed on 13 June 2023).
- Chen, Q.; Yuan, S.; Sun, H.; Peng, L. CD3+ CD20+ T Cells and Their Roles in Human Diseases. Hum. Immunol. 2019, 80, 191–194. [Google Scholar] [CrossRef] [PubMed]
- Florou, D.; Katsara, M.; Feehan, J.; Dardiotis, E.; Apostolopoulos, V. Anti-Cd20 Agents for Multiple Sclerosis: Spotlight on Ocrelizumab and Ofatumumab. Brain Sci. 2020, 10, 758. [Google Scholar] [CrossRef] [PubMed]
- Uchida, J.; Lee, Y.; Hasegawa, M.; Liang, Y.; Bradney, A.; Oliver, J.A.; Bowen, K.; Steeber, D.A.; Haas, K.M.; Poe, J.C.; et al. Mouse CD20 Expression and Function. Int. Immunol. 2004, 16, 119–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henry, C.; Deschamps, M.; Rohrlich, P.S.; Pallandre, J.R.; Rémy-Martin, J.P.; Callanan, M.; Traverse-Glehen, A.; GrandClément, C.; Garnache-Ottou, F.; Gressin, R.; et al. Identification of an Alternative CD20 Transcript Variant in B-Cell Malignancies Coding for a Novel Protein Associated to Rituximab Resistance. Blood 2010, 115, 2420–2429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gamonet, C.; Bole-Richard, E.; Delherme, A.; Aubin, F.; Toussirot, E.; Garnache-Ottou, F.; Godet, Y.; Ysebaert, L.; Tournilhac, O.; Caroline, D.; et al. New CD20 Alternative Splice Variants: Molecular Identification and Differential Expression within Hematological B Cell Malignancies. Exp. Hematol. Oncol. 2016, 5, 7. [Google Scholar] [CrossRef] [Green Version]
- Pavlasova, G.; Borsky, M.; Svobodova, V.; Oppelt, J.; Cerna, K.; Novotna, J.; Seda, V.; Fojtova, M.; Fajkus, J.; Brychtova, Y.; et al. Rituximab Primarily Targets an Intra-Clonal BCR Signaling Proficient CLL Subpopulation Characterized by High CD20 Levels. Leukemia 2018, 32, 2028–2031. [Google Scholar] [CrossRef]
- Pavlasova, G.; Mraz, M. The Regulation and Function of CD20: An “Enigma” of B-Cell Biology and Targeted Therapy. Haematologica 2020, 105, 1494–1506. [Google Scholar] [CrossRef]
- Schuh, E.; Berer, K.; Mulazzani, M.; Feil, K.; Meinl, I.; Lahm, H.; Krane, M.; Lange, R.; Pfannes, K.; Subklewe, M.; et al. Features of Human CD3+ CD20+ T Cells. J. Immunol. 2016, 197, 1111–1117. [Google Scholar] [CrossRef] [Green Version]
- Kang, C. Mosunetuzumab: First Approval. Drugs 2022, 82, 1229–1234. [Google Scholar] [CrossRef]
- Fischer, L. Oncology Nursing News. Available online: https://www.oncnursingnews.com/view/epcoritamab-obtains-accelerated-approval-for-relapsed-refractory-dlbcl (accessed on 19 May 2023).
- Faloso, A.; Gianni, L. Introduction and Background Biology. In Handbook of HER2-Targeted Agents in Breast Cancer; Alvarez, R., Cortés, J., Falzon, M., Gandy, M., Gianni, L., Harbeck, N., Piccart, M., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 1–13. [Google Scholar]
- Connell, C.M.; Doherty, G.J. Activating HER2 Mutations as Emerging Targets in Multiple Solid Cancers. ESMO Open 2017, 2, e000279. [Google Scholar] [CrossRef] [Green Version]
- Albagoush, S.A.; Limaiem, F. HER2; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Slamon, D.J.; Clark, G.M.; Wong, S.G.; Levin, W.J.; Mcguire, W.L. American Association for the Advancement of Science. Science 1987, os-2, 341–342. [Google Scholar] [CrossRef] [Green Version]
- Sjögren, S.; Inganäs, M.; Lindgren, A.; Holmberg, L.; Bergh, J. Prognostic and Predictive Value of C-ErbB-2 Overexpression in Primary Breast Cancer, Alone and in Combination with Other Prognostic Markers. J. Clin. Oncol. 1998, 16, 462–469. [Google Scholar] [CrossRef]
- Li, X.; Ding, Y.; Zi, M.; Sun, L.; Zhang, W.; Chen, S.; Xu, Y. CD19, from Bench to Bedside. Immunol. Lett. 2017, 183, 86–95. [Google Scholar] [CrossRef]
- Bradbury, L.E.; Goldmacher, V.S.; Tedder, T.F. The CD19 Signal Transduction Complex of B Lymphocytes. Deletion of the CD19 Cytoplasmic Domain Alters Signal Transduction but Not Complex Formation with TAPA-1 and Leu 13. J. Immunol. 1993, 151, 2915–2927. [Google Scholar] [CrossRef]
- Zhou, L.J.; Ord, D.C.; Hughes, A.L.; Tedder, T.F. Structure and Domain Organization of the CD19 Antigen of Human, Mouse, and Guinea Pig B Lymphocytes. Conservation of the Extensive Cytoplasmic Domain. J. Immunol. 1991, 147, 1424–1432. [Google Scholar] [CrossRef]
- Baba, Y.; Kurosaki, T. Impact of Ca2+ Signaling on B Cell Function. Trends Immunol. 2011, 32, 589–594. [Google Scholar] [CrossRef]
- Rickert, R.C.; Rajewsky, K.; Roes, J. Impairment of T-Cell-Dependent B-Cell Responses and B-l Cell Development in CD19-Deficient Mice. Nature 1995, 376, 352–355. [Google Scholar] [CrossRef]
- Yoshizaki, A.; Iwata, Y.; Komura, K.; Ogawa, F.; Hara, T.; Muroi, E.; Takenaka, M.; Shimizu, K.; Hasegawa, M.; Fujimoto, M.; et al. CD19 Regulates Skin and Lung Fibrosis via Toll-like Receptor Signaling in a Model of Bleomycin-Induced Scleroderma. Am. J. Pathol. 2008, 172, 1650–1663. [Google Scholar] [CrossRef] [Green Version]
- Sato, S.; Hasegawa, M.; Fujimoto, M.; Tedder, T.F.; Takehara, K. Quantitative Genetic Variation in CD19 Expression Correlates with Autoimmunity. J. Immunol. 2000, 165, 6635–6643. [Google Scholar] [CrossRef]
- Nazha, B.; Inal, C.; Owonikoko, T.K. Disialoganglioside GD2 Expression in Solid Tumors and Role as a Target for Cancer Therapy. Front. Oncol. 2020, 10, 1000. [Google Scholar] [CrossRef]
- Svennerholm, L.; Bostrom, K.; Fredman, P.; Jungbjer, B.; Lekman, A.; Jan-eric, M.; Rynmark, B. Gangliosides and Allied Glycosphingolipids in Human Peripheral Nerve and Spinal Cord. Biochim. Et Biophys. Acta 1994, 1214, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, M.; Yoshimura, S.; Yu, R.K. Expression of GD2 and GD3 Gangliosides in Human Embryonic Neural Stem Cells. ASN Neuro 2011, 3, AN20110006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulz, G.; Cheresh, D.A.; Varki, N.M.; Staffilano, L.K.; Reisfeld, R.A.; Yu, A. Detection of Ganglioside GD2 in Tumor Tissues and Sera of Neuroblastoma Patients. Cancer Res. 1984, 44, 5914–5920. [Google Scholar] [PubMed]
- Chen, L.C.; Brown, A.B.; Cheung, I.Y.; Cheung, N.K.V.; Kris, M.G.; Krug, L.M. Analysis of GD2/GM2 Synthase MRNA as a Biomarker for Small Cell Lung Cancer. Lung Cancer 2010, 67, 216–220. [Google Scholar] [CrossRef]
- Tsuchida, T.; Irie, R.F.; Ishibashi, Y. Gangliosides of Human Melanoma. Pigment. Cell Res. 1987, 3, 147–150. [Google Scholar] [CrossRef]
- Casey, D.L.; Lin, T.Y.; Cheung, N.K.V. Exploiting Signaling Pathways and Immune Targets beyond the Standard of Care for Ewing Sarcoma. Front. Oncol. 2019, 9, 537. [Google Scholar] [CrossRef]
- Roth, M.; Linkowski, M.; Tarim, J.; Piperdi, S.; Sowers, R.; Geller, D.; Gill, J.; Gorlick, R. Ganglioside GD2 as a Therapeutic Target for Antibody-Mediated Therapy in Patients with Osteosarcoma. Cancer 2014, 120, 548–554. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.; Cordon-Cardo, C.; Houghton, A.; Cheung, N.; Brennan, M. Expression of Disialogangliosides GD2 and GD3 on Human Soft Tissue Sarcomas. Cancer 1992, 70, 633–638. [Google Scholar] [CrossRef]
- Longee, D.C.; Wikstrand, C.J.; Månsson, J.E.; He, X.; Fuller, G.N.; Bigner, S.H.; Fredman, P.; Svennerholm, L.; Bigner, D.D. Disialoganglioside GD2 in Human Neuroectodermal Tumor Cell Lines and Gliomas. Acta Neuropathol. 1991, 82, 45–54. [Google Scholar] [CrossRef]
- Andersch, L.; Radke, J.; Klaus, A.; Schwiebert, S.; Winkler, A.; Schumann, E.; Grunewald, L.; Zirngibl, F.; Flemmig, C.; Jensen, M.C.; et al. CD171- and GD2-Specific CAR-T Cells Potently Target Retinoblastoma Cells in Preclinical in Vitro Testing. BMC Cancer 2019, 19, 895. [Google Scholar] [CrossRef] [Green Version]
- Orsi, G.; Barbolini, M.; Ficarra, G.; Tazzioli, G.; Manni, P.; Petrachi, T.; Mastrolia, I.; Orvieto, E.; Spano, C.; Prapa, M.; et al. GD2 Expression in Breast Cancer. Oncotarget 2017, 8, 31592–31600. [Google Scholar] [CrossRef]
- Vantaku, V.; Donepudi, S.R.; Ambati, C.R.; Jin, F.; Putluri, V.; Nguyen, K.; Rajapakshe, K.; Coarfa, C.; Battula, V.L.; Lotan, Y.; et al. Expression of Ganglioside GD2, Reprogram the Lipid Metabolism and EMT Phenotype in Bladder Cancer. Oncotarget 2017, 8, 95620–95631. [Google Scholar] [CrossRef] [Green Version]
- Dobrenkov, K.; Cheung, N.-K.V. GD2-Targeted Immunotherapy and Radioimmunotherapy. Semin. Oncol. 2014, 41, 589–612. [Google Scholar] [CrossRef] [Green Version]
- Hung, J.; Yu, A. GD2-Targeted Immunotherapy of Neuroblastoma. In Neuroblastoma: Molecular Mechanisms and Therapeutic Interventions; Swapan, R., Ed.; Elsevier Academic Press: Amsterdam, The Netherlands, 2019; pp. 62–78. [Google Scholar]
- Cavdarli, S.; Groux-Degroote, S.; Delannoy, P. Gangliosides: The Double-Edge Sword of Neuro-Ectodermal Derived Tumors. Biomolecules 2019, 9, 311. [Google Scholar] [CrossRef] [Green Version]
- Esaki, N.; Ohkawa, Y.; Hashimoto, N.; Tsuda, Y.; Ohmi, Y.; Bhuiyan, R.H.; Kotani, N.; Honke, K.; Enomoto, A.; Takahashi, M.; et al. ASC Amino Acid Transporter 2, Defined by Enzyme-Mediated Activation of Radical Sources, Enhances Malignancy of GD2-Positive Small-Cell Lung Cancer. Cancer Sci. 2018, 109, 141–153. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Wondimu, A.; Yan, S.; Bobb, D.; Ladisch, S. Tumor Gangliosides Accelerate Murine Tumor Angiogenesis. Angiogenesis 2014, 17, 563–571. [Google Scholar] [CrossRef]
- Avery, D.T.; Kalled, S.L.; Ellyard, J.I.; Ambrose, C.; Bixler, S.A.; Thien, M.; Brink, R.; Mackay, F.; Hodgkin, P.D.; Tangye, S.G. BAFF Selectively Enhances the Survival of Plasmablasts Generated from Human Memory B Cells. J. Clin. Investig. 2003, 112, 286–297. [Google Scholar] [CrossRef]
- O’Connor, B.P.; Raman, V.S.; Erickson, L.D.; Cook, W.J.; Weaver, L.K.; Ahonen, C.; Lin, L.-L.; Mantchev, G.T.; Bram, R.J.; Noelle, R.J. BCMA Is Essential for the Survival of Long-Lived Bone Marrow Plasma Cells. J. Exp. Med. 2004, 199, 91–98. [Google Scholar] [CrossRef]
- Tai, Y.T.; Anderson, K.C. B Cell Maturation Antigen (BCMA)-Based Immunotherapy for Multiple Myeloma. Expert Opin. Biol. Ther. 2019, 19, 1143–1156. [Google Scholar] [CrossRef]
- Dogan, A.; Siegel, D.; Tran, N.; Fu, A.; Fowler, J.; Belani, R.; Landgren, O. B-Cell Maturation Antigen Expression across Hematologic Cancers: A Systematic Literature Review. Blood Cancer J. 2020, 10, 73. [Google Scholar] [CrossRef]
- Guadagnoli, M.; Kimberley, F.C.; Phan, U.; Cameron, K.; Vink, P.M.; Rodermond, H.; Eldering, E.; Kater, A.P.; van Eenennaam, H.; Medema, J.P. Development and Characterization of APRIL Antagonistic Monoclonal Antibodies for Treatment of B-Cell Lymphomas. Blood 2011, 117, 6856–6865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belnoue, E.; Pihlgren, M.; McGaha, T.L.; Tougne, C.; Rochat, A.-F.; Bossen, C.; Schneider, P.; Huard, B.; Lambert, P.-H.; Siegrist, C.-A. APRIL Is Critical for Plasmablast Survival in the Bone Marrow and Poorly Expressed by Early-Life Bone Marrow Stromal Cells. Blood 2008, 111, 2755–2764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khattar, P.; Pichardo, J.; Jungbluth, A.; Gao, Q.; Smith, E.; Roshal, M.; Dogan, A. B-Cell Maturation Antigen Is Exclusively Expressed in a Wide Range of B-Cell and Plasma Cell Neoplasm and in a Potential Therapeutic Target for Bcma Directed Therapies. In Proceedings of the Lymphoma Biology Non Genetic Studies: Poster II; Elsevier: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Maia, S.; Pelletier, M.; Ding, J.; Hsu, Y.-M.; Sallan, S.E.; Rao, S.P.; Nadler, L.M.; Cardoso, A.A. Aberrant Expression of Functional BAFF-System Receptors by Malignant B-Cell Precursors Impacts Leukemia Cell Survival. PLoS ONE 2011, 6, e20787. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, E.; Tanenbaum, E.J.; Patil, S.; Li, M.; Soof, C.M.; Vidisheva, A.; Waterman, G.N.; Hekmati, T.; Tang, G.; Wang, C.S.; et al. The Clinical Significance of B-Cell Maturation Antigen as a Therapeutic Target and Biomarker. Expert Rev. Mol. Diagn. 2018, 18, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Kaplon, H.; Muralidharan, M.; Schneider, Z.; Reichert, J.M. Antibodies to Watch in 2020. mAbs 2020, 12, 1703531. [Google Scholar] [CrossRef] [Green Version]
- Zaman, S.; Jadid, H.; Denson, A.C.; Gray, J.E. Targeting Trop-2 in Solid Tumors: Future Prospects. OncoTargets Ther. 2019, 12, 1781–1790. [Google Scholar] [CrossRef] [Green Version]
- Cubas, R.; Li, M.; Chen, C.; Yao, Q. Trop2: A Possible Therapeutic Target for Late Stage Epithelial Carcinomas. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2009, 1796, 309–314. [Google Scholar] [CrossRef]
- Shvartsur, A.; Bonavida, B. Trop2 and Its Overexpression in Cancers: Regulation and Clinical/Therapeutic Implications. Genes Cancer 2014, 6, 84–105. [Google Scholar] [CrossRef] [Green Version]
- Basu, A.; Goldenberg, D.M.; Stein, R. The Epithelial/Carcinoma Antigen EGP-1, Recognized by Monoclonal Antibody RS7–3G11, Is Phosphorylated on Serine 303. Int. J. Cancer 1995, 62, 472–479. [Google Scholar] [CrossRef]
- Goldenberg, D.M.; Stein, R.; Sharkey, R.M. The Emergence of Trophoblast Cell-Surface Antigen 2 (TROP-2) as a Novel Cancer Target. Oncotarget 2018, 9, 28989–29006. [Google Scholar] [CrossRef] [Green Version]
- Martini, M.; De Santis, M.C.; Braccini, L.; Gulluni, F.; Hirsch, E. PI3K/AKT Signaling Pathway and Cancer: An Updated Review. Ann. Med. 2014, 46, 372–383. [Google Scholar] [CrossRef]
- Lim, H.J.; Crowe, P.; Yang, J.L. Current Clinical Regulation of PI3K/PTEN/Akt/MTOR Signalling in Treatment of Human Cancer. J. Cancer Res. Clin. Oncol. 2015, 141, 671–689. [Google Scholar] [CrossRef]
- Li, X.; Teng, S.; Zhang, Y.; Zhang, W.; Zhang, X.; Xu, K.; Yao, H.; Yao, J.; Wang, H.; Liang, X.; et al. TROP2 Promotes Proliferation, Migration and Metastasis of Gallbladder Cancer Cells by Regulating PI3K/AKT Pathway and Inducing EMT. Oncotarget 2017, 8, 47052–47063. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.; Zhang, H.; Wang, J.; Lu, M.; Zheng, F.; Wang, C.; Tang, X.; Xu, N.; Chen, R.; Zhang, D.; et al. A Novel Human Fab Antibody for Trop2 Inhibits Breast Cancer Growth in Vitro and in Vivo. Int. J. Cancer 2014, 134, 1239–1249. [Google Scholar] [CrossRef]
- Mao, Y.; Wang, X.; Zheng, F.; Wang, C.; Tang, Q.; Tang, X.; Xu, N.; Zhang, H.; Zhang, D.; Xiong, L.; et al. The Tumor-Inhibitory Effectiveness of a Novel Anti-Trop2 Fab Conjugate in Pancreatic Cancer. Oncotarget 2016, 7, 24810–24823. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Yang, D.; Yin, Z.; Gao, M.; Tong, H.; Su, Y.; Zhu, J.; Ye, C.; Zhang, H. A Novel Human Monoclonal Trop2-IgG Antibody Inhibits Ovarian Cancer Growth in Vitro and in Vivo. Biochem. Biophys. Res. Commun. 2019, 512, 276–282. [Google Scholar] [CrossRef]
- Crowell, P.D.; Goldstein, A.S. Functional Evidence That Progenitor Cells near Sites of Inflammation Are Precursors for Aggressive Prostate Cancer. Mol. Cell. Oncol. 2017, 4, e1279723. [Google Scholar] [CrossRef] [Green Version]
- Krejcik, J.; Casneuf, T.; Nijhof, I.S.; Verbist, B.; Bald, J.; Plesner, T.; Syed, K.; Liu, K.; Van De Donk, N.W.C.J.; Weiss, B.M.; et al. Daratumumab Depletes CD38+ Immune Regulatory Cells, Promotes T-Cell Expansion, and Skews T-Cell Repertoire in Multiple Myeloma. Blood 2016, 128, 384–394. [Google Scholar] [CrossRef] [Green Version]
- Kotlikoff, M.I.; Kannan, M.S.; Solway, J.; Deng, K.Y.; Deshpande, D.A.; Dowell, M.; Feldman, M.; Green, K.S.; Ji, G.; Johnston, R.; et al. Methodologic Advancements in the Study of Airway Smooth Muscle. J. Allergy Clin. Immunol. 2004, 114, 18–31. [Google Scholar] [CrossRef]
- Horenstein, A.L.; Sizzano, F.; Lusso, R.; Besso, F.G.; Ferrero, E.; Deaglio, S.; Corno, F.; Malavasi, F. CD38 and CD157 Ectoenzymes Mark Cell Subsets in the Human Corneal Limbus. Mol. Med. 2009, 15, 76–84. [Google Scholar] [CrossRef]
- Chini, E. CD38 as a Regulator of Cellular NAD: A Novel Potential Pharmacological Target for Metabolic Conditions. Curr. Pharm. Des. 2009, 15, 57–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kar, A.; Mehrotra, S.; Chatterjee, S. CD38: T Cell Immuno-Metabolic Modulator. Cells 2020, 9, 1716. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.K.; Bolton, E.L.; Cortopassi, W.A.; Wang, Y.; O’Brien, F.; Maciejewska, M.; Jacobson, M.P.; Garnham, C.; Ruas, M.; Parrington, J.; et al. Synthesis of the Ca2+-Mobilizing Messengers NAADP and CADPR by Intracellular CD38 Enzyme in the Mouse Heart: Role in -Adrenoceptor Signaling. J. Biol. Chem. 2017, 292, 13243–13257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van De Donk, N.W.C.J.; Richardson, P.G.; Malavasi, F. CD38 Antibodies in Multiple Myeloma: Back to the Future. Blood 2018, 131, 13–29. [Google Scholar] [CrossRef] [Green Version]
- Hogan, K.A.; Chini, C.C.S.; Chini, E.N. The Multi-Faceted Ecto-Enzyme CD38: Roles in Immunomodulation, Cancer, Aging, and Metabolic Diseases. Front. Immunol. 2019, 10, 1187. [Google Scholar] [CrossRef] [Green Version]
- Ben Baruch, B.; Mantsur, E.; Franco-Barraza, J.; Blacher, E.; Cukierman, E.; Stein, R. CD38 in Cancer-Associated Fibroblasts Promotes pro-Tumoral Activity. Lab. Investig. 2020, 100, 1517–1531. [Google Scholar] [CrossRef]
- Sanofi. Phase 3 Trial of Isatuximab Combination Therapy Showed 40% Reduction in the Risk of Disease Progression or Death for Patients with Relapsed/Refractory Multiple Myeloma; Sanofi: Paris, France, 2019. [Google Scholar]
- Sanofi. FDA to Review Isatuximab as a Potential Treatment for Relapsed/Refractory Multiple Myeloma July 10, 2019 Press Release; Sanofi: Paris, France, 2019. [Google Scholar]
- Reymond, N.; Fabre, S.; Lecocq, E.; Adelaide, J.; Dubreuil, P.; Lopez, M. Nectin4/PRR4, a New Afadin-Associated Member of the Nectin Family That Trans-Interacts with Nectin1/PRR1 through V Domain Interaction. J. Biol. Chem. 2001, 276, 43205–43215. [Google Scholar] [CrossRef] [Green Version]
- Takai, Y.; Miyoshi, J.; Ikeda, W.; Ogita, H. Nectins and Nectin-like Molecules: Roles in Contact Inhibition of Cell Movement and Proliferation. Nat. Rev. Mol. Cell Biol. 2008, 9, 603–615. [Google Scholar] [CrossRef]
- Brancati, F.; Fortugno, P.; Bottillo, I.; Lopez, M.; Josselin, E.; Boudghene-Stambouli, O.; Agolini, E.; Bernardini, L.; Bellacchio, E.; Iannicelli, M.; et al. Mutations in PVRL4, Encoding Cell Adhesion Molecule Nectin-4, Cause Ectodermal Dysplasia-Syndactyly Syndrome. Am. J. Hum. Genet. 2010, 87, 265–273. [Google Scholar] [CrossRef] [Green Version]
- Fabre, S.; Reymond, N.; Cocchi, F.; Menotti, L.; Dubreuil, P.; Campadelli-Fiume, G.; Lopez, M. Prominent Role of the Ig-like V Domain in Trans-Interactions of Nectins. Nectin3 and Nectin4 Bind to the Predicted C-C′-C″-D β-Strands of the Nectin1 V Domain. J. Biol. Chem. 2002, 277, 27006–27013. [Google Scholar] [CrossRef] [Green Version]
- Nakanishi, H.; Takai, Y. Roles of Nectins in Cell Adhesion, Migration and Polarization. Biol. Chem. 2004, 385, 885–892. [Google Scholar] [CrossRef]
- Challita-Eid, P.M.; Satpayev, D.; Yang, P.; An, Z.; Morrison, K.; Shostak, Y.; Raitano, A.; Nadell, R.; Liu, W.; Lortie, D.R.; et al. Enfortumab Vedotin Antibody-Drug Conjugate Targeting Nectin-4 Is a Highly Potent Therapeutic Agent in Multiple Preclinical Cancer Models. Cancer Res. 2016, 76, 3003–3013. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Chen, P.; Yin, W.; Ji, Y.; Shen, Q.; Ni, Q. Nectin-4 Promotes Gastric Cancer Progression via the PI3K/AKT Signaling Pathway. Hum. Pathol. 2018, 72, 107–116. [Google Scholar] [CrossRef]
- M-Rabet, M.; Cabaud, O.; Josselin, E.; Finetti, P.; Castellano, R.; Farina, A.; Agavnian-Couquiaud, E.; Saviane, G.; Collette, Y.; Viens, P.; et al. Nectin-4: A New Prognostic Biomarker for Efficient Therapeutic Targeting of Primary and Metastatic Triple-Negative Breast Cancer. Ann. Oncol. 2017, 28, 769–776. [Google Scholar] [CrossRef]
- Heath, E.I.; Rosenberg, J.E. The Biology and Rationale of Targeting Nectin-4 in Urothelial Carcinoma. Nat. Rev. Urol. 2021, 18, 93–103. [Google Scholar] [CrossRef]
- Erturk, K.; Karaman, S.; Dagoglu, N.; Serilmez, M.; Duranyildiz, D.; Tas, F. Serum Nectin-2 and Nectin-4 Are Diagnostic in Lung Cancer: Which Is Superior? Wien. Klin. Wochenschr. 2019, 131, 419–426. [Google Scholar] [CrossRef]
- Johnson, S.A.; Pleiman, C.M.; Pao, L.; Schneringer, J.; Hippen, K.; Cambier, J.C. Phosphorylated Immunoreceptor Signaling Motifs (ITAMs) Exhibit Unique Abilities to Bind and Activate Lyn and Syk Tyrosine Kinases. J. Immunol. 1995, 155, 4596–4603. [Google Scholar] [CrossRef]
- Bourbon, E.; Salles, G. Polatuzumab Vedotin: An Investigational Anti-CD79b Antibody Drug Conjugate for the Treatment of Diffuse Large B-Cell Lymphoma. Expert Opin. Investig. Drugs 2020, 29, 1079–1088. [Google Scholar] [CrossRef]
- RETH, M. Antigen Receptor Tail Clue. Nature 1989, 338, 383–384. [Google Scholar] [CrossRef]
- Hombach, J.; Tsubata, T.; Leclercq, L.; Stappert, H.; Reth, M. Molecular Components of the B-Cell Antigen Receptor Complex of the IgM Class. Nature 1990, 343, 760–762. [Google Scholar] [CrossRef]
- Pelanda, R.; Braun, U.; Hobeika, E.; Nussenzweig, M.C.; Reth, M. B Cell Progenitors Are Arrested in Maturation but Have Intact VDJ Recombination in the Absence of Ig-α and Ig-β. J. Immunol. 2002, 169, 865–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitz, R.; Wright, G.W.; Huang, D.W.; Johnson, C.A.; Phelan, J.D.; Wang, J.Q.; Roulland, S.; Kasbekar, M.; Young, R.M.; Shaffer, A.L.; et al. Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2018, 378, 1396–1407. [Google Scholar] [CrossRef] [PubMed]
- Naeim, F.; Nagesh Rao, P.; Song, S.X.; Phan, R.T. Principles of Immunophenotyping. In Atlas of Hematopathology; Elsevier: Amsterdam, The Netherlands, 2018; pp. 29–56. [Google Scholar]
- Young, R.M.; Shaffer, A.L.; Phelan, J.D.; Staudt, L.M. B-Cell Receptor Signaling in Diffuse Large B-Cell Lymphoma. Semin. Hematol. 2015, 52, 77–85. [Google Scholar] [CrossRef]
- Pfeifer, M.; Zheng, B.; Erdmann, T.; Koeppen, H.; McCord, R.; Grau, M.; Staiger, A.; Chai, A.; Sandmann, T.; Madle, H.; et al. Anti-CD22 and Anti-CD79B Antibody Drug Conjugates Are Active in Different Molecular Diffuse Large B-Cell Lymphoma Subtypes. Leukemia 2015, 29, 1578–1586. [Google Scholar] [CrossRef] [PubMed]
- Visco, C.; Tanasi, I.; Quaglia, F.M.; Ferrarini, I.; Fraenza, C.; Krampera, M. Oncogenic Mutations of MYD88 and CD79B in Diffuse Large B-Cell Lymphoma and Implications for Clinical Practice. Cancers 2020, 12, 2913. [Google Scholar] [CrossRef]
- Deeks, E.D. Polatuzumab Vedotin: First Global Approval. Drugs 2019, 79, 1467–1475. [Google Scholar] [CrossRef] [Green Version]
- Lanza, F.; Maffini, E.; Rondoni, M.; Massari, E.; Faini, A.C.; Malavasi, F. CD22 Expression in B-Cell Acute Lymphoblastic Leukemia: Biological Significance and Implications for Inotuzumab Therapy in Adults. Cancers 2020, 12, 303. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Gil, A.; Schnaar, R.L. Siglec Ligands. Cells 2021, 10, 1260. [Google Scholar] [CrossRef]
- Clark, E.A.; Giltiay, N.V. CD22: A Regulator of Innate and Adaptive B Cell Responses and Autoimmunity. Front. Immunol. 2018, 9, 2235. [Google Scholar] [CrossRef]
- Shah, N.N.; Sokol, L. Targeting CD22 for the Treatment of B-Cell Malignancies. ImmunoTargets Ther. 2021, 10, 225–236. [Google Scholar] [CrossRef]
- Tedder, T.F.; Sato, S.; Poe, J.C.; Fujimoto, M. CD19 and CD22 Regulate a B Lymphocyte Signal Transduction Pathway That Contributes to Autoimmunity. Keio J. Med. 2000, 49, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Melissaropoulos, K.; Liossis, S.-N. Decreased CD22 Expression and Intracellular Signaling Aberrations in B Cells of Patients with Systemic Sclerosis. Rheumatol. Int. 2018, 38, 1225–1234. [Google Scholar] [CrossRef]
- Lee, W.S.; Amengual, O. B Cells Targeting Therapy in the Management of Systemic Lupus Erythematosus. Immunol. Med. 2020, 43, 16–35. [Google Scholar] [CrossRef]
- Yurkiewicz, I.R.; Muffly, L.; Liedtke, M. Inotuzumab Ozogamicin: A CD22 MAb–Drug Conjugate for Adult Relapsed or Refractory B-Cell Precursor Acute Lymphoblastic Leukemia. Drug Des. Dev. Ther. 2018, 12, 2293–2300. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.; Li, W.; Mellors, J.W.; Orentas, R.; Dimitrov, D.S. Construction of a Large Size Human Immunoglobulin Heavy Chain Variable (VH) Domain Library, Isolation and Characterization of Novel Human Antibody VH Domains Targeting PD-L1 and CD22. Front. Immunol. 2022, 13, 869825. [Google Scholar] [CrossRef]
- Rousseau, J.; Lau, J.; Bénard, F. Radiolabeled Antibodies for Cancer Radioimmunotherapy. In Nuclear Medicine and Immunology; Springer International Publishing: Cham, Switzerland, 2022; pp. 297–345. [Google Scholar]
- Spiegel, J.Y.; Patel, S.; Muffly, L.; Hossain, N.M.; Oak, J.; Baird, J.H.; Frank, M.J.; Shiraz, P.; Sahaf, B.; Craig, J.; et al. CAR T Cells with Dual Targeting of CD19 and CD22 in Adult Patients with Recurrent or Refractory B Cell Malignancies: A Phase 1 Trial. Nat. Med. 2021, 27, 1419–1431. [Google Scholar] [CrossRef]
- Schneider, D.; Xiong, Y.; Wu, D.; Hu, P.; Alabanza, L.; Steimle, B.; Mahmud, H.; Anthony-Gonda, K.; Krueger, W.; Zhu, Z.; et al. Trispecific CD19-CD20-CD22–Targeting DuoCAR-T Cells Eliminate Antigen-Heterogeneous B Cell Tumors in Preclinical Models. Sci. Transl. Med. 2021, 13, eabc6401. [Google Scholar] [CrossRef]
- Dhillon, S. Moxetumomab Pasudotox: First Global Approval. Drugs 2018, 78, 1763–1767. [Google Scholar] [CrossRef] [Green Version]
- Zamarin, D.; Hamid, O.; Nayak-Kapoor, A.; Sahebjam, S.; Sznol, M.; Collaku, A.; Fox, F.E.; Marshall, M.A.; Hong, D.S. Mogamulizumab in Combination with Durvalumab or Tremelimumab in Patients with Advanced Solid Tumors: A Phase I Study. Clin. Cancer Res. 2020, 26, 4531–4541. [Google Scholar] [CrossRef]
- Yoshie, O.; Matsushima, K. CCR4 and Its Ligands: From Bench to Bedside. Int. Immunol. 2015, 27, 11–20. [Google Scholar] [CrossRef]
- Marshall, L.A.; Marubayashi, S.; Jorapur, A.; Jacobson, S.; Zibinsky, M.; Robles, O.; Hu, D.X.; Jackson, J.J.; Pookot, D.; Sanchez, J.; et al. Tumors Establish Resistance to Immunotherapy by Regulating T Reg Recruitment via CCR4. J. ImmunoTher. Cancer 2020, 8, e000764. [Google Scholar] [CrossRef] [PubMed]
- Ketcham, J.M.; Marshall, L.A.; Talay, O. CCR4 Antagonists Inhibit Treg Trafficking into the Tumor Microenvironment. ACS Med. Chem. Lett. 2018, 9, 953–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, M.; Kanao, K.; Suzuki, S.; Muramatsu, H.; Morinaga, S.; Kajikawa, K.; Kobayashi, I.; Nishikawa, G.; Kato, Y.; Zannami, K.; et al. Increased Infiltration of CCR4-Positive Regulatory T Cells in Prostate Cancer Tissue Is Associated with a Poor Prognosis. Prostate 2019, 79, 1658–1665. [Google Scholar] [CrossRef] [PubMed]
- Nicolay, J.P.; Albrecht, J.D.; Alberti-Violetti, S.; Berti, E. CCR4 in Cutaneous T-Cell Lymphoma: Therapeutic Targeting of a Pathogenic Driver. Eur. J. Immunol. 2021, 51, 1660–1671. [Google Scholar] [CrossRef]
- Hu, M.; Kang, G.; Cheng, X.; Wang, J.; Li, R.; Bai, Z.; Yang, D.; Huang, H. In Vitro Affinity Maturation to Improve the Efficacy of a Hypoxia-Inducible Factor 1α Single-Domain Intrabody. Biochem. Biophys. Res. Commun. 2020, 529, 936–942. [Google Scholar] [CrossRef]
- Zhao, H.; Bo, Q.; Wang, W.; Wang, R.; Li, Y.; Chen, S.; Xia, Y.; Wang, W.; Wang, Y.; Zhu, K.; et al. CCL17-CCR4 Axis Promotes Metastasis via ERK/MMP13 Pathway in Bladder Cancer. J. Cell. Biochem. 2019, 120, 1979–1989. [Google Scholar] [CrossRef]
- Ling, Z.; Li, W.; Hu, J.; Li, Y.; Deng, M.; Zhang, S.; Ren, X.; Wu, T.; Xia, J.; Cheng, B.; et al. Targeting CCL2-CCR4 Axis Suppress Cell Migration of Head and Neck Squamous Cell Carcinoma. Cell Death Dis. 2022, 13, 158. [Google Scholar] [CrossRef]
- Kohli, K.; Pillarisetty, V.G.; Kim, T.S. Key Chemokines Direct Migration of Immune Cells in Solid Tumors. Cancer Gene Ther. 2022, 29, 10–21. [Google Scholar] [CrossRef]
- Shimizu, Y.; Koyasu, S.; Suzukida, M.; Izumi, K.; Kidera, E.; Shindo, T.; Saga, T.; Ono, M.; Takaori-Kondo, A.; Nakamoto, Y. Development of a Novel Indium-111 Radiolabeled Mogamulizumab Targeting CCR4 for Imaging Adult T-Cell Leukemia/Lymphoma in Vivo. Ann. Nucl. Med. 2022, 36, 319–326. [Google Scholar] [CrossRef]
- Nazarenko, I.; Hede, S.M.; He, X.; Hedrén, A.; Thompson, J.; Lindström, M.S.; Nistér, M. PDGF and PDGF Receptors in Glioma. Upsala J. Med. Sci. 2012, 117, 99–112. [Google Scholar] [CrossRef] [Green Version]
- Fletcher, C.D.M.; Berman, J.J.; Corless, C.; Gorstein, F.; Lasota, J.; Longley, B.J.; Miettinen, M.; O’Leary, T.J.; Remotti, H.; Rubin, B.P.; et al. Diagnosis of Gastrointestinal Stromal Tumors: A Consensus Approach. Hum. Pathol. 2002, 33, 459–465. [Google Scholar] [CrossRef] [Green Version]
- Lemmon, M.A.; Schlessinger, J. Cell Signaling by Receptor Tyrosine Kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [Green Version]
- Miettinen, M.; Lasota, J. Gastrointestinal Stromal Tumors: Review on Morphology, Molecular Pathology, Prognosis, and Differential Diagnosis. Arch. Pathol. Lab. Med. 2006, 130, 1466–1478. [Google Scholar] [CrossRef]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved Seed Pairing, Often Flanked by Adenosines, Indicates That Thousands of Human Genes Are MicroRNA Targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef] [Green Version]
- Andrae, J.; Gallini, R.; Betsholtz, C. Role of Platelet-Derived Growth Factors in Physiology and Medicine. Genes Dev. 2008, 22, 1276–1312. [Google Scholar] [CrossRef] [Green Version]
- Mavroeidis, L.; Metaxa-Mariatou, V.; Papoudou-Bai, A.; Lampraki, A.M.; Kostadima, L.; Tsinokou, I.; Zarkavelis, G.; Papadaki, A.; Petrakis, D.; Gκoura, S.; et al. Comprehensive Molecular Screening by next Generation Sequencing Reveals a Distinctive Mutational Profile of KIT/PDGFRA Genes and Novel Genomic Alterations: Results from a 20-Year Cohort of Patients with GIST from North-Western Greece. ESMO Open 2018, 3, e000335. [Google Scholar] [CrossRef] [Green Version]
- Chang, K.K.; Yoon, C.; Yi, B.C.; Tap, W.D.; Simon, M.C.; Yoon, S.S. Platelet-Derived Growth Factor Receptor-α and -β Promote Cancer Stem Cell Phenotypes in Sarcomas. Oncogenesis 2018, 7, 47. [Google Scholar] [CrossRef] [Green Version]
- Pantaleo, M.A.; Tarantino, G.; Agostinelli, C.; Urbini, M.; Nannini, M.; Saponara, M.; Castelli, C.; Stacchiotti, S.; Fumagalli, E.; Gatto, L.; et al. Immune Microenvironment Profiling of Gastrointestinal Stromal Tumors (GIST) Shows Gene Expression Patterns Associated to Immune Checkpoint Inhibitors Response. OncoImmunology 2019, 8, e1617588. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Cui, R.; Clement, C.G.; Nawgiri, R.; Powell, D.W.; Pinchuk, I.V.; Watts, T.L. Activation PDGFR-α/AKT Mediated Signaling Pathways in Oral Squamous Cell Carcinoma by Mesenchymal Stem/Stromal Cells Promotes Anti-Apoptosis and Decreased Sensitivity to Cisplatin. Front. Oncol. 2020, 10, 552. [Google Scholar] [CrossRef]
- Maleddu, A.; Pantaleo, M.A.; Nannini, M.; Biasco, G. The Role of Mutational Analysis of KIT and PDGFRA in Gastrointestinal Stromal Tumors in a Clinical Setting. J. Transl. Med. 2011, 9, 75. [Google Scholar] [CrossRef] [Green Version]
- Poveda, A.; García del Muro, X.; López-Guerrero, J.A.; Cubedo, R.; Martínez, V.; Romero, I.; Serrano, C.; Valverde, C.; Martín-Broto, J. GEIS Guidelines for Gastrointestinal Sarcomas (GIST). Cancer Treat. Rev. 2017, 55, 107–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wozniak, A.; Rutkowski, P.; Piskorz, A.; Ciwoniuk, M.; Osuch, C.; Bylina, E.; Sygut, J.; Chosia, M.; Rys, J.; Urbanczyk, K.; et al. Prognostic Value of KIT/PDGFRA Mutations in Gastrointestinal Stromal Tumours (GIST): Polish Clinical GIST Registry Experience. Ann. Oncol. 2012, 23, 353–360. [Google Scholar] [CrossRef]
- Shi, L.; Chen, H.; Qin, Y.-Y.; Gan, T.-Q.; Wei, K.-L. Clinical and Biologic Roles of PDGFRA in Papillary Thyroid Cancer: A Study Based on Immunohistochemical and in Vitro Analyses. Int. J. Clin. Exp. Pathol. 2020, 13, 1094–1107. [Google Scholar] [PubMed]
- Lin, C.-L.; Tsai, M.-L.; Chen, Y.-H.; Liu, W.-N.; Lin, C.-Y.; Hsu, K.-W.; Huang, C.-Y.; Chang, Y.-J.; Wei, P.-L.; Chen, S.-H.; et al. Platelet-Derived Growth Factor Receptor-α Subunit Targeting Suppresses Metastasis in Advanced Thyroid Cancer In Vitro and In Vivo. Biomol. Ther. 2021, 29, 551–561. [Google Scholar] [CrossRef]
- Gonzáles, M.F.; Islas, A.E. Receptor SLAMF7 Asociado a Cáncer. Alianzas Y Tend.-BUAP 2019, 4, 15–21. [Google Scholar]
- Detre, C.; Keszei, M.; Romero, X.; Tsokos, G.C.; Terhorst, C. SLAM Family Receptors and the SLAM-Associated Protein (SAP) Modulate T Cell Functions. Semin. Immunopathol. 2010, 32, 157–171. [Google Scholar] [CrossRef] [Green Version]
- Cannons, J.L.; Tangye, S.G.; Schwartzberg, P.L. SLAM Family Receptors and SAP Adaptors in Immunity. Annu. Rev. Immunol. 2011, 29, 665–705. [Google Scholar] [CrossRef]
- Pérez-Quintero, L.-A.; Roncagalli, R.; Guo, H.; Latour, S.; Davidson, D.; Veillette, A. EAT-2, a SAP-like Adaptor, Controls NK Cell Activation through Phospholipase Cγ, Ca++, and Erk, Leading to Granule Polarization. J. Exp. Med. 2014, 211, 727–742. [Google Scholar] [CrossRef]
- Gutierrez-Guerrero, A.; Mancilla-Herrera, I.; Maravillas-Montero, J.L.; Martinez-Duncker, I.; Veillette, A.; Cruz-Munoz, M.E. SLAMF7 Selectively Favors Degranulation to Promote Cytotoxicity in Human NK Cells. Eur. J. Immunol. 2022, 52, 62–74. [Google Scholar] [CrossRef]
- Cruz-Munoz, M.-E.; Dong, Z.; Shi, X.; Zhang, S.; Veillette, A. Influence of CRACC, a SLAM Family Receptor Coupled to the Adaptor EAT-2, on Natural Killer Cell Function. Nat. Immunol. 2009, 10, 297–305. [Google Scholar] [CrossRef]
- O’Connell, P.; Hyslop, S.; Blake, M.K.; Godbehere, S.; Amalfitano, A.; Aldhamen, Y.A. SLAMF7 Signaling Reprograms T Cells toward Exhaustion in the Tumor Microenvironment. J. Immunol. 2021, 206, 193–205. [Google Scholar] [CrossRef]
- Zamagni, E.; Tacchetti, P.; Pantani, L.; Cavo, M. Anti-CD38 and Anti-SLAMF7: The Future of Myeloma Immunotherapy. Expert Rev. Hematol. 2018, 11, 423–435. [Google Scholar] [CrossRef]
- Araldi, R.P.; Prezoto, B.C.; Gonzaga, V.; Policiquio, B.; Mendes, T.B.; D’Amélio, F.; Vigerelli, H.; Viana, M.; Valverde, C.W.; Pagani, E.; et al. Advanced Cell Therapy with Low Tissue Factor Loaded Product NestaCell® Does Not Confer Thrombogenic Risk for Critically Ill COVID-19 Heparin-Treated Patients. Biomed. Pharmacother. 2022, 149, 112920. [Google Scholar] [CrossRef]
- Gao, Q.; Chen, Z.; He, Y.; Hou, Z.; Ye, R.; Xue, W.; Lin, J.; Tu, X. CD142 Plays an Important Role in the Mobility of Colorectal Cancer Cells. Biosci. Biotechnol. Biochem. 2020, 84, 1856–1860. [Google Scholar] [CrossRef]
- Xu, W.; Chen, B.; Ke, D.; Chen, X. CD142 Plays a Key Role in the Carcinogenesis of Gastric Adenocarcinoma by Inhibiting BCL2 -Dependent Autophagy. Biochem. Cell Biol. 2022, 100, 17–27. [Google Scholar] [CrossRef]
- Arderiu, G.; Peña, E.; Aledo, R.; Juan-Babot, O.; Badimon, L. Tissue Factor Regulates Microvessel Formation and Stabilization by Induction of Chemokine (C-C Motif) Ligand 2 Expression. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2607–2615. [Google Scholar] [CrossRef] [Green Version]
- Queiroz, K.C.S.; van’t Veer, C.; van den Berg, Y.; Duitman, J.; Versteeg, H.H.; Aberson, H.L.; Groot, A.P.; Verstege, M.I.; Roelofs, J.J.T.H.; te Velde, A.A.; et al. Tissue Factor-Dependent Chemokine Production Aggravates Experimental Colitis. Mol. Med. 2011, 17, 1119–1126. [Google Scholar] [CrossRef]
- Chanakira, A.; Westmark, P.R.; Ong, I.M.; Sheehan, J.P. Tissue Factor-Factor VIIa Complex Triggers Protease Activated Receptor 2-Dependent Growth Factor Release and Migration in Ovarian Cancer. Gynecol. Oncol. 2017, 145, 167–175. [Google Scholar] [CrossRef] [Green Version]
- Bhanvadia, R.R.; VanOpstall, C.; Brechka, H.; Barashi, N.S.; Gillard, M.; McAuley, E.M.; Vasquez, J.M.; Paner, G.; Chan, W.-C.; Andrade, J.; et al. MEIS1 and MEIS2 Expression and Prostate Cancer Progression: A Role For HOXB13 Binding Partners in Metastatic Disease. Clin. Cancer Res. 2018, 24, 3668–3680. [Google Scholar] [CrossRef] [Green Version]
- Pasquier, J.; Thomas, B.; Hoarau-Véchot, J.; Odeh, T.; Robay, A.; Chidiac, O.; Dargham, S.R.; Turjoman, R.; Halama, A.; Fakhro, K.; et al. Circulating Microparticles in Acute Diabetic Charcot Foot Exhibit a High Content of Inflammatory Cytokines, and Support Monocyte-to-Osteoclast Cell Induction. Sci. Rep. 2017, 7, 16450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stojkovic, S.; Kaun, C.; Basilio, J.; Rauscher, S.; Hell, L.; Krychtiuk, K.A.; Bonstingl, C.; de Martin, R.; Gröger, M.; Ay, C.; et al. Tissue Factor Is Induced by Interleukin-33 in Human Endothelial Cells: A New Link between Coagulation and Inflammation. Sci. Rep. 2016, 6, 25171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holnthoner, W.; Bonstingl, C.; Hromada, C.; Muehleder, S.; Zipperle, J.; Stojkovic, S.; Redl, H.; Wojta, J.; Schöchl, H.; Grillari, J.; et al. Endothelial Cell-Derived Extracellular Vesicles Size-Dependently Exert Procoagulant Activity Detected by Thromboelastometry. Sci. Rep. 2017, 7, 3707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markham, A. Tisotumab Vedotin: First Approval. Drugs 2021, 81, 2141–2147. [Google Scholar] [CrossRef]
- Liu, J.-N.; Kong, X.-S.; Huang, T.; Wang, R.; Li, W.; Chen, Q.-F. Clinical Implications of Aberrant PD-1 and CTLA4 Expression for Cancer Immunity and Prognosis: A Pan-Cancer Study. Front. Immunol. 2020, 11, 2048. [Google Scholar] [CrossRef]
- Xu, F.; Jin, T.; Zhu, Y.; Dai, C. Immune Checkpoint Therapy in Liver Cancer. J. Exp. Clin. Cancer Res. 2018, 37, 110. [Google Scholar] [CrossRef] [Green Version]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Dai, Z.; Wu, W.; Wang, Z.; Zhang, N.; Zhang, L.; Zeng, W.-J.; Liu, Z.; Cheng, Q. Regulatory Mechanisms of Immune Checkpoints PD-L1 and CTLA-4 in Cancer. J. Exp. Clin. Cancer Res. 2021, 40, 184. [Google Scholar] [CrossRef]
- Rowshanravan, B.; Halliday, N.; Sansom, D.M. CTLA-4: A Moving Target in Immunotherapy. Blood 2018, 131, 58–67. [Google Scholar] [CrossRef]
- Pai, C.-C.S.; Simons, D.M.; Lu, X.; Evans, M.; Wei, J.; Wang, Y.; Chen, M.; Huang, J.; Park, C.; Chang, A.; et al. Tumor-Conditional Anti-CTLA4 Uncouples Antitumor Efficacy from Immunotherapy-Related Toxicity. J. Clin. Investig. 2018, 129, 349–363. [Google Scholar] [CrossRef] [Green Version]
- Rotte, A. Combination of CTLA-4 and PD-1 Blockers for Treatment of Cancer. J. Exp. Clin. Cancer Res. 2019, 38, 255. [Google Scholar] [CrossRef]
- Carreau, N.A.; Pavlick, A.C. Nivolumab and Ipilimumab: Immunotherapy for Treatment of Malignant Melanoma. Future Oncol. 2019, 15, 349–358. [Google Scholar] [CrossRef]
- Shitara, K.; Ajani, J.A.; Moehler, M.; Garrido, M.; Gallardo, C.; Shen, L.; Yamaguchi, K.; Wyrwicz, L.; Skoczylas, T.; Bragagnoli, A.C.; et al. Nivolumab plus Chemotherapy or Ipilimumab in Gastro-Oesophageal Cancer. Nature 2022, 603, 942–948. [Google Scholar] [CrossRef]
- Overman, M.J.; Lonardi, S.; Wong, K.Y.M.; Lenz, H.-J.; Gelsomino, F.; Aglietta, M.; Morse, M.A.; Van Cutsem, E.; McDermott, R.; Hill, A.; et al. Durable Clinical Benefit With Nivolumab Plus Ipilimumab in DNA Mismatch Repair–Deficient/Microsatellite Instability–High Metastatic Colorectal Cancer. J. Clin. Oncol. 2018, 36, 773–779. [Google Scholar] [CrossRef]
- Owonikoko, T.K.; Park, K.; Govindan, R.; Ready, N.; Reck, M.; Peters, S.; Dakhil, S.R.; Navarro, A.; Rodríguez-Cid, J.; Schenker, M.; et al. Nivolumab and Ipilimumab as Maintenance Therapy in Extensive-Disease Small-Cell Lung Cancer: CheckMate 451. J. Clin. Oncol. 2021, 39, 1349–1359. [Google Scholar] [CrossRef]
- Tremelimumab. Drugs RD 2010, 10, 123–132. [CrossRef]
- Ricard-Blum, S. The Collagen Family. Cold Spring Harb. Perspect. Biol. 2011, 3, a004978. [Google Scholar] [CrossRef] [Green Version]
- Nallanthighal, S.; Heiserman, J.P.; Cheon, D.-J. Collagen Type XI Alpha 1 (COL11A1): A Novel Biomarker and a Key Player in Cancer. Cancers 2021, 13, 935. [Google Scholar] [CrossRef]
- Kadler, K.E.; Hill, A.; Canty-Laird, E.G. Collagen Fibrillogenesis: Fibronectin, Integrins, and Minor Collagens as Organizers and Nucleators. Curr. Opin. Cell Biol. 2008, 20, 495–501. [Google Scholar] [CrossRef]
- Morris, N.P.; Bächinger, H.P. Type XI Collagen Is a Heterotrimer with the Composition (1 Alpha, 2 Alpha, 3 Alpha) Retaining Non-Triple-Helical Domains. J. Biol. Chem. 1987, 262, 11345–11350. [Google Scholar] [CrossRef]
- Yoshioka, H.; Inoguchi, K.; Khaleduzzaman, M.; Ninomiya, Y.; Andrikopoulos, K.; Ramirez, F. Coding Sequence and Alternative Splicing of the Mouse A1(XI) Collagen Gene (Col11a1). Genomics 1995, 28, 337–340. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.Y.; Karsdal, M.A. Type XI Collagen. In Biochemistry of Collagens, Laminins and Elastin; Elsevier: Amsterdam, The Netherlands, 2016; pp. 77–80. [Google Scholar]
- Vázquez-Villa, F.; García-Ocaña, M.; Galván, J.A.; García-Martínez, J.; García-Pravia, C.; Menéndez-Rodríguez, P.; Rey, C.G.; Barneo-Serra, L.; de los Toyos, J.R. COL11A1/(pro)Collagen 11A1 Expression Is a Remarkable Biomarker of Human Invasive Carcinoma-Associated Stromal Cells and Carcinoma Progression. Tumor Biol. 2015, 36, 2213–2222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, H.; Li, J.; Lin, L.; Wang, L. COL11A1 Was Involved in Cell Proliferation, Apoptosis and Migration in Non-Small Cell Lung Cancer Cells. J. Investig. Surg. 2021, 34, 664–669. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Lai, J.; Jiang, H.; Ma, C.; Huang, H. Collagen XI Alpha 1 Chain, a Potential Therapeutic Target for Cancer. FASEB J. 2021, 35, e21603. [Google Scholar] [CrossRef]
- Wu, Y.-H.; Chang, T.-H.; Huang, Y.-F.; Huang, H.-D.; Chou, C.-Y. COL11A1 Promotes Tumor Progression and Predicts Poor Clinical Outcome in Ovarian Cancer. Oncogene 2014, 33, 3432–3440. [Google Scholar] [CrossRef]
- Giussani, M.; Landoni, E.; Merlino, G.; Turdo, F.; Veneroni, S.; Paolini, B.; Cappelletti, V.; Miceli, R.; Orlandi, R.; Triulzi, T.; et al. Extracellular Matrix Proteins as Diagnostic Markers of Breast Carcinoma. J. Cell. Physiol. 2018, 233, 6280–6290. [Google Scholar] [CrossRef]
- Zhang, J.; Dong, R.; Shen, L. Evaluation and Reflection on Claudin 18.2 Targeting Therapy in Advanced Gastric Cancer. Chin. J. Cancer Res. 2020, 32, 263–270. [Google Scholar] [CrossRef]
- Sweerus, K.; Lachowicz-Scroggins, M.; Gordon, E.; LaFemina, M.; Huang, X.; Parikh, M.; Kanegai, C.; Fahy, J.V.; Frank, J.A. Claudin-18 Deficiency Is Associated with Airway Epithelial Barrier Dysfunction and Asthma. J. Allergy Clin. Immunol. 2017, 139, 72–81.e1. [Google Scholar] [CrossRef] [Green Version]
- Kojima, T.; Kyuno, D.; Sawada, N. Targeting Claudin-4 in Human Pancreatic Cancer. Expert Opin. Ther. Targets 2012, 16, 881–887. [Google Scholar] [CrossRef]
- Kotton, D.N. Claudin-18: Unexpected Regulator of Lung Alveolar Epithelial Cell Proliferation. J. Clin. Investig. 2018, 128, 903–905. [Google Scholar] [CrossRef] [Green Version]
- Athauda, A.; Chau, I. Claudin 18.2—A FAST-Moving Target in Gastric Cancer? Ann. Oncol. 2021, 32, 584–586. [Google Scholar] [CrossRef]
- Hollande, F.; Blanc, E.M.; Bali, J.P.; Whitehead, R.H.; Pelegrin, A.; Baldwin, G.S.; Choquet, A. HGF Regulates Tight Junctions in New Nontumorigenic Gastric Epithelial Cell Line. Am. J. Physiol.-Gastrointest. Liver Physiol. 2001, 280, G910–G921. [Google Scholar] [CrossRef] [Green Version]
- Hong, J.Y.; An, J.Y.; Lee, J.; Park, S.H.; Park, J.O.; Park, Y.S.; Lim, H.Y.; Kim, K.-M.; Kang, W.K.; Kim, S.T. Claudin 18.2 Expression in Various Tumor Types and Its Role as a Potential Target in Advanced Gastric Cancer. Transl. Cancer Res. 2020, 9, 3367–3374. [Google Scholar] [CrossRef]
- Yang, J.; Liu, Z.; Zeng, B.; Hu, G.; Gan, R. Epstein–Barr Virus-Associated Gastric Cancer: A Distinct Subtype. Cancer Lett. 2020, 495, 191–199. [Google Scholar] [CrossRef]
- Dottermusch, M.; Krüger, S.; Behrens, H.-M.; Halske, C.; Röcken, C. Expression of the Potential Therapeutic Target Claudin-18.2 Is Frequently Decreased in Gastric Cancer: Results from a Large Caucasian Cohort Study. Virchows Arch. 2019, 475, 563–571. [Google Scholar] [CrossRef] [Green Version]
- Oue, N.; Sentani, K.; Sakamoto, N.; Yasui, W. Clinicopathologic and Molecular Characteristics of Gastric Cancer Showing Gastric and Intestinal Mucin Phenotype. Cancer Sci. 2015, 106, 951–958. [Google Scholar] [CrossRef] [Green Version]
- Bednarz-Misa, I.; Fortuna, P.; Diakowska, D.; Jamrozik, N.; Krzystek-Korpacka, M. Distinct Local and Systemic Molecular Signatures in the Esophageal and Gastric Cancers: Possible Therapy Targets and Biomarkers for Gastric Cancer. Int. J. Mol. Sci. 2020, 21, 4509. [Google Scholar] [CrossRef]
- Sahin, U.; Schuler, M.; Richly, H.; Bauer, S.; Krilova, A.; Dechow, T.; Jerling, M.; Utsch, M.; Rohde, C.; Dhaene, K.; et al. A Phase I Dose-Escalation Study of IMAB362 (Zolbetuximab) in Patients with Advanced Gastric and Gastro-Oesophageal Junction Cancer. Eur. J. Cancer 2018, 100, 17–26. [Google Scholar] [CrossRef] [Green Version]
- Türeci, O.; Sahin, U.; Schulze-Bergkamen, H.; Zvirbule, Z.; Lordick, F.; Koeberle, D.; Thuss-Patience, P.; Ettrich, T.; Arnold, D.; Bassermann, F.; et al. A Multicentre, Phase IIa Study of Zolbetuximab as a Single Agent in Patients with Recurrent or Refractory Advanced Adenocarcinoma of the Stomach or Lower Oesophagus: The MONO Study. Ann. Oncol. 2019, 30, 1487–1495. [Google Scholar] [CrossRef] [Green Version]
- Sahin, U.; Türeci, Ö.; Manikhas, G.; Lordick, F.; Rusyn, A.; Vynnychenko, I.; Dudov, A.; Bazin, I.; Bondarenko, I.; Melichar, B.; et al. FAST: A Randomised Phase II Study of Zolbetuximab (IMAB362) plus EOX versus EOX Alone for First-Line Treatment of Advanced CLDN18.2-Positive Gastric and Gastro-Oesophageal Adenocarcinoma. Ann. Oncol. 2021, 32, 609–619. [Google Scholar] [CrossRef]
- Horta, E.; Bongiorno, C.; Ezzeddine, M.; Neil, E.C. Neurotoxicity of Antibodies in Cancer Therapy: A Review. Clin. Neurol. Neurosurg. 2020, 188, 105566. [Google Scholar] [CrossRef] [PubMed]
- Minor, M.; Alcedo, K.P.; Battaglia, R.A.; Snider, N.T. Cell Type- and Tissue-Specific Functions of Ecto-5′-Nucleotidase (CD73). Am. J. Physiol.-Cell Physiol. 2019, 317, C1079–C1092. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Pu, N.; Yin, H.; Zhang, J.; Zhao, G.; Lou, W.; Wu, W. CD73 Acts as a Prognostic Biomarker and Promotes Progression and Immune Escape in Pancreatic Cancer. J. Cell. Mol. Med. 2020, 24, 8674–8686. [Google Scholar] [CrossRef] [PubMed]
- Ghalamfarsa, G.; Kazemi, M.H.; Raoofi Mohseni, S.; Masjedi, A.; Hojjat-Farsangi, M.; Azizi, G.; Yousefi, M.; Jadidi-Niaragh, F. CD73 as a Potential Opportunity for Cancer Immunotherapy. Expert Opin. Ther. Targets 2019, 23, 127–142. [Google Scholar] [CrossRef] [PubMed]
- Roh, M.; Wainwright, D.A.; Wu, J.D.; Wan, Y.; Zhang, B. Targeting CD73 to Augment Cancer Immunotherapy. Curr. Opin. Pharmacol. 2020, 53, 66–76. [Google Scholar] [CrossRef]
- Borea, P.A.; Gessi, S.; Merighi, S.; Vincenzi, F.; Varani, K. Pharmacology of Adenosine Receptors: The State of the Art. Physiol. Rev. 2018, 98, 1591–1625. [Google Scholar] [CrossRef] [Green Version]
- Hay, C.M.; Sult, E.; Huang, Q.; Mulgrew, K.; Fuhrmann, S.R.; McGlinchey, K.A.; Hammond, S.A.; Rothstein, R.; Rios-Doria, J.; Poon, E.; et al. Targeting CD73 in the Tumor Microenvironment with MEDI9447. OncoImmunology 2016, 5, e1208875. [Google Scholar] [CrossRef]
- Perrot, I.; Michaud, H.-A.; Giraudon-Paoli, M.; Augier, S.; Docquier, A.; Gros, L.; Courtois, R.; Déjou, C.; Jecko, D.; Becquart, O.; et al. Blocking Antibodies Targeting the CD39/CD73 Immunosuppressive Pathway Unleash Immune Responses in Combination Cancer Therapies. Cell Rep. 2019, 27, 2411–2425.e9. [Google Scholar] [CrossRef] [Green Version]
- Samanta, D.; Park, Y.; Ni, X.; Li, H.; Zahnow, C.A.; Gabrielson, E.; Pan, F.; Semenza, G.L. Chemotherapy Induces Enrichment of CD47+/CD73+/PDL1+ Immune Evasive Triple-Negative Breast Cancer Cells. Proc. Natl. Acad. Sci. USA 2018, 115, E1239–E1248. [Google Scholar] [CrossRef] [Green Version]
- Chapoval, A.I.; Ni, J.; Lau, J.S.; Wilcox, R.A.; Flies, D.B.; Liu, D.; Dong, H.; Sica, G.L.; Zhu, G.; Tamada, K.; et al. B7-H3: A Costimulatory Molecule for T Cell Activation and IFN-γ Production. Nat. Immunol. 2001, 2, 269–274. [Google Scholar] [CrossRef]
- Liu, J.; Yang, S.; Cao, B.; Zhou, G.; Zhang, F.; Wang, Y.; Wang, R.; Zhu, L.; Meng, Y.; Hu, C.; et al. Targeting B7-H3 via Chimeric Antigen Receptor T Cells and Bispecific Killer Cell Engagers Augments Antitumor Response of Cytotoxic Lymphocytes. J. Hematol. Oncol. 2021, 14, 21. [Google Scholar] [CrossRef]
- Feng, R.; Chen, Y.; Liu, Y.; Zhou, Q.; Zhang, W. The Role of B7-H3 in Tumors and Its Potential in Clinical Application. Int. Immunopharmacol. 2021, 101, 108153. [Google Scholar] [CrossRef]
- Picarda, E.; Galbo, P.M.; Zong, H.; Rajan, M.R.; Wallenius, V.; Zheng, D.; Börgeson, E.; Singh, R.; Pessin, J.; Zang, X. The Immune Checkpoint B7-H3 (CD276) Regulates Adipocyte Progenitor Metabolism and Obesity Development. Sci. Adv. 2022, 8, eabm7012. [Google Scholar] [CrossRef]
- MacGregor, H.L.; Sayad, A.; Elia, A.; Wang, B.X.; Katz, S.R.; Shaw, P.A.; Clarke, B.A.; Crome, S.Q.; Robert-Tissot, C.; Bernardini, M.Q.; et al. High Expression of B7-H3 on Stromal Cells Defines Tumor and Stromal Compartments in Epithelial Ovarian Cancer and Is Associated with Limited Immune Activation. J. ImmunoTher. Cancer 2019, 7, 357. [Google Scholar] [CrossRef] [Green Version]
- Han, S.; Wang, Y.; Shi, X.; Zong, L.; Liu, L.; Zhang, J.; Qian, Q.; Jin, J.; Ma, Y.; Cui, B.; et al. Negative Roles of B7-H3 and B7-H4 in the Microenvironment of Cervical Cancer. Exp. Cell Res. 2018, 371, 222–230. [Google Scholar] [CrossRef]
- Hu, X.; Xu, M.; Hu, Y.; Li, N.; Zhou, L. B7-H3, Negatively Regulated by MiR-128, Promotes Colorectal Cancer Cell Proliferation and Migration. Cell Biochem. Biophys. 2021, 79, 397–405. [Google Scholar] [CrossRef]
- Zhou, W.T.; Jin, W.L. B7-H3/CD276: An Emerging Cancer Immunotherapy. Front. Immunol. 2021, 12, 701006. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, J.; Li, C.; Xu, H.; Dong, R.; Chen, C.C.; Hua, W. Survival Association and Cell Cycle Effects of B7H3 in Neuroblastoma. J. Korean Neurosurg. Soc. 2020, 63, 707–716. [Google Scholar] [CrossRef]
- Flem-Karlsen, K.; Fodstad, Ø.; Tan, M.; Nunes-Xavier, C.E. B7-H3 in Cancer—Beyond Immune Regulation. Trends Cancer 2018, 4, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Wei, W.; Zhao, Q. B7-H3, a Checkpoint Molecule, as a Target for Cancer Immunotherapy. Int. J. Biol. Sci. 2020, 16, 1767–1773. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Quan, Y.; Che, F.; Wang, L. B7-H3 in Tumors: Friend or Foe for Tumor Immunity? Cancer Chemother. Pharmacol. 2018, 81, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Memorial Sloan Kettering Cancer Center. MSK Kids–Neuroblastoma Treatments. Available online: https://www.mskcc.org/pediatrics/cancer-care/types/neuroblastoma/treatment (accessed on 13 July 2023).
- McCormick, S.M.; Heller, N.M. Commentary: IL-4 and IL-13 Receptors and Signaling. Cytokine 2015, 75, 38–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Junttila, I.S. Tuning the Cytokine Responses: An Update on Interleukin (IL)-4 and IL-13 Receptor Complexes. Front. Immunol. 2018, 9, 888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandhi, N.A.; Pirozzi, G.; Graham, N.M.H. Commonality of the IL-4/IL-13 Pathway in Atopic Diseases. Expert Rev. Clin. Immunol. 2017, 13, 425–437. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, C.; Zhang, M.; Li, Z. IL-13 and IL-13Rα1 Are Overexpressed in Extranodal Natural Killer/T Cell Lymphoma and Mediate Tumor Cell Proliferation. Biochem. Biophys. Res. Commun. 2018, 503, 2715–2720. [Google Scholar] [CrossRef]
- Suzuki, A.; Leland, P.; Joshi, B.H.; Puri, R.K. Targeting of IL-4 and IL-13 Receptors for Cancer Therapy. Cytokine 2015, 75, 79–88. [Google Scholar] [CrossRef]
- Song, X.; Traub, B.; Shi, J.; Kornmann, M. Possible Roles of Interleukin-4 and -13 and Their Receptors in Gastric and Colon Cancer. Int. J. Mol. Sci. 2021, 22, 727. [Google Scholar] [CrossRef]
- Jaén, M.; Martín-Regalado, Á.; Bartolomé, R.A.; Robles, J.; Casal, J.I. Interleukin 13 Receptor Alpha 2 (IL13Rα2): Expression, Signaling Pathways and Therapeutic Applications in Cancer. Biochim. Biophys. Acta BBA- Rev. Cancer 2022, 1877, 188802. [Google Scholar] [CrossRef]
- Kornmann, M.; Kleeff, J.; Debinski, W.; Korc, M. Pancreatic Cancer Cells Express Interleukin-13 and -4 Receptors, and Their Growth Is Inhibited by Pseudomonas Exotoxin Coupled to Interleukin-13 and -4. Anticancer. Res. 1999, 19, 125–131. [Google Scholar]
- Geng, B.; Pan, J.; Zhao, T.; Ji, J.; Zhang, C.; Che, Y.; Yang, J.; Shi, H.; Li, J.; Zhou, H.; et al. Chitinase 3-like 1-CD44 Interaction Promotes Metastasis and Epithelial-to-Mesenchymal Transition through β-Catenin/Erk/Akt Signaling in Gastric Cancer. J. Exp. Clin. Cancer Res. 2018, 37, 208. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, S.; Wang, Q.; Zhang, X. Tumor-Recruited M2 Macrophages Promote Gastric and Breast Cancer Metastasis via M2 Macrophage-Secreted CHI3L1 Protein. J. Hematol. Oncol. 2017, 10, 36. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.; Liu, H.; Zhang, H.; He, H.; Li, H.; Shen, Z.; Qin, J.; Qin, X.; Xu, J.; Sun, Y. Interleukin-13 Receptor A2 Is Associated with Poor Prognosis in Patients with Gastric Cancer after Gastrectomy. Oncotarget 2016, 7, 49281–49288. [Google Scholar] [CrossRef] [Green Version]
- Manso, T.; Kushwaha, A.; Abdollahi, N.; Duroux, P.; Giudicelli, V.; Kossida, S. Mechanisms of Action of Monoclonal Antibodies in Oncology Integrated in IMGT/MAb-DB. Front. Immunol. 2023, 14, 1129323. [Google Scholar] [CrossRef]
- O’Mahony, D.; Bishop, M. Monoclonal Antibody Therapy. Front. Biosci. 2006, 11, 1620–1635. [Google Scholar] [CrossRef]
- Saldarriaga-Valiente, T. Monoclonal Antibodies: Mechanisms of Actions. Diagnostico 2021, 60, 213–217. [Google Scholar] [CrossRef]
- Baltazar, E.; Christen, G. Conjugados Anticuerpo-Farmaco: El Estado de Arte. Rev. Mex. De Cienc. Farm. 2011, 42, 7–16. [Google Scholar]
- Fu, Z.; Li, S.; Han, S.; Shi, C.; Zhang, Y. Antibody Drug Conjugate: The “Biological Missile” for Targeted Cancer Therapy. Signal Transduct. Target. Ther. 2022, 7, 93. [Google Scholar] [CrossRef]
- Tsao, L.; Force, J.; Hartman, Z. Mechanisms of Therapeutic Antitumor Monoclonal Antibodies. Cancer Res. 2021, 81, 4641–4651. [Google Scholar] [CrossRef]
- Rodríguez-Nava, C.; Ortuño-Pineda, C.; Illades-Aguiar, B.; Flores-Alfaro, E.; Leyva-Vázquez, M.; Parra-Rojas, I.; Moral-Hernández, O.; Vences-Velázquez, A.; Cortés-Sarabia, K.; Alarcón-Romero, O. Mechanisms of Action and Limitations of Monoclonal Antibodies and Single Chain Fragment Variable (ScFv) in the Treatment of Cancer. Biomedicines 2023, 11, 1610. [Google Scholar] [CrossRef]
- Jin, S.; Sun, Y.; Liang, X.; Gu, X.; Ning, J.; Xu, Y.; Chen, S.; Pan, L. Emerging New Therapeutic Antibody Derivatives for Cancer Treatment. Nature 2022, 7, 39. [Google Scholar] [CrossRef]
- Bayer, V. An Overview of Monoclonal Antibodies. Semin. Oncol. Nurs. 2019, 35, 15092. [Google Scholar] [CrossRef] [PubMed]
- Redman, J.; Hill, E.; AlDeghaither, D.; Weiner, L. Mechanisms of Action of Therapeutic Antibodies for Cancer. Mol. Immunol. 2015, 67, 28–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Marks, J. Mechanism of Action for Therapeutic Antibodies. In Biosimilars of Monoclonal Antibodies: A Practical Guide to Manufacturing, Preclinical, and Clinical Development; Wiley: New York, NY, USA, 2016. [Google Scholar]
- Weiner, G. Monoclonal Antibody Mechanisms of Action in Cancer. Immunol. Res. 2007, 39, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Yip, H.; Papa, A. Signaling Pathways in Cancer: Therapeutic Targets, Combinatorial Treatments, and New Developments. Cells 2021, 10, 659. [Google Scholar] [CrossRef]
- Mezynski, M.; Farrelly, A.; Cremona, M.; Carr, A.; Morgan, C.; Workman, J.; Armstrong, P.; McAuley, J.; Madden, S.; Fay, J.; et al. Targeting the PI3K and MAPK Pathways to Improve Response to HER2-Targeted Therapies in HER2-Positive Gastric Cancer. J. Transl. Med. 2021, 19, 184. [Google Scholar] [CrossRef]
- Costa, R.; Czerniecki, B. Clinical Development of Immunotherapies for HER2+ Breast Cancer: A Review of HER2-Directed Monoclonal Antibodies and Beyond. NPJ Breast Cancer 2020, 6, 10. [Google Scholar] [CrossRef]
- Min, H.; Lee, H. Molecular Targeted Therapy for Anticancer Treatment. Exp. Mol. Med. 2022, 54, 1670–1694. [Google Scholar] [CrossRef]
- Li, X.; Shao, C.; Shi, Y.; Han, W. Lessons Learned from the Blockade of Immune Checkpoints in Cancer Immunotherapy. J. Hematol. Oncol. 2018, 11, 31. [Google Scholar] [CrossRef] [Green Version]
- Forthal, D.; Finzi, A. Antibody-Dependent Cellular Cytotoxicity (ADCC) in HIV Infection. AIDS 2018, 32, 2439–2451. [Google Scholar] [CrossRef]
- Tay, M.; Wuihe, K.; Pollara, J. Antibody-Dependent Cellular Phagocytosis in Antiviral Immune Responses. Front. Immunol. 2019, 10, 332. [Google Scholar] [CrossRef] [Green Version]
- Mortensen, S.A.; Sander, B.; Jensen, R.K.; Pedersen, J.S.; Golas, M.M.; Jensenius, J.C. Structure and Activation of C1, the Complex Initiating the Classical Pathway of the Complement Cascade. Proc. Natl. Acad. Sci. USA 2017, 114, 986–991. [Google Scholar] [CrossRef]
- Fishelson, Z.; Kirschfink, M. Complement C5b-9 and Cancer: Mechanisms of Cell Damage, Cancer Counteractions, and Approaches for Intervention. Front. Immunol. 2019, 10, 752. [Google Scholar] [CrossRef] [Green Version]
- Natsume, A.; Niwa, R.; Satoh, M. Improving Effector Functions of Antibodies for Cancer Treatment: Enhancing ADCC and CDC. Drug Des. Dev. Ther. 2009, 3, 7–16. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Woods, C.; He, F.; Han, H.; Treuheit, M.; Volkin, D. Structural Changes and Aggregation Mechanisms of Two Different Dimers of an IgG2 Monoclonal Antibody. Biochemistry 2018, 57, 5466–5479. [Google Scholar] [CrossRef]
- Stapleton, N.; Andersen, J.; Stemerding, A.; Bjarnarson, S.; Verheul, R.; Gerritsen, J.; Zhao, Y.; Kleijer, M.; Sandlie, I.; Jonsdottir, M.; et al. Competition for FcRn-Mediated Transport Gives Rise to Short Half-Life of Human IgG3 and Offers Therapeutic Potential. Nat. Commun. 2011, 2, 599. [Google Scholar] [CrossRef] [Green Version]
- Spiess, C.; Bevers, J.; Jackman, J.; Chiang, N.; Nakamura, G.; Dillon, M.; Liu, H.; Molina, P.; Elliott, J.; Shatz, W.; et al. Development of a Human IgG4 Bispecific Antibody for Dual Targeting of Interleukin-4 (IL-4) and Interleukin-13 (IL-13) Cytokines. J. Biol. Chem. 2013, 288, 26583–26593. [Google Scholar] [CrossRef] [Green Version]
- Markham, A. Dostarlimab: First Approval. Drugs 2021, 81, 1213–1219. [Google Scholar] [CrossRef]
- Markham, A.; Duggan, S. Cemiplimab: First Global Approval. Drugs 2018, 78, 1841–1846. [Google Scholar] [CrossRef]
- Antonia, S.J.; Villegas, A.; Daniel, D.; Vicente, D.; Murakami, S.; Hui, R.; Yokoi, T.; Chiappori, A.; Lee, K.H.; de Wit, M.; et al. Durvalumab after Chemoradiotherapy in Stage III Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 377, 1919–1929. [Google Scholar] [CrossRef] [Green Version]
- Powles, T.; Park, S.H.; Voog, E.; Caserta, C.; Valderrama, B.P.; Gurney, H.; Kalofonos, H.; Radulović, S.; Demey, W.; Ullén, A.; et al. Avelumab Maintenance Therapy for Advanced or Metastatic Urothelial Carcinoma. N. Engl. J. Med. 2020, 383, 1218–1230. [Google Scholar] [CrossRef]
- Collins, J.; Gulley, J. Product Review: Avelumab, an Anti-PD-L1 Antibody. Hum. Vaccines Immunother. 2019, 15, 891–908. [Google Scholar] [CrossRef] [PubMed]
- Markham, A. Atezolizumab: First Global Approval. Drugs 2016, 76, 1227–1232. [Google Scholar] [CrossRef]
- Hosseini, I.; Gadkar, K.; Stefanich, E.; Li, C.; Sun, L.; Chu, Y.; Ramanujan, S. Mitigating the Risk of Cytokine Release Syndrome in a Phase I Trial of CD20/CD3 Bispecific Antibody Mosunetuzumab in NHL: Impact of Translational System Modeling. NPJ Syst. Biol. Appl. 2020, 6, 28. [Google Scholar] [CrossRef] [PubMed]
- Sehn, L.; Duell, J.; Avivi, I.; Brody, J.; Yoon, D.; Elliot, B.; Siddani, S.; Bai, Y.; Parikh, A.; Seliem, M.; et al. Subcutaneous Epcoritamab in Novel Combinations with Antineoplastic Agents Among Patients with B-Cell Non-Hodgkin Lymphoma in a Phase 1b/2, Multicenter, Open-Label Study: Assessing Safety, Tolerability, and Preliminary Efficacy (EPCORE NHL-5). Blood 2022, 140 (Suppl. 1), 12108–12109. [Google Scholar] [CrossRef]
- Markham, A. Margetuximab: First Approval. Drugs 2021, 81, 599–604. [Google Scholar] [CrossRef]
- Keam, S. Trastuzumab Deruxtecan: First Approval. Drugs 2020, 80, 501–508. [Google Scholar] [CrossRef]
- Lee, A. Loncastuximab Tesirine: First Approval. Drugs 2021, 81, 1229–1233. [Google Scholar] [CrossRef]
- Hartley, J.A.; Flynn, M.J.; Bingham, J.P.; Corbett, S.; Reinert, H.; Tiberghien, A.; Masterson, L.A.; Antonow, D.; Adams, L.; Chowdhury, S.; et al. Pre-Clinical Pharmacology and Mechanism of Action of SG3199, the Pyrrolobenzodiazepine (PBD) Dimer Warhead Component of Antibody-Drug Conjugate (ADC) Payload Tesirine. Sci. Rep. 2018, 8, 10479. [Google Scholar] [CrossRef] [Green Version]
- Hoy, S.M. Tafasitamab: First Approval. Drugs 2020, 80, 1731–1737. [Google Scholar] [CrossRef]
- Salles, G.; Długosz-Danecka, M.; Ghesquières, H.; Jurczak, W. Tafasitamab for the Treatment of Relapsed or Refractory Diffuse Large B-Cell Lymphoma. Expert Opin. Biol. Ther. 2021, 21, 455–463. [Google Scholar] [CrossRef]
- Markham, A. Naxitamab: First Approval. Drugs 2021, 81, 291–296. [Google Scholar] [CrossRef]
- Kang, C. Teclistamab: First Approval. Drugs 2022, 82, 1613–1619. [Google Scholar] [CrossRef]
- Markham, A. Belantamab Mafodotin: First Approval. Drugs 2020, 80, 1607–1613. [Google Scholar] [CrossRef]
- Syed, Y.Y. Sacituzumab Govitecan: First Approval. Drugs 2020, 80, 1019–1025. [Google Scholar] [CrossRef]
- Bardia, A.; Hurvitz, S.A.; Tolaney, S.M.; Loirat, D.; Punie, K.; Oliveira, M.; Brufsky, A.; Sardesai, S.D.; Kalinsky, K.; Zelnak, A.B.; et al. Sacituzumab Govitecan in Metastatic Triple-Negative Breast Cancer. N. Engl. J. Med. 2021, 384, 1529–1541. [Google Scholar] [CrossRef]
- Dhillon, S. Isatuximab: First Approval. Drugs 2020, 80, 905–912. [Google Scholar] [CrossRef]
- Feng, X.; Zhang, L.; Acharya, C.; An, G.; Wen, K.; Qiu, L.; Munshi, N.; Tai, Y.; Anderson, K. Targeting CD38 Suppresses Induction and Function of T Regulatory Cells to Mitigate Immunosuppression in Multiple Myeloma. Clin. Cancer Res. 2017, 23, 4290–4300. [Google Scholar] [CrossRef] [Green Version]
- Shen, F.; Shen, W. Isatuximab in the Treatment of Multiple Myeloma: A Review and Comparison With Daratumumab. Technol. Cancer Res. Treat. 2022, 21, 15330338221106563. [Google Scholar] [CrossRef]
- Alt, M.; Stecca, C.; Tobin, S.; Jiang, D.M.; Sridhar, S.S. Enfortumab Vedotin in Urothelial Cancer. Ther. Adv. Urol. 2020, 12, 175628722098019. [Google Scholar] [CrossRef]
- Internet. LiverTox: Clinical and Research Information on Drug-Induced Liver Injury; Internet, 2019. [Google Scholar]
- Lamb, Y.N. Inotuzumab Ozogamicin: First Global Approval. Drugs 2017, 77, 1603–1610. [Google Scholar] [CrossRef]
- Subramaniam, J.M.; Whiteside, G.; McKeage, K.; Croxtall, J.C. Mogamulizumab. Drugs 2012, 72, 1293–1298. [Google Scholar] [CrossRef]
- Duvic, M.; Evans, M.; Wang, C. Mogamulizumab for the Treatment of Cutaneous T-Cell Lymphoma: Recent Advances and Clinical Potential. Ther. Adv. Hematol. 2016, 7, 171–174. [Google Scholar] [CrossRef] [PubMed]
- Shirley, M. Olaratumab: First Global Approval. Drugs 2017, 77, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Markham, A. Elotuzumab: First Global Approval. Drugs 2016, 76, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Castet, F.; Heikenwalder, M.; Maini, M.K.; Mazzaferro, V.; Pinato, D.J.; Pikarsky, E.; Zhu, A.X.; Finn, R.S. Immunotherapies for Hepatocellular Carcinoma. Nat. Rev. Clin. Oncol. 2022, 19, 151–172. [Google Scholar] [CrossRef] [PubMed]
- Keam, S. Tremelimumab: First Approval. Drugs 2023, 83, 93–102. Available online: https://link.springer.com/article/10.1007/s40265-022-01827-8 (accessed on 13 July 2023). [CrossRef]
- Farkona, S.; Diamandis, E.P.; Blasutig, I.M. Cancer Immunotherapy: The Beginning of the End of Cancer? BMC Med. 2016, 14, 73. [Google Scholar] [CrossRef] [Green Version]
- Cercek, A.; Lumish, M.; Sinopoli, J.; Weiss, J.; Shia, J.; Lamendola-Essel, M.; El Dika, I.H.; Segal, N.; Shcherba, M.; Sugarman, R.; et al. PD-1 Blockade in Mismatch Repair–Deficient, Locally Advanced Rectal Cancer. N. Engl. J. Med. 2022, 386, 2363–2376. [Google Scholar] [CrossRef]
- Pasello, M.; Mallano, A.; Flego, M.; Zamboni, S.; Giudice, A.; Scotlandi, K. Construction of Human Naive Antibody Gene Liraries. In Antibody Engineering; Nevoltris, D., Patrick, C., Eds.; Springer: Berlin/Heidelberg, Germany, 2018; pp. 73–91. ISBN 978-1-4939-8648-4. [Google Scholar]
- Caucheteur, D.; Robin, G.; Parez, V.; Martineau, P. Construction of Synthetic Antibody Libraries. In Antibody Engineering; Nevoltris, D., Chames, P., Eds.; Springer: Berlin/Heidelberg, Germany, 2018; pp. 93–108. [Google Scholar]
- Kugler, J.; Tomszak, F.; Frenzel, A.; Hust, M. Construction of Human Immune and Naive ScFv Libraries. In Phage Display Methods and Protocols; Hust, M., Lim, T.S., Eds.; Springer: Berlin/Heidelberg, Germany, 2018; Volume 1701, pp. 3–24. ISBN 978-1-4939-7446-7. [Google Scholar]
- Scholler, N. Selection of Antibody Fragments by Yeast Display. In Antibody Engineering Methods and Protocols; Nevoltris, D., Chames, P., Eds.; Springer: Berlin/Heidelberg, Germany, 2018; pp. 211–233. [Google Scholar]
- Dreier, B.; Pluckthun, A. Rapid Selection of High-Affinity Antibody ScFv Fragments Using Ribosome Display. In Antibody Engineering; Nevoltris, D., Chames, P., Eds.; Springer: Berlin/Heidelberg, Germany, 2018; pp. 235–268. [Google Scholar]
- Nemoto, N.; Kumachi, S.; Arai, H. In Vitro Selection of Single-Domain Antibody (VHH) Using CDNA Display. In Antibody Engineering Methods and Protocols; Nevoltris, D., Chames, P., Eds.; Springer: Berlin/Heidelberg, Germany, 2018; pp. 269–285. [Google Scholar]
- Smith, G.P. Phage Display: Simple Evolution in a Petri Dish (Nobel Lecture). Angew. Chem.-Int. Ed. 2019, 58, 14428–14437. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
| mAbs | Antigen that Recognizes | Format | Cancer for It Was Approved | Mechanism of Action | Reference |
|---|---|---|---|---|---|
| Dostarlimab | PD1 | IgG4-humanized | Endometrial Cancer | Produced from a mouse hybridoma that acts as a PD-1 blocker via steric impediment with PD-L1 and PD-L2, thereby normalizing the immune response. | [279] |
| Cemiplimab | PD1 | IgG4-humanized | Cutaneous squamous cell carcinoma | This mAb binds to PD1 on T-cells, blocking the interaction with PDL-1 and PDL-2 ligands and activating the immune response. | [280] |
| Durvalumab | PD-L1 | IgG1-human | Bladder cancer | This is a human mAb with high affinity by PD-L1 and CD80. | [281] |
| Avelumab | PD-L1 | IgG1-human | Merkel cell carcinoma | Regulates cytotoxicity mediated by antibody-dependent cells, due to the fact that it presents a native Fc region. | [282,283] |
| Atezolizumab | PD-L1 | IgG1-humanized | Bladder cancer | Inhibits the interaction between PD-1 and B7.1, restoring the antitumor function of T cells. | [284] |
| Retifanlimab | PD-L1 | IgG4-humanized | Merkel cell carcinoma | Blocks PD-1 interaction with its PD-L1 and PD-L2 ligands. | [30] |
| Mosunetuzumab | CD20-CD3 | IgG1-humanized bispecific | Follicular lymphoma | Simultaneously binds to CD20 on malignant B cells and CD3 on T cells, causing T-cell activation and B-cell elimination. | [39,285] |
| Epcoritamab | CD20-CD3 | IgG1-humanized bispecific | Diffuse large B-cell lymphoma | Induces T cells to kill CD20+ tumor cells through a unique mechanism of action (MOA). | [40,286] |
| Margetuximab | HER2 | IgG1-chimeric | HER2+ breast cancer | Designed against HER2 to decrease binding to the inhibitory receptor Fcγ IIB (CD32B) and to increase binding to the receptor Fcγ activation IIA (CD16A). | [287] |
| Fam-Trastuzumab Deruxtecan | HER2 | IgG1-humanized antibody–drug conjugate | HER2+ breast cancer | Designed against HER2 and conjugated to a cytotoxic topoisomerase 1 inhibitor | [80,288] |
| Loncastuximab Tesirine | CD19 | IgG1-humanized antibody–drug conjugate | Diffuse large B-cell lymphoma | Designed to target CD19 and conjugated to a pyrrolobenzodiazepine DNA-alkylating warhead. It produces DNA interstrand crosslinks with high efficiency, leading to triggering of cell death. | [289,290] |
| Tafasitamab | CD19 | IgG1-humanized | Diffuse large B-cell lymphoma | Produces antibody-dependent cellular cytotoxicity and antibody-dependent cell-mediated phagocytosis. | [291,292] |
| Naxitamab | GD2 | IgG1-humanized | Neuroblastoma | Induces complement-dependent and cell-mediated antibody-dependent cytotoxicity | [293] |
| Teclistamab | BCMA-CD3 | IgG4-humanized bispecific | Multiple myeloma | Redirects CD3-positive cells to BCMA-expressing tumor cells, inducing cytotoxicity and apoptosis. | [294] |
| Belantamab mafodotin | BCMA | IgG1-humanized antibody–drug conjugate | Multiple myeloma | Composed of an antibody that targets B-cell maturation antigen (BCMA), conjugated to the microtubule inhibitor monomethyl auristatin F (MMAF). The other part of the antibody binds to BCMA on the surface of the tumor cell, delivering the cytotoxic microtubule inhibitor MMAF to the therapeutic target. | [295] |
| Sacituzumab govitecan | TROP-2 | IgG1-humanized antibody–drug conjugate | Triple-negative breast cancer | This mAb acts against TROP-2 conjugate with the active metabolite of irinotecan and topoisomerase 1 inhibitor. | [296,297] |
| Isatuximab | CD38 | IgG1-chimeric | Multiple myeloma | By binding to CD38, this mAb causes apoptosis via multiple mechanisms such as antibody-dependent cellular phagocytosis, complement-dependent cytotoxicity, and effects that depend on the Fc region. | [298,299,300] |
| Enfortumab vedotin | Nectin-4 | IgG1-human antibody–drug conjugate | Urothelial cancer | Works by releasing monomethyl auristatin E (MMAE) into cells that express nectin-4, causing apoptosis. | [301] |
| Polatuzumab vedotin | CD79b | IgG1-humanized antibody–drug conjugate | Diffuse large B-cell lymphoma | Works by binding to CD79b upon entering the cell, releasing MMAE, inhibiting cell division, and inducing apoptosis. | [124] |
| Moxetumomab pasudotox | CD22 | IgG1-murine dsFv | Hairy cell leukemia | This mAb is conjugated with a toxic fragment of A exotoxin from Pseudomonas aeruginosa, which is internalized, resulting in apoptotic cell death. | [137,302] |
| Inotuzumab ozogamicin | CD22 | IgG4-humanized antibody–drug conjugate | Hematological malignancy | By binding to CD22, the cytotoxic derivative of calicheamicin enters the cell, causing apoptosis. | [303] |
| Mogamulizumab | CCR4 | IgG1-humanized | Cutaneous T-cell lymphoma | Has a defucosylated Fc region that enhances its antibody-dependent cellular cytotoxicity. | [304,305] |
| Olaratumab | PDGRFα | IgG1-human | Soft-tissue sarcoma | Blocks PDGF ligand biding and inhibits PDGFRα. | [306] |
| Elotuzumab | SLAMF7 | IgG1-humanized | Multiple myeloma | Induces antibody-dependent cellular cytotoxicity and the activation of natural killer cells. | [307] |
| Tisotumab vedotin | CD142 | IgG1-human antibody–drug conjugate | Cervical cancer | This is a human mAb conjugated with an antimitotic monomethyl auristatin E, which inhibits cell division by blocking polymerization of tubulin. | [183] |
| Tremelimumab | CTLA-4 | IgG2A-human | Liver cancer | Blocks the union of B7-1 and B7-2 to CTLA4. | [308,309] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Effer, B.; Perez, I.; Ulloa, D.; Mayer, C.; Muñoz, F.; Bustos, D.; Rojas, C.; Manterola, C.; Vergara-Gómez, L.; Dappolonnio, C.; et al. Therapeutic Targets of Monoclonal Antibodies Used in the Treatment of Cancer: Current and Emerging. Biomedicines 2023, 11, 2086. https://doi.org/10.3390/biomedicines11072086
Effer B, Perez I, Ulloa D, Mayer C, Muñoz F, Bustos D, Rojas C, Manterola C, Vergara-Gómez L, Dappolonnio C, et al. Therapeutic Targets of Monoclonal Antibodies Used in the Treatment of Cancer: Current and Emerging. Biomedicines. 2023; 11(7):2086. https://doi.org/10.3390/biomedicines11072086
Chicago/Turabian StyleEffer, Brian, Isabela Perez, Daniel Ulloa, Carolyn Mayer, Francisca Muñoz, Diego Bustos, Claudio Rojas, Carlos Manterola, Luis Vergara-Gómez, Camila Dappolonnio, and et al. 2023. "Therapeutic Targets of Monoclonal Antibodies Used in the Treatment of Cancer: Current and Emerging" Biomedicines 11, no. 7: 2086. https://doi.org/10.3390/biomedicines11072086
APA StyleEffer, B., Perez, I., Ulloa, D., Mayer, C., Muñoz, F., Bustos, D., Rojas, C., Manterola, C., Vergara-Gómez, L., Dappolonnio, C., Weber, H., & Leal, P. (2023). Therapeutic Targets of Monoclonal Antibodies Used in the Treatment of Cancer: Current and Emerging. Biomedicines, 11(7), 2086. https://doi.org/10.3390/biomedicines11072086