
Perry Disease: Bench to Bedside Circulation and a Team Approach


Abstract

1. Introduction
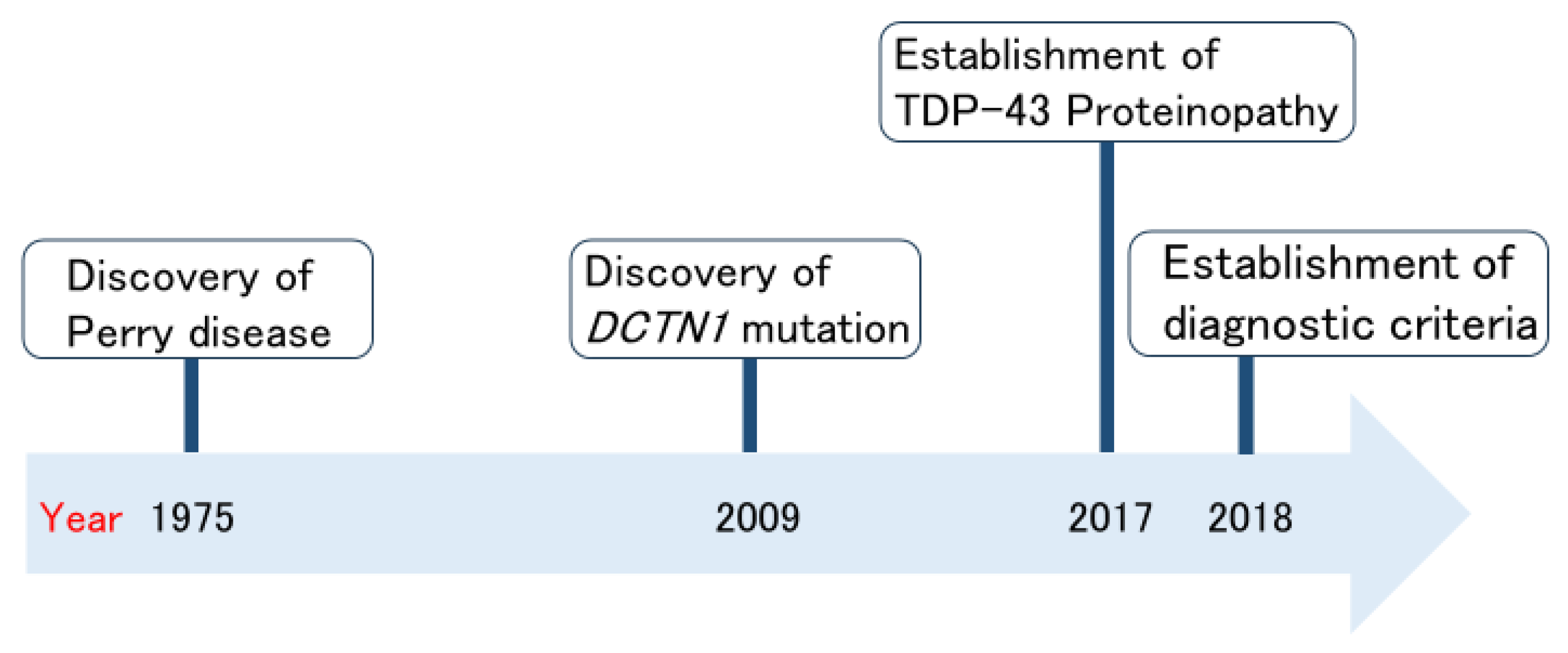
2. History of Perry Disease Research
3. Importance of Early Diagnosis in Perry Disease
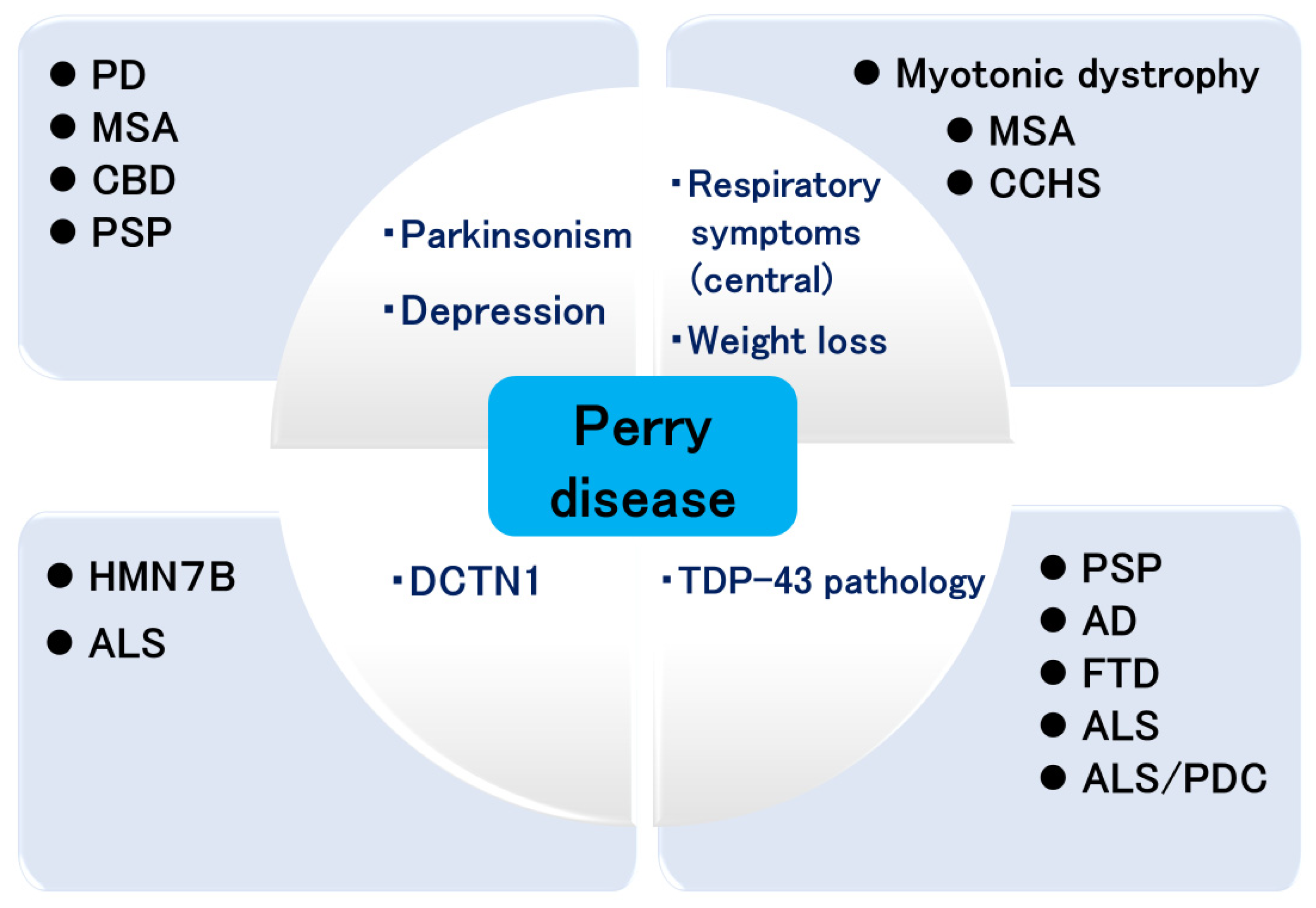
4. Pathology of Perry Disease
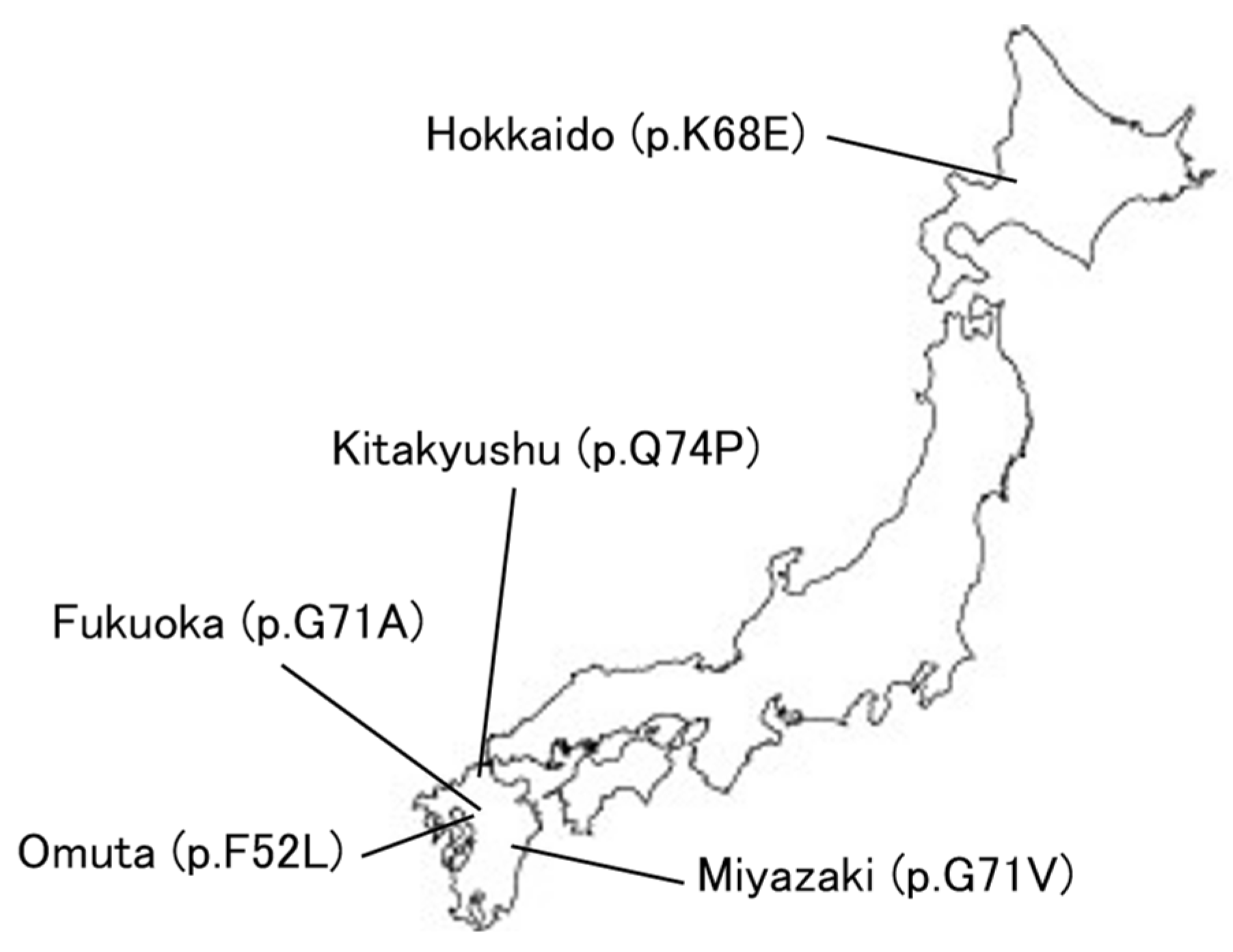
5. Expansion of DCTN1 Mutations
6. Basic Research of Perry Disease
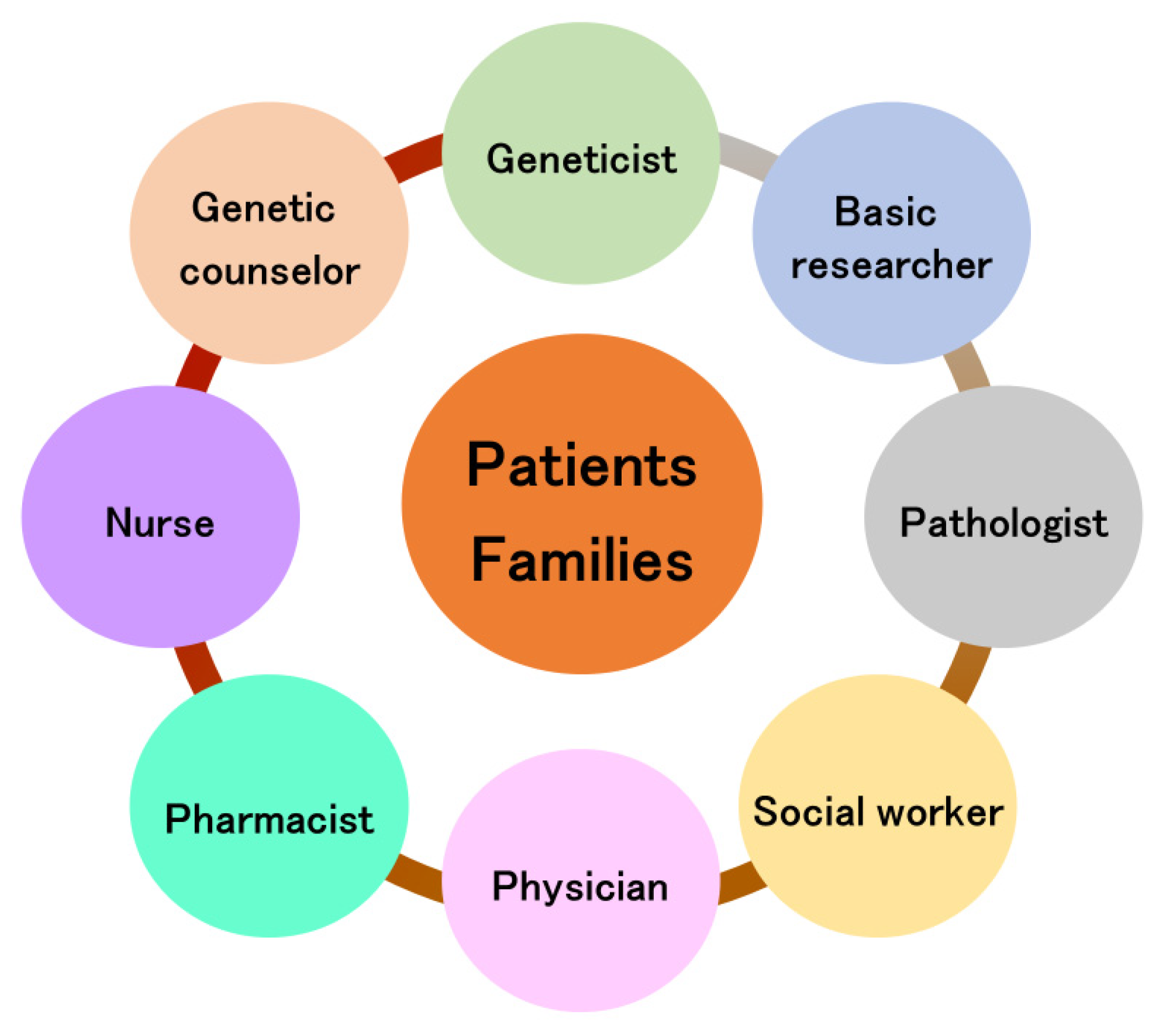
7. Team Approach for Perry Disease
8. Discussion
9. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Haendel, M.; Vasilevsky, N.; Unni, D.; Bologa, C.; Harris, N.; Rehm, H.; Hamosh, A.; Baynam, G.; Groza, T.; McMurry, J.; et al. How many rare diseases are there? Nat. Rev. Drug Discov. 2020, 19, 77–78. [Google Scholar] [CrossRef] [PubMed]
- Tsuboi, Y.; Mishima, T.; Fujioka, S. Perry Disease: Concept of a new disease and clinical diagnostic criteria. J. Mov. Disord. 2021, 14, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Mishima, T.; Fujioka, S.; Tsuboi, Y. Perry Disease: Recent advances and perspectives. Expert Opin. Orphan Drugs 2019, 7, 253–259. [Google Scholar] [CrossRef]
