
A Comparative Study on Cyclodextrin Derivatives in Improving Oral Bioavailability of Etoricoxib as a Model Drug: Formulation and Evaluation of Solid Dispersion-Based Fast-Dissolving Tablets

 , , and
, , and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
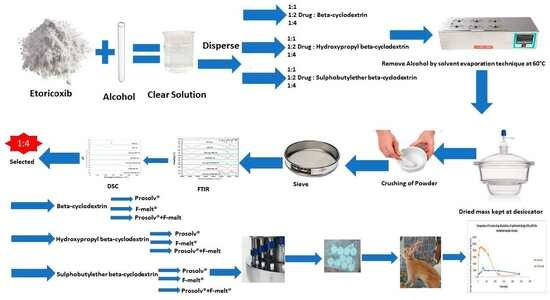
2.2. Preparation of Etoricoxib Solid Dispersions and Their Physical Mixtures Using Cyclodextrin Derivatives
2.3. Characterization of Etoricoxib Solid Dispersion and Their Physical Mixtures Using Cyclodextrin Derivatives
2.3.1. Determination of Percentage Yield
2.3.2. Determination of Percent Drug Content
2.3.3. Determination of Saturation Solubility
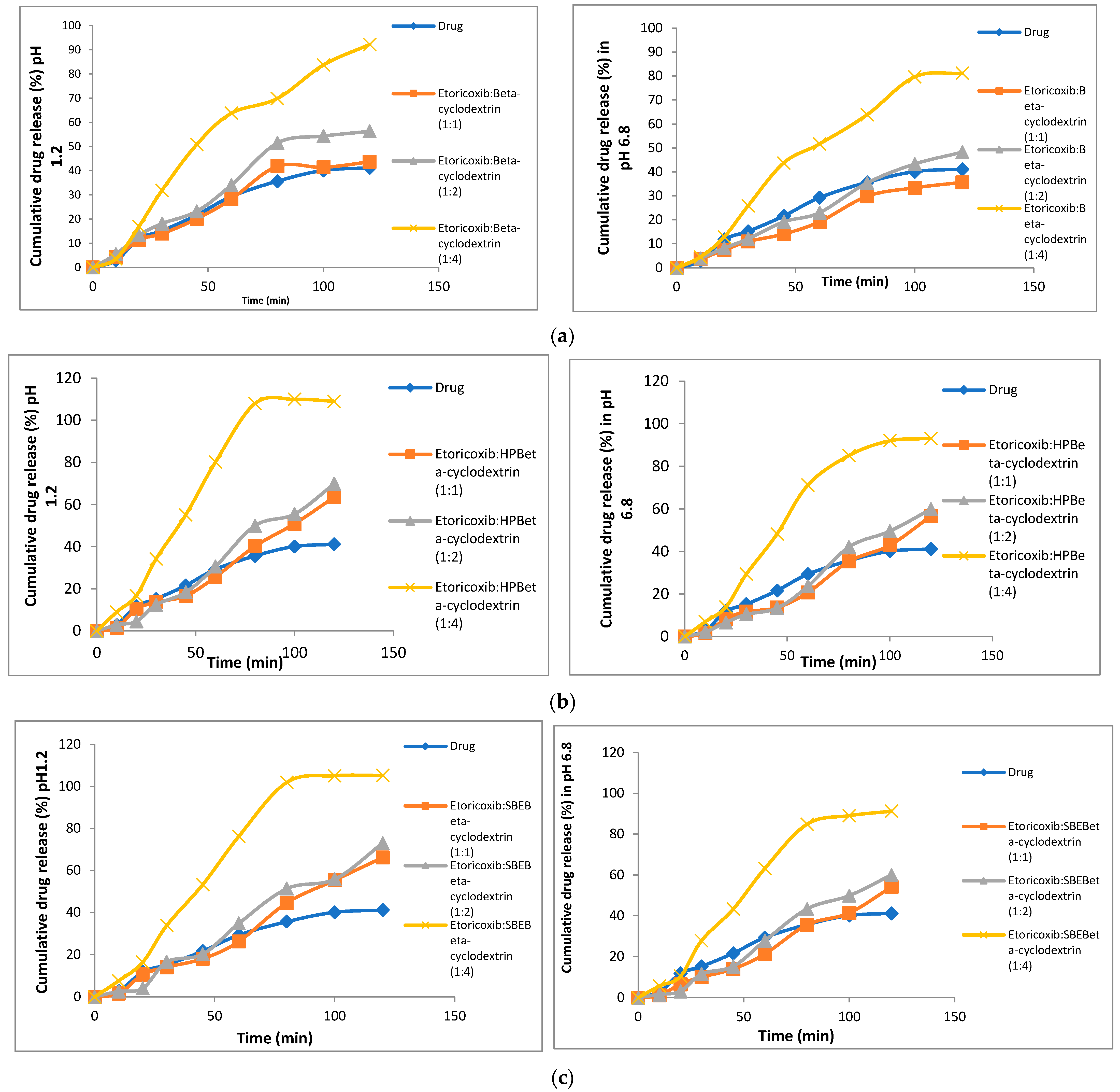
2.3.4. In vitro Dissolution Studies
2.3.5. Fourier Transform-Infrared (FTIR) Spectroscopy
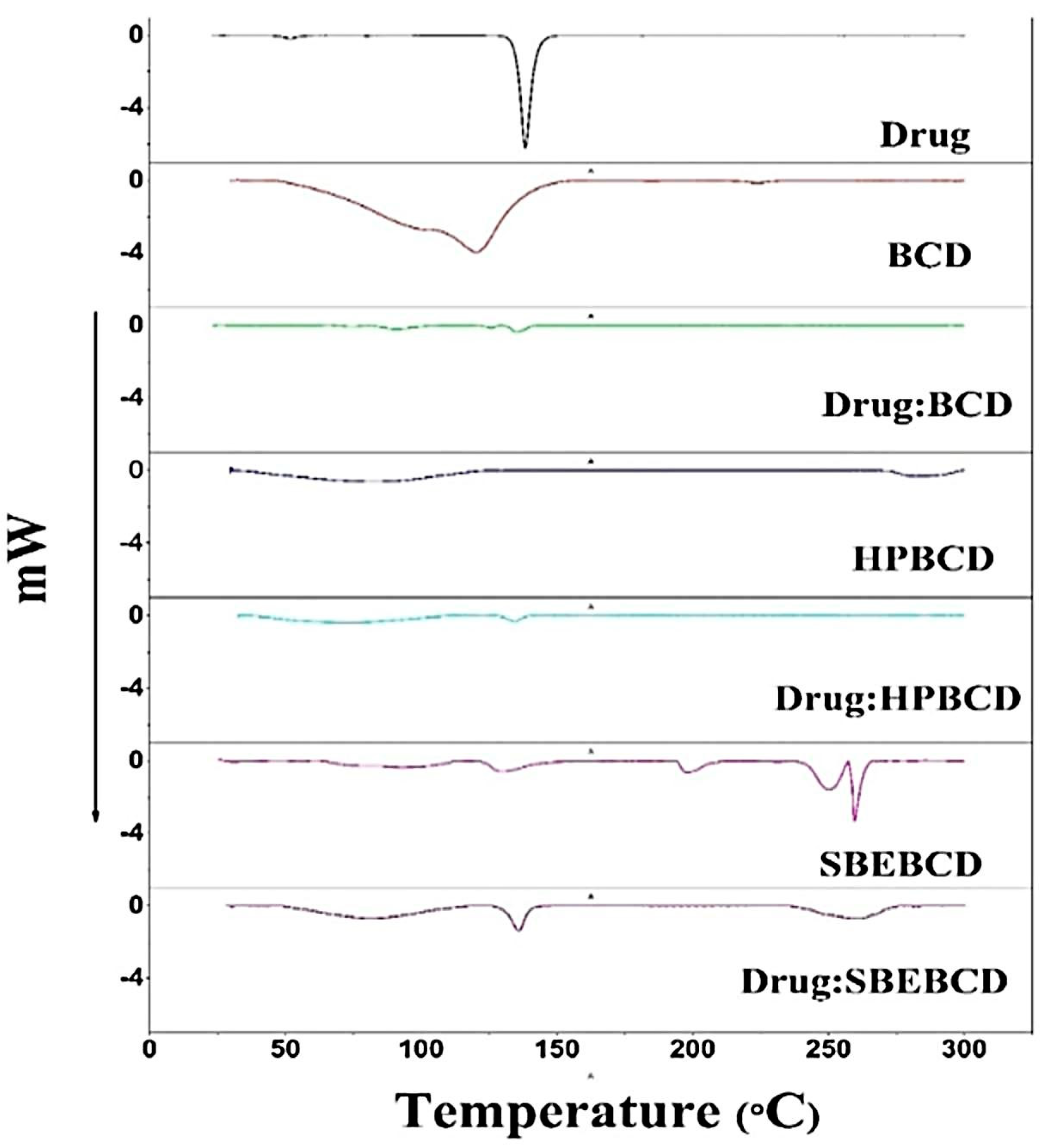
2.3.6. Differential Scanning Calorimetry (DSC)
2.4. Pre-Compression Studies of Etoricoxib Solid Dispersion
2.4.1. Preparation of Powder Blend
2.4.2. Characterization of the Powder Blend
2.5. Preparation of Etoricoxib Tablets by Direct Compression
2.6. Evaluation of Fast-Dissolving Tablets
2.6.1. In Vitro Disintegration Time
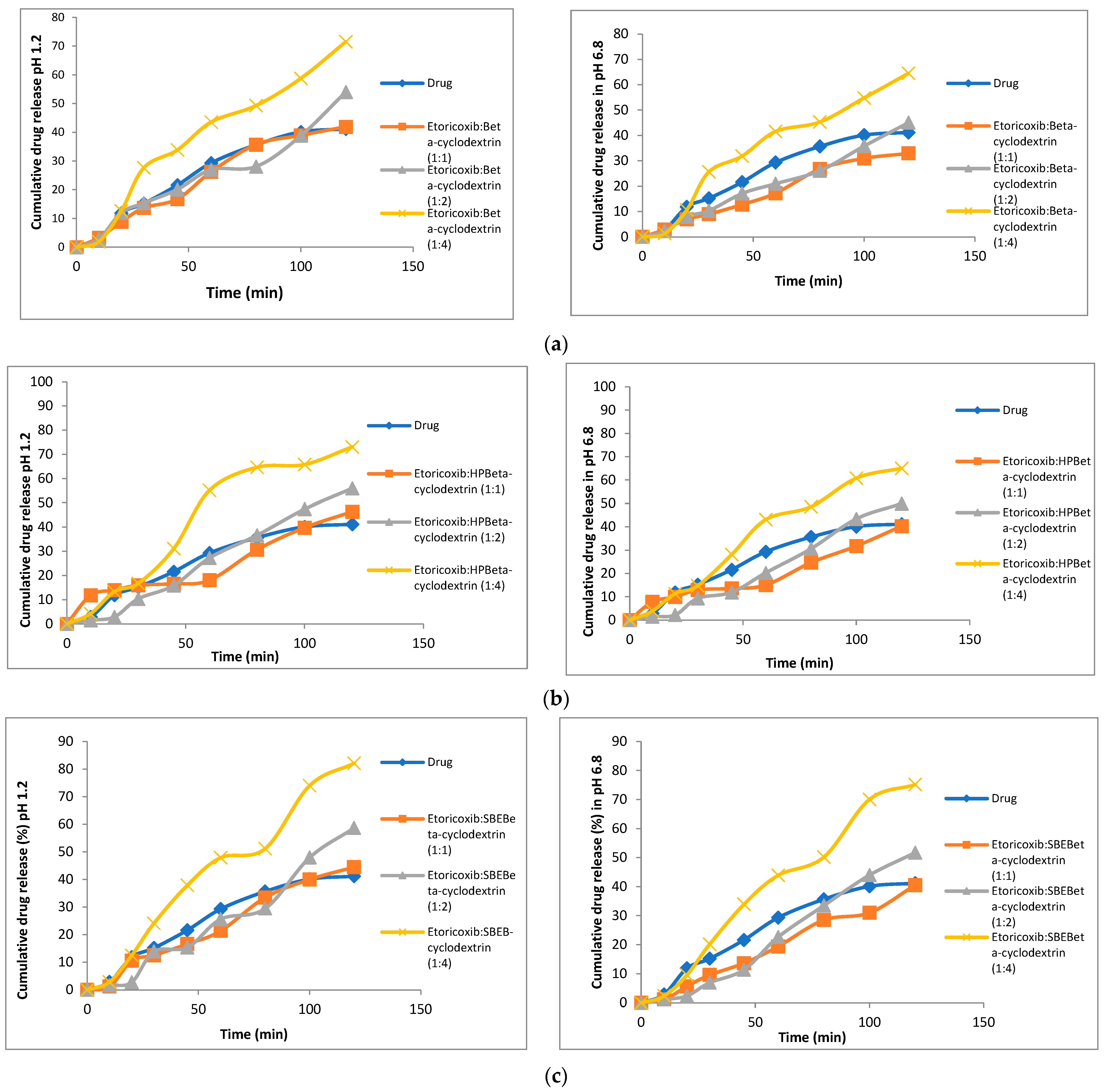
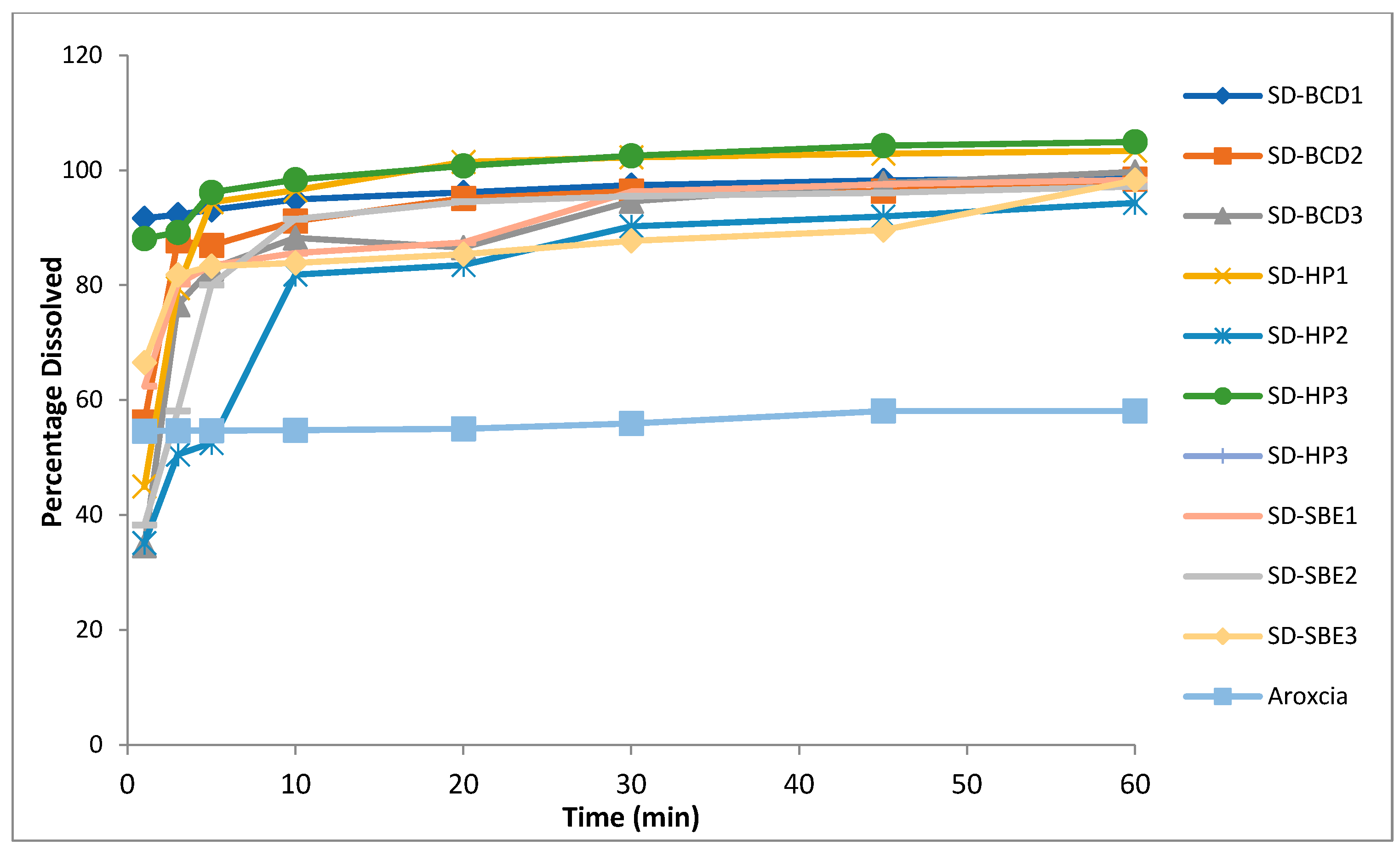
2.6.2. In Vitro Drug Dissolution
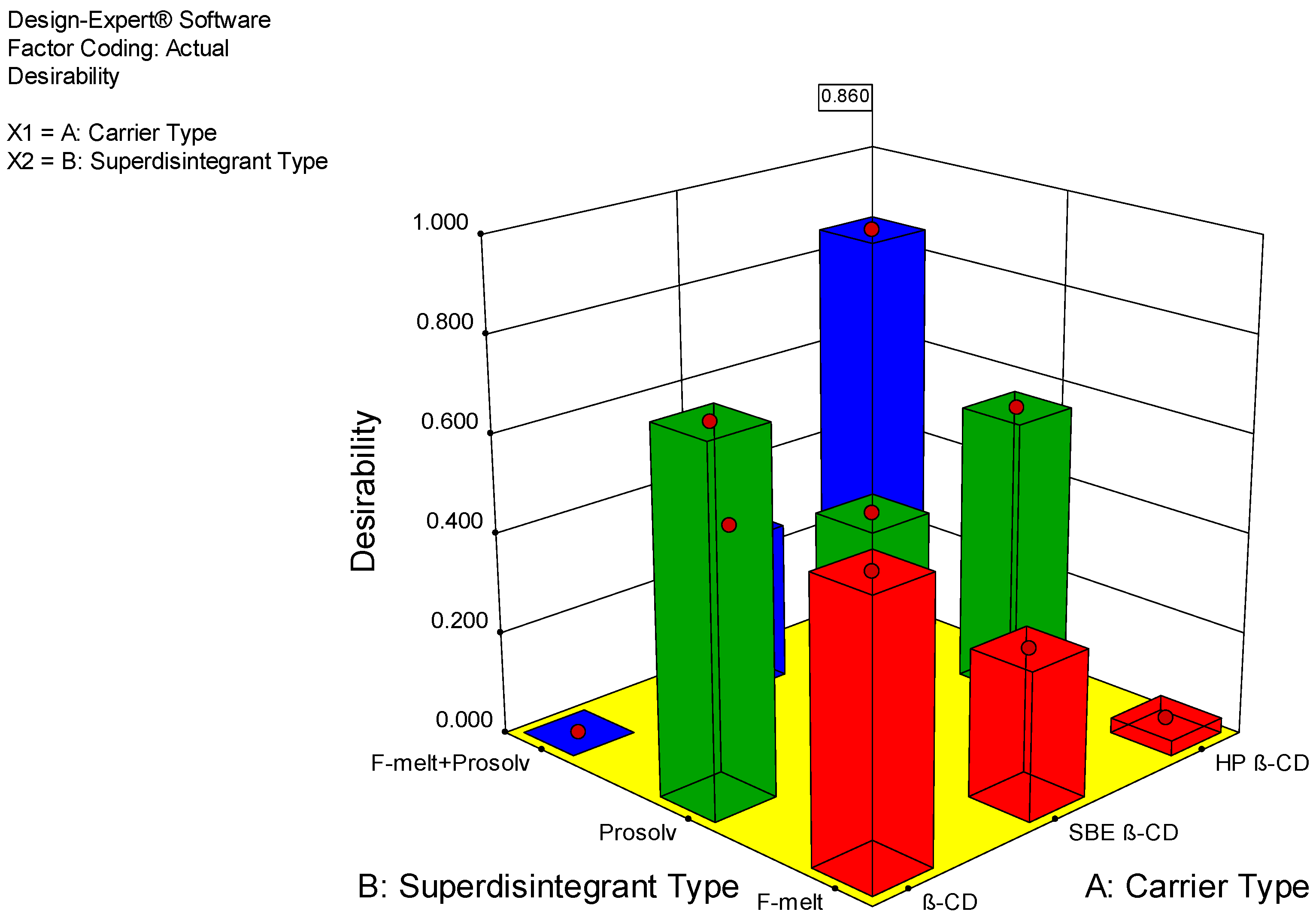
2.6.3. Optimization of Fast-Dissolving Etoricoxib Tablet
2.7. In vivo Bioavailability Study
2.8. Statistical Analysis of Results
3. Results
3.1. Percentage Yield and Percent Drug Content
3.2. Saturation Solubility
3.3. FTIR Spectroscopy Analysis
3.4. DSC Analysis
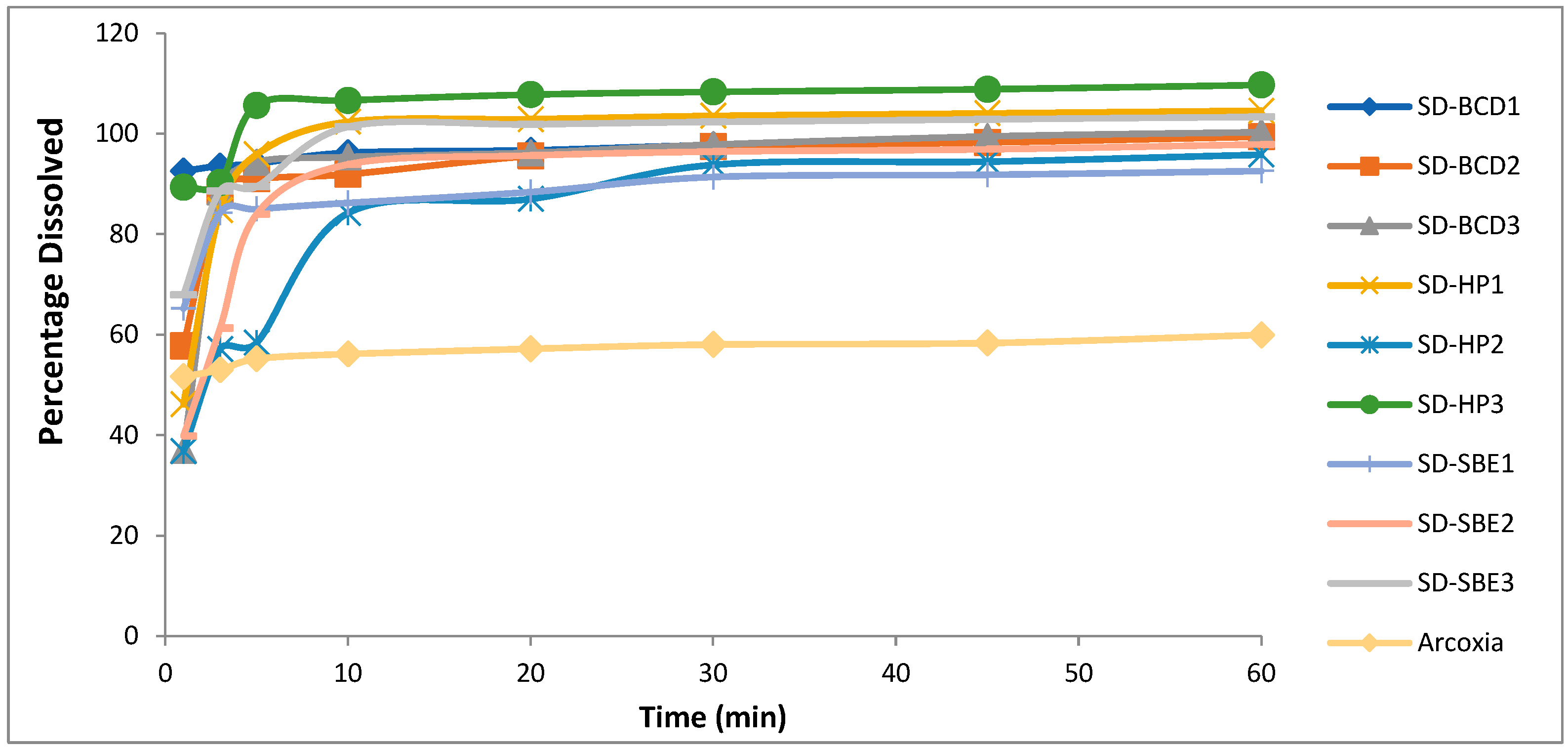
3.5. In Vitro Dissolution
3.6. Characterization of Fast-Dissolving Tablets of Etoricoxib
Evaluation of the Powder Blend for Tablet Formulations
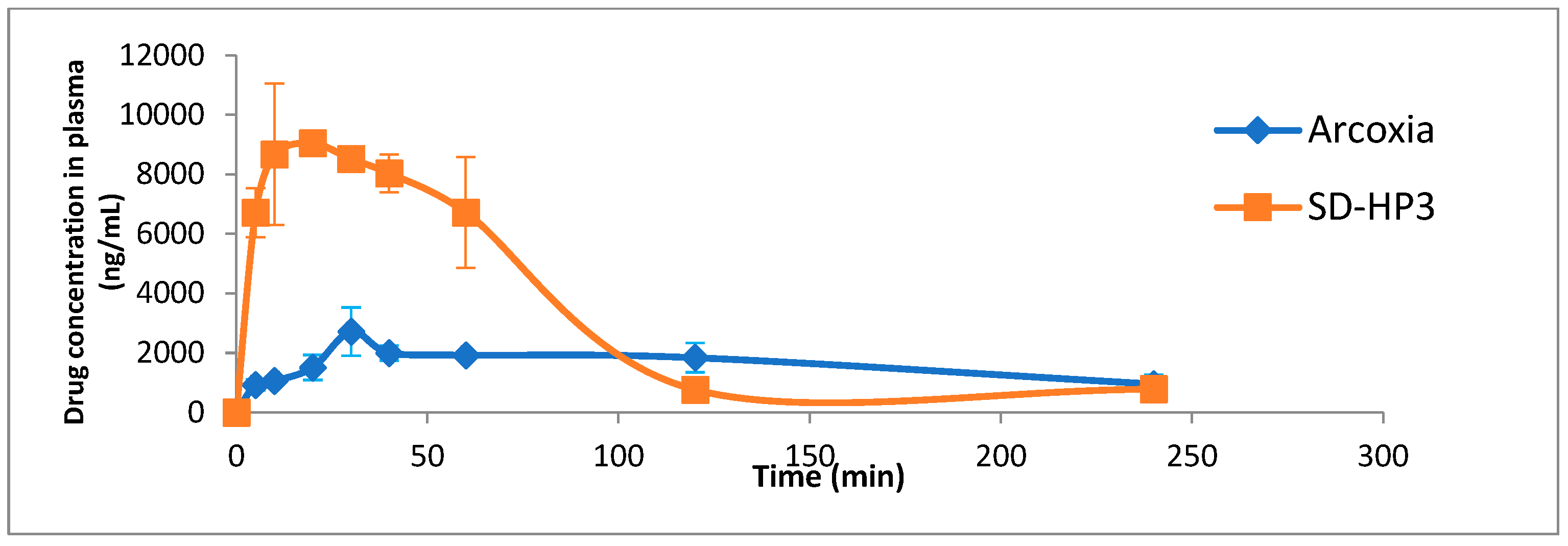
3.7. In Vivo Study
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Azhar Danish, K.; Lubhan, S. Various techniques of bioavailability enhancement: A review. J. Drug Deliv. Ther. 2016, 6, 34. [Google Scholar]
- Younis, M.A.; Singh, S.; Singh Baghel, R.; Yadav, L. A review on solid dispersion. Int. J. Pharm. Life Sci. 2011, 2, 1078–1095. [Google Scholar]
- Chiou, W.L.; Riegelman, S. Pharmaceutical applications of solid dispersion systems. J. Pharm. Sci. 1971, 60, 1281–1302. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.A.; Ali, R.; Al-Jenoobi, F.I.; Al-Mohizea, A.M. Solid dispersions: A strategy for poorly aqueous soluble drugs and technology updates. Expert Opin. Drug Deliv. 2012, 9, 1419–1440. [Google Scholar] [CrossRef]
- Nayak, A.K.; Panigrahi, P.P. Solubility enhancement of etoricoxib by cosolvency approach. ISRN Phys. Chem. 2012, 2012, 1–5. [Google Scholar] [CrossRef]
- Razi, M.; Khan, U.; Musaddiq, H.; Raza, S.M.; Nazir, S. Formulation development, evaluation and anti-inflammatory effects of etoricoxib cream. Indo Am. J. Pharm. Sci. 2014, 4, 3945–3952. [Google Scholar]
- Capone, M.L.; Tacconelli, S.; Patrignani, P. Clinical pharmacology of etoricoxib. Expert Opin Drug Metab. Toxicol. 2005, 1, 269–282. [Google Scholar] [CrossRef]
- DMartina, S.; Vesta, K.S.; Ripley, T.L. Etoricoxib: A highly selective COX-2 inhibitor. Ann. Pharmacother. 2005, 39, 854–862. [Google Scholar] [CrossRef]
- van der Heijde, D.; Baraf, H.S.; Ramos-Remus, C.; Calin, A.; Weaver, A.L.; Schiff, M.; Dougados, M. Evaluation of the efficacy of etoricoxib in ankylosing spondylitis: Results of a fifty-two-week, randomized, controlled study. Arthritis Rheum. 2005, 52, 1205–1215. [Google Scholar] [CrossRef]
- Chowdary, K.P.R.; Tarakamarao, C. Factorial study on the evaluation of formulation variables on the dissolution rate of etoricoxib tablets. Asian J. Chem. 2011, 23, 958–960. [Google Scholar]
- Patel, H.M.; Suhagia, B.N.; Shah, S.A.; Rathod, I.S.; Parmar, V.K. Preparation and characterization of etoricoxib-β-cyclodextrin complexes prepared by the kneading method. Acta Pharm. 2007, 57, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Nayak, A.K.; Jana, S. A Solid Self-Emulsifying System for Dissolution Enhancement of Etoricoxib. J. PharmaSciTech 2013, 2, 87–90. [Google Scholar]
- Nardello-Rataj, V.; Leclercq, L. Encapsulation of biocides by cyclodextrins: Toward synergistic effects against pathogens. Beilstein J. Org. Chem. 2014, 10, 2603–2622. [Google Scholar] [CrossRef]
- Chowdary, K.P.R.; Venkata Reddy, M. Formulation Development studies on Enhancement of Solubility and Dissolution Rate of Etoricoxib by cyclodextrin Complexation. Asian J. Chem. 2011, 23, 1445–1448. [Google Scholar]
- Serno, T.; Geidobler, R.; Winter, G. Protein stabilization by cyclodextrins in the liquid and dried state. Adv. Drug Deliv. Rev. 2011, 63, 1086–1106. [Google Scholar] [CrossRef] [PubMed]
- Siva, S.; Venkatesh, G.; Prabhu, A.A.M.; Sankaranarayanan, R.K.; Rajendiran, N. Absorption and fluorescence spectral characteristics of norepinephrine, epinephrine, isoprenaline, methyl dopa, terbutaline and orciprenaline drugs. Phys. Chem. Liq. 2012, 50, 434–452. [Google Scholar] [CrossRef]
- Rasheed, A. Cyclodextrins as drug carrier molecule: A review. Sci. Pharm. 2008, 76, 567–598. [Google Scholar] [CrossRef]
- Uekama, K. Recent Aspects of Pharmaceutical Application of Cyclodextrins. J. Incl. Phenom. Macrocycl. Chem. 2002, 44, 3–7. [Google Scholar] [CrossRef]
- Hirayama, F.; Uekama, K. Cyclodextrin-based controlled drug release system. Adv. Drug Deliv. Rev. 1999, 36, 125–141. [Google Scholar] [CrossRef]
- Mura, P. Advantages of the combined use of cyclodextrins and nanocarriers in drug delivery: A review. Int. J. Pharm. 2020, 579, e119181. [Google Scholar] [CrossRef]
- Cid-Samamed, A.; Rakmai, J.; Mejuto, J.C.; Simal-Gandara, J.; Astray, G. Cyclodextrins inclusion complex: Preparation methods, analytical techniques and food industry applications. Food Chem. 2022, 384, e132467. [Google Scholar] [CrossRef] [PubMed]
- Muralidhar, S.; Rao, G.D.; Murthy, M.K.; Kumar, K.K.; Teja, K.K.; Nawaj, S.K.; Narayana, T.V. Enhancement of dissolution rate of etoricoxib through solid dispersion technique. J. Appl. Pharm. Sci. 2011, 1, 129–132. [Google Scholar]
- Sapkal, S.B.; Adhao, V.S.; Thenge, R.R.; Darakhe, R.A.; Shinde, S.A.; Shrikhande, V.N. Formulation and characterization of solid dispersions of etoricoxib using natural polymers. Turk. J. Pharm. Sci. 2020, 17, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, B.; Shimpi, S.; Paradkar, A. Preparation and characterization of etoricoxib solid dispersions using lipid carriers by spray drying technique. AAPS PharmaSciTech 2005, 6, E405–E409. [Google Scholar] [CrossRef]
- Kumudhavalli, M.; Margret Chandira, R.; Venkataeswarlu, B.; Jayakar, B. Formulation and evaluation of mouth dissolving tablets of the Etoricoxib. Pak. J. Pharm. Sci. 2010, 23, 178–181. [Google Scholar]
- Manjula, S.; Shashidhara, S.; Anitha, S.; Shilpa, S. Design development and evaluation of herbal tablets containing andrographis paniculata and phyllanthus amarus. Pharma. Sci. Monit. 2012, 3, 4. [Google Scholar]
- Singh, I.; Kaur, B.; Kumar, P.; Arora, S. Masking the unpleasant taste of Etoricoxib by crosslinked acrylic polymer based ion-exchange resin complexation. Polim. Med. 2010, 40, 19–26. [Google Scholar]
- Shivakumar, H.N.; Somasekhar, V.; Roy, A. Preparation and characterization of directly compressible spherical agglomerates of etoricoxib. Ind. J. Pharm. Edu. Res. 2020, 54, 983–990. [Google Scholar] [CrossRef]
- Saker, A.; Cares-Pacheco, M.G.; Marchal, P.; Falk, V. Powders flowability assessment in granular compaction: What about the consistency of Hausner ratio? Powder Technol. 2019, 354, 52–63. [Google Scholar] [CrossRef]
- Konapure, S.A.; Chaudhari, P.S.; Oswal, R.J.; Kshirsagar, S.S.; Antre, R.V.; Chorage, T.V. “Mouth dissolving tablets” an innovative technology. Int. J. Appl. Biol. Pharm. 2011, 2, 496–503. [Google Scholar]
- Siddiqui, M.N.; Garg, G.; Sharma, K. Fast dissolving tablets: Preparation, characterization and evaluation: An overview. Int. J. Pharm. Sci. Rev. Res. 2010, 4, 87–96. [Google Scholar]
- Singh Panwar, A.; Chauhan, J.K.; Darwhekar, G. Formulation and evaluation of fast dissolving tablet of Piroxicam. Am. J. PharmTech Res. 2011, 1, 255–273. [Google Scholar]
- Council of Europe, European Pharmacopoeia Commission, European Directorate for the Quality of Medicines & Healthcare; European Pharmacopoeia: Strasbourg, France, 2019.
- Radke, R.S.; Jadhav, J.K.; Chajeed, M.R. Formulation and evaluation of orodispersible tablets of baclofen. Int. J. Chem. Tech. Res. 2009, 1, 517–521. [Google Scholar]
- Klancke, J. Dissolution testing of orally disintegrating tablets. Dissolut. Technol. 2003, 10, 6–8. [Google Scholar] [CrossRef]
- Annepogu, H.; Ahad, H.A. Unique Solid Dispersions by Microwave Fusion Technique with Etoricoxib and Thiocolchicoside: In vivo Evaluation and Comparison with Marketed Formulation. Adv. Pharmacol. Pharm. 2022, 10, 104–113. [Google Scholar] [CrossRef]
- Attia, I.A.; El-Gizawy, S.A.; Fouda, M.A.; Donia, A.M. Influence of a Niosomal Formulation on the Oral Bioavailability of Acyclovir in Rabbits. AAPS PharmSciTech 2007, 8, 106. [Google Scholar] [CrossRef]
- Loftsson, T.; Brewster, M.E. Cyclodextrins as functional excipients: Methods to enhance complexation efficiency. J. Pharm. Sci. 2012, 101, 3019–3032. [Google Scholar] [CrossRef]
- Pitha, J.; Milecki, J.; Fales, H.; Pannell, L.; Uekama, K. Hydroxypropyl-β-cyclodextrin: Preparation and characterization; effects on solubility of drugs. Int. J. Pharm. 1986, 29, 73–82. [Google Scholar] [CrossRef]
- Saokham, P.; Muankaew, C.; Jansook, P.; Loftsson, T. Solubility of cyclodextrins and drug/cyclodextrin complexes. Molecules 2018, 23, 1161. [Google Scholar] [CrossRef]
- Saleh, A.; McGarry, K.; Chaw, C.S.; Elkordy, A.A. Feasibility of using gluconolactone, trehalose and hydroxy-propyl gamma cyclodextrin to enhance bendroflumethiazide dissolution using lyophilisation and physical mixing techniques. Pharmaceutics 2018, 10, 22. [Google Scholar] [CrossRef]
- Ma, Y.H.; Zhu, M.M.; Zhang, C.N.; Tang, X.S.; Zhang, W.G.; Ma, W.J. The co-crystal structure of etoricoxib-phthalic acid (1/1), C18H15ClN2O2S·C8H6O4. Z. Fur Krist. New Cryst. Struct. 2023, 238, 641–643. [Google Scholar] [CrossRef]
- Kang, B.K.; Lee, J.S.; Chon, S.K.; Jeong, S.Y.; Yuk, S.H.; Khang, G.; Cho, S.H. Development of self-microemulsifying drug delivery systems (SMEDDS) for oral bioavailability enhancement of simvastatin in beagle dogs. Int. J. Pharm. 2004, 274, 65–73. [Google Scholar] [CrossRef]
- Pokharkar, V.B.; Mandpe, L.P.; Padamwar, M.N.; Ambike, A.A.; Mahadik, K.R.; Paradkar, A. Development, characterization and stabilization of amorphous form of a low Tg drug. Powder Technol. 2006, 167, 20–25. [Google Scholar] [CrossRef]
- Karavas, E.; Ktistis, G.; Xenakis, A.; Georgarakis, E. Effect of hydrogen bonding interactions on the release mechanism of felodipine from nanodispersions with polyvinylpyrrolidone. Eur. J. Pharm. Biopharm. 2006, 63, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Krupa, A.; Jachowicz, R.; Pędzich, Z.; Wodnicka, K. The influence of the API properties on the ODTs manufacturing from co-processed excipient systems. AAPS PharmSciTech. 2012, 13, 1120–1129. [Google Scholar] [CrossRef] [PubMed]
- Gohel, M.C. A review of co-processed directly compressible excipients. J. Pharm. Sci. 2005, 16, 76–93. [Google Scholar]
- Fouad, S.A.; Teaima, M.H.; Gebril, M.I.; Abd Allah, F.I.; El-Nabarawi, M.A.; Elhabal, S.F. Formulation of novel niosomal repaglinide chewable tablets using coprocessed excipients: In vitro characterization, optimization and enhanced hypoglycemic activity in rats. Drug Deliv. 2023, 30, 1747. [Google Scholar] [CrossRef]
- Dahiya, S.; Kaushik, A.; Pathak, K. Improved pharmacokinetics of aceclofenac immediate release tablets incorporating its inclusion complex with hydroxypropyl-β-cyclodextrin. Sci. Pharm. 2015, 83, 501–510. [Google Scholar] [CrossRef]
- Chay, S.K.; Keating, A.V.; James, C.; Aliev, A.E.; Haider, S.; Craig, D.Q.M. Evaluation of the taste-masking effects of (2-hydroxypropyl)-β-cyclodextrin on ranitidine hydrochloride; A combined biosensor, spectroscopic and molecular modelling assessment. RSC Adv. 2018, 8, 3564–3573. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Factors (Independent Variables) | Levels | ||
|---|---|---|---|
| Type of cyclodextrin | β-CD | HP β-CD | SBE β-CD |
| Directly compressible vehicle | Prosolv® ODT | Prosolv® ODT and F-melt | F-melt |
| Formulation | Formulation | Etoricoxib mg | BCD mg | HPBCD mg | SBEBCD mg | Prosolv® mg | Fmelt® mg | Total Weight mg |
|---|---|---|---|---|---|---|---|---|
| F1 | SD-BCD1 | 60 | 240 | 300 | 600 | |||
| F2 | SD-BCD2 | 60 | 240 | 300 | 600 | |||
| F3 | SD-BCD3 | 60 | 240 | 150 | 150 | 600 | ||
| F4 | SD-HP1 | 60 | 240 | 300 | 600 | |||
| F5 | SD-HP2 | 60 | 240 | 300 | 600 | |||
| F6 | SD-HP3 | 60 | 240 | 150 | 150 | 600 | ||
| F7 | SD-SBE1 | 60 | 240 | 300 | 600 | |||
| F8 | SD-SBE2 | 60 | 240 | 300 | 600 | |||
| F9 | SD-SBE3 | 60 | 240 | 150 | 150 | 600 |
| Response (Independent Variable) | Constraint |
|---|---|
| Hardness | Maximize |
| Friability | Minimize |
| Disintegration time | Minimize |
| Dissolution after one minute in phosphate buffer (pH 1.2 and 6.8) | Maximize |
| Dissolution after ten minutes in phosphate buffer (pH 1.2 and 6.8) | Maximize |
| Dissolution Efficiency | Maximize |
| Solid Dispersion Type | Ratio | Percentage Yield % ± S.D | Percentage Drug Content % ± S.D |
|---|---|---|---|
| Etoricoxib: β-CD | 1:1 | 82.5 ± 0.6 | 81.11 ± 1.41 |
| 1:2 | 87.5 ± 0.4 | 83.03 ± 0.04 | |
| 1:4 | 96.3 ± 0.7 | 90.48 ± 0.03 | |
| Etoricoxib: HP β-CD | 1:1 | 91.6 ± 0.8 | 94.71 ± 0.14 |
| 1:2 | 88.8 ± 0.1 | 96.21 ± 0.41 | |
| 1:4 | 97.0 ± 1.4 | 98.32 ± 0.02 | |
| Etoricoxib: SBE β-CD | 1:1 | 91.6 ± 0.8 | 96.11 ± 0.01 |
| 1:2 | 93.3 ± 0.1 | 97.72 ± 0.42 | |
| 1:4 | 98.3 ± 0.8 | 98.73 ± 0.04 |
| Mixture | Ratio | Saturation Solubility μg/mL | |||
|---|---|---|---|---|---|
| Physical Mixture pH 1.2 | Physical Mixture pH 6.8 | Solid Dispersion pH 1.2 | Solid Dispersion pH 6 | ||
| Etoricoxib: β-CD | 1:1 | 78.62 ± 0.82 | 76.30 ± 0.85 | 82.47 ± 2.02 | 81.36 ± 1.29 |
| 1:2 | 80.00 ± 1.56 | 78.31 ± 0.86 | 86.51 ± 3.52 | 85.14 ± 0.76 | |
| 1:4 | 82.47 ± 0.75 | 81.11 ± 1.12 | 90.27 ± 2.58 | 88.63 ± 3.08 | |
| Etoricoxib: HP β-CD | 1:1 | 90.13 ± 0.09 | 88.33 ± 0.01 | 110.91 ± 1.41 | 107.60 ± 1.84 |
| 1:2 | 92.61 ± 0.57 | 91.79 ± 0.55 | 116.28 ± 4.78 | 115.30 ± 2.12 | |
| 1:4 | 105.41 ± 5.50 | 101.54 ± 0.76 | 131.55 ± 3.32 | 129.10 ± 1.27 | |
| Etoricoxib: SBE β-CD | 1:1 | 93.44 ± 1.05 | 93.00 ± 1.27 | 115.18 ± 1.95 | 112.98 ± 1.99 |
| 1:2 | 96.19 ± 0.07 | 95.51 ± 0.55 | 126.74 ± 2.97 | 111.36 ± 1.92 | |
| 1:4 | 103.89 ± 2.90 | 100.40 ± 0.71 | 123.85 ± 1.06 | 110.20 ± 0.71 | |
| Pure Etoricoxib in 0.1 N HCL pH(1.2) | 73.85 μg/mL ± 2.20 | ||||
| Pure Etoricoxib in Phosphate buffer pH(6.8) | 71.12 μg/mL ± 1.23 | ||||
| Formulation | Bulk Density | Tapped Density | Carr’s Index | Hausner’s Ratio | Angle of Repose |
|---|---|---|---|---|---|
| SD-BCD1 | 0.46 ± 0.03 | 0.5 ± 0.0 | 8.0 ± 0.6 | 1.094 ± 0.042 | 22.0 ± 2.8 |
| SD-BCD2 | 0.44 ± 0.03 | 0.48 ± 0.01 | 8.33 ± 0.47 | 1.07 ± 0.02 | 21.0 ± 0.0 |
| SD-BCD3 | 0.46 ± 0.02 | 0.49 ± 0.06 | 6.12 ± 0.03 | 1.05 ± 0.04 | 23.0 ± 0.7 |
| SD-HP1 | 0.42 ± 0.02 | 0.44 ± 0.01 | 4.45 ± 0.07 | 1.06 ± 0.01 | 19.0 ± 1.4 |
| SD-HP2 | 0.46 ± 0.01 | 0.49 ± 0.01 | 6.12 ± 0.17 | 1.048 ± 0.049 | 20.0 ± 0.7 |
| SD-HP3 | 0.48 ± 0.01 | 0.52 ± 0.03 | 7.69 ± 0.18 | 1.068 ± 0.049 | 20.0 ± 0.0 |
| SD-SBE1 | 0.45 ± 0.03 | 0.49 ± 0.01 | 8.163 ± 0.231 | 1.086 ± 0.003 | 19.0 ± 1.4 |
| SD-SBE2 | 0.41 ± 0.02 | 0.45 ± 0.028 | 8.88 ± 0.62 | 1.067 ± 0.005 | 21.0 ± 0.7 |
| SD-SBE3 | 0.45 ± 0.01 | 0.490 ± 0.028 | 8.16 ± 0.08 | 1.080 ± 0.113 | 22.0 ± 0.7 |
| Formulation | Weight Variation | Hardness (Kg) | Drug Content (%) | Friability (% Weight Loss) |
|---|---|---|---|---|
| SD-BCD1 | Passes | 5.20 ± 0.28 | 98.1 ± 1.3 | 0.44 ± 0.03 |
| SD-BCD2 | Passes | 4.25 ± 0.14 | 99.6 ± 0.9 | 0.425 ± 0.028 |
| SD-BCD3 | Passes | 4.90 ± 0.28 | 98.3 ± 0.4 | 0.51 ± 0.01 |
| SD-HP1 | Passes | 5.00 ± 0.42 | 98.9 ± 1.4 | 0.40 ± 0.01 |
| SD-HP2 | Passes | 4.85 ± 0.28 | 100.3 ± 0.4 | 0.525 ± 0.028 |
| SD-HP3 | Passes | 4.50 ± 0.14 | 98.7 ± 0.3 | 0.46 ± 0.01 |
| SD-SBE1 | Passes | 5.40 ± 0.42 | 98.7 ± 0.7 | 0.380 ± 0.014 |
| SD-SBE2 | Passes | 4.60 ± 0.28 | 99.1 ± 0.4 | 0.440 ± 0.028 |
| SD-SBE3 | Passes | 4.60 ± 0.71 | 99.3 ± 1.8 | 0.470 ± 0.028 |
| Formulation | Disintegration Time in 0.1 N HCL pH 1.2 (sec) | Disintegration Time in Phosphate Buffer pH 6.8 (sec) | Percentage Drug Dissolved after 1 min in 0.1 N HCL Buffer pH 1.2 | Percentage Drug Dissolved after 1 min in Phosphate Buffer pH 6.8 | Percentage Drug Dissolved after 10 min in 0.1 M HCL Buffer pH 1.2 | Percentage Drug Dissolved after 10 min in Phosphate Buffer pH 6.8 |
|---|---|---|---|---|---|---|
| SD-BCD1 | 37.0 ± 0.4 | 43.0 ± 1.4 | 92.6 ± 0.7 | 91.6 ± 1.3 | 96.3 ± 1.3 | 94.9 ± 0.7 |
| SD-BCD2 | 45.0 ± 1.4 | 47.5 ± 1.4 | 57.8 ± 2.0 | 56.4 ± 1.6 | 92 ± 0.4 | 91.1 ± 0.3 |
| SD-BCD3 | 45.0 ± 2.8 | 49.0 ± 4.2 | 37.0 ± 0.3 | 34.7 ± 3.1 | 95.4 ± 0.7 | 88.2± 1.8 |
| SD-HP1 | 60.0 ± 4.2 | 52.0 ± 2.8 | 46.2 ± 3.7 | 44.9 ± 2.8 | 102.0 ± 2.0 | 96.5± 2.0 |
| SD-HP2 | 80.0 ± 2.8 | 72.0 ± 1.4 | 36.8 ± 2.8 | 35.1 ± 1.4 | 84.3 ± 1.7 | 81.8± 0.4 |
| SD-HP3 | 25.0 ± 0.0 | 30.0 ± 7.1 | 89.4 ± 4.0 | 88.1 ± 0.3 | 100.7 ± 0.4 | 98.3± 7.1 |
| SD-SBE1 | 33.0 ± 1.4 | 42.0 ± 8.5 | 65.2 ± 4.8 | 62.4 ± 0.6 | 86.2 ± 1.8 | 85.6± 2.8 |
| SD-SBE2 | 70.0 ± 7.1 | 66.0 ± 7.1 | 39.8 ± 0. 6 | 38.3 ± 4.0 | 93.9 ± 0.7 | 91.5± 1.0 |
| SD-SBE3 | 52.0 ± 1.4 | 48.0 ± 8.5 | 67.9 ± 1.4 | 66.5 ± 0.4 | 101.0 ± 2.0 | 83.9± 3.5 |
| Arcoxia® | 120.0 ± 2.7 | 135.0 ± 1.5 | 51.7 ± 1.0 | 54.6 ± 2.6 | 56.2 ± 1.7 | 54.8± 1.3 |
| Formulation | Taste Masking | Thickness (mm) | Dissolution Efficiency % | Desirability |
|---|---|---|---|---|
| SD-BCD1 | Accepted | 4 | 93.936 | 0.734 |
| SD-BCD2 | Accepted | 4 | 90.855 | 0.561 |
| SD-BCD3 | Accepted | 4 | 80.370 | Was excluded |
| SD-HP1 | Accepted | 4 | 96.487 | 0.573 |
| SD-HP2 | Accepted | 4 | 81.979 | 0.031 |
| SD-HP3 | Accepted | 4 | 98.344 | 0.860 |
| SD-SBE1 | Accepted | 4 | 85.920 | 0.456 |
| SD-SBE2 | Accepted | 4 | 91.917 | 0.298 |
| SD-SBE3 | Accepted | 4 | 83.851 | 0.324 |
| Bioavailability Parameters | SD-HP3 ± S.D | Arcoxia® ± S.D |
|---|---|---|
| Tmax (minutes) | 13.333 ± 5.773 | 40.0 ± 17.3 |
| Cmax (μg/mL) | 9122.156 ± 225.508 | 2747.15 ± 767.48 |
| AUC 0–240 (μg.min/mL) | 541,863.4 | 375818.3 |
| AUC 0-∞ μg.min/mL | 854,200.1 | 665460.5 |
| MRT (minutes) | 62.070 ± 8.031 | 101.398 ± 7.106 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elsegaie, D.; El-Nabarawi, M.A.; Mahmoud, H.A.; Teaima, M.; Louis, D. A Comparative Study on Cyclodextrin Derivatives in Improving Oral Bioavailability of Etoricoxib as a Model Drug: Formulation and Evaluation of Solid Dispersion-Based Fast-Dissolving Tablets. Biomedicines 2023, 11, 2440. https://doi.org/10.3390/biomedicines11092440
Elsegaie D, El-Nabarawi MA, Mahmoud HA, Teaima M, Louis D. A Comparative Study on Cyclodextrin Derivatives in Improving Oral Bioavailability of Etoricoxib as a Model Drug: Formulation and Evaluation of Solid Dispersion-Based Fast-Dissolving Tablets. Biomedicines. 2023; 11(9):2440. https://doi.org/10.3390/biomedicines11092440
Chicago/Turabian StyleElsegaie, Doaa, Mohamed A. El-Nabarawi, Hanaa Abdelmonem Mahmoud, Mahmoud Teaima, and Dina Louis. 2023. "A Comparative Study on Cyclodextrin Derivatives in Improving Oral Bioavailability of Etoricoxib as a Model Drug: Formulation and Evaluation of Solid Dispersion-Based Fast-Dissolving Tablets" Biomedicines 11, no. 9: 2440. https://doi.org/10.3390/biomedicines11092440
APA StyleElsegaie, D., El-Nabarawi, M. A., Mahmoud, H. A., Teaima, M., & Louis, D. (2023). A Comparative Study on Cyclodextrin Derivatives in Improving Oral Bioavailability of Etoricoxib as a Model Drug: Formulation and Evaluation of Solid Dispersion-Based Fast-Dissolving Tablets. Biomedicines, 11(9), 2440. https://doi.org/10.3390/biomedicines11092440