- Perry, T.L.; Bratty, P.J.; Hansen, S.; Kennedy, J.; Urquhart, N.; Dolman, C.L. Hereditary mental depression and Parkinsonism with taurine deficiency. Arch. Neurol. 1975, 32, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Farrer, M.J.; Hulihan, M.M.; Kachergus, J.M.; Dächsel, J.C.; Stoessl, A.J.; Grantier, L.L.; Calne, S.; Calne, D.B.; Lechevalier, B.; Chapon, F.; et al. DCTN1 Mutations in Perry syndrome. Nat. Genet. 2009, 41, 163–165. [Google Scholar] [CrossRef]
- Wider, C.; Dickson, D.W.; Stoessl, A.J.; Tsuboi, Y.; Chapon, F.; Gutmann, L.; Lechevalier, B.; Calne, D.B.; Personett, D.A.; Hulihan, M.; et al. Pallidonigral TDP-43 Pathology in Perry syndrome. Parkinsonism Relat. Disord. 2009, 15, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Mishima, T.; Koga, S.; Lin, W.L.; Kasanuki, K.; Castanedes-Casey, M.; Wszolek, Z.K.; Oh, S.J.; Tsuboi, Y.; Dickson, D.W. Perry Syndrome: A distinctive type of TDP-43 proteinopathy. J. Neuropathol. Exp. Neurol. 2017, 76, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Mishima, T.; Fujioka, S.; Tomiyama, H.; Yabe, I.; Kurisaki, R.; Fujii, N.; Neshige, R.; Ross, O.A.; Farrer, M.J.; Dickson, D.W.; et al. Establishing diagnostic criteria for Perry syndrome. J. Neurol. Neurosurg. Psychiatry 2018, 89, 482–487. [Google Scholar] [CrossRef]
- Gustavsson, E.K.; Trinh, J.; Guella, I.; Szu-Tu, C.; Khinda, J.; Lin, C.H.; Wu, R.M.; Stoessl, J.; Appel-Cresswell, S.; McKeown, M.; et al. DCTN1 p.K56R in Progressive supranuclear palsy. Parkinsonism Relat. Disord. 2016, 28, 56–61. [Google Scholar] [CrossRef]
- Rexach, J.; Lee, H.; Martinez-Agosto, J.A.; Németh, A.H.; Fogel, B.L. Clinical application of next-generation sequencing to the practice of neurology. Lancet Neurol. 2019, 18, 492–503. [Google Scholar] [CrossRef]
- Koyama, S.; Okabe, Y.; Suzuki, Y.; Igari, R.; Sato, H.; Iseki, C.; Tanji, K.; Suzuki, K.; Ohta, Y. Differing clinical features between Japanese siblings with cerebrotendinous xanthomatosis with a novel compound heterozygous CYP27A1 mutation: A case report. BMC Neurol. 2022, 22, 193. [Google Scholar] [CrossRef]
- Klein, C.J.; Foroud, T.M. Neurology individualized medicine: When to use next-generation sequencing panels. Mayo Clin. Proc. 2017, 92, 292–305. [Google Scholar] [CrossRef] [PubMed]
- Naruse, H.; Ishiura, H.; Mitsui, J.; Takahashi, Y.; Matsukawa, T.; Toda, T.; Tsuji, S. Juvenile amyotrophic lateral sclerosis with complex phenotypes associated with novel SYNE1 mutations. Amyotroph Lateral Scler. Front. Degener. 2021, 22, 576–578. [Google Scholar] [CrossRef] [PubMed]
- Yamada, H.; Neshige, S.; Morino, H.; Maruyama, H. Extubation failure due to atypical Parkinsonism with negligible motor and variable non-motor symptoms associated with a variant of DCTN1. Intern. Emerg. Med. 2023, 18, 329–331. [Google Scholar] [CrossRef] [PubMed]
- Magalhães, J.D.; Fão, L.; Vilaça, R.; Cardoso, S.M.; Rego, A.C. Macroautophagy and mitophagy in neurodegenerative disorders: Focus on therapeutic interventions. Biomedicines 2021, 9, 1625. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.J.; Lin, C.H.; Lane, H.Y. From Menopause to Neurodegeneration—Molecular Basis and Potential Therapy. Int. J. Mol. Sci. 2021, 22, 8654. [Google Scholar] [CrossRef]
- Perry, T.L.; Wright, J.M.; Berry, K.; Hansen, S.; Perry, T.L., Jr. Dominantly inherited apathy, central hypoventilation, and Parkinson’s syndrome: Clinical, biochemical, and neuropathologic studies of 2 new cases. Neurology 1990, 40, 1882–1887. [Google Scholar] [CrossRef]
- Roy, E.P., III; Riggs, J.E.; Martin, J.D.; Ringel, R.A.; Gutmann, L. Familial Parkinsonism, apathy, weight loss, and central hypoventilation: Successful long-term management. Neurology 1988, 38, 637–639. [Google Scholar] [CrossRef]
- Bhatia, K.P.; Daniel, S.E.; Marsden, C.D. Familial Parkinsonism with depression: A clinicopathological study. Ann. Neurol. 1993, 34, 842–847. [Google Scholar] [CrossRef]
- Tsuboi, Y.; Wszolek, Z.K.; Kusuhara, T.; Doh-ura, K.; Yamada, T. Japanese family with Parkinsonism, depression, weight loss, and central hypoventilation. Neurology 2002, 58, 1025–1030. [Google Scholar] [CrossRef]
- Kim, D.D.; Alghefari, H.; Jenkins, M.; Ang, L.C.; Pasternak, S.H. Neuropathology of Perry syndrome: Evidence of medullary and hypothalamic involvement. Mov. Disord. Clin. Pract. 2021, 8, 713–716. [Google Scholar] [CrossRef] [PubMed]
- Dulski, J.; Koga, S.; Prudencio, M.; Tipton, P.W.; Ali, S.; Strongosky, A.J.; Rose, J.H.; Parrales, Z.A.; Dunmore, J.A.; Jansen-West, K.; et al. Perry syndrome: Novel DCTN1 mutation in a large kindred and first observation of prodromal disease. Parkinsonism Relat. Disord. 2023, 112, 105481. [Google Scholar] [CrossRef] [PubMed]
- Dulski, J.; Koga, S.; Liberski, P.P.; Sitek, E.J.; Butala, A.A.; Sławek, J.; Dickson, D.W.; Wszolek, Z.K. Perry disease: Expanding the genetic basis. Mov. Disord. Clin. Pract. 2023, 10, 1136–1142. [Google Scholar] [CrossRef] [PubMed]
- Tsuboi, Y.; Dickson, D.W.; Nabeshima, K.; Schmeichel, A.M.; Wszolek, Z.K.; Yamada, T.; Benarroch, E.E. Neurodegeneration involving putative respiratory neurons in Perry syndrome. Acta Neuropathol. 2008, 115, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Puls, I.; Jonnakuty, C.; LaMonte, B.H.; Holzbaur, E.L.; Tokito, M.; Mann, E.; Floeter, M.K.; Bidus, K.; Drayna, D.; Oh, S.J.; et al. Mutant dynactin in motor neuron disease. Nat. Genet. 2003, 33, 455–456. [Google Scholar] [CrossRef] [PubMed]
- Puls, I.; Oh, S.J.; Sumner, C.J.; Wallace, K.E.; Floeter, M.K.; Mann, E.A.; Kennedy, W.R.; Wendelschafer-Crabb, G.; Vortmeyer, A.; Powers, R.; et al. Distal spinal and bulbar muscular atrophy caused by dynactin mutation. Ann. Neurol. 2005, 57, 687–694. [Google Scholar] [CrossRef]
- Alafuzoff, I.; Libard, S. Mixed brain pathology is the most common cause of cognitive impairment in the elderly. J. Alzheimers Dis. 2020, 78, 453–465. [Google Scholar] [CrossRef] [PubMed]
- Stein, T.D.; Crary, J.F. Chronic traumatic encephalopathy and neuropathological comorbidities. Semin. Neurol. 2020, 40, 384–393. [Google Scholar] [CrossRef]
- Yu, L.; Boyle, P.A.; Dawe, R.J.; Bennett, D.A.; Arfanakis, K.; Schneider, J.A. Contribution of TDP and hippocampal sclerosis to hippocampal volume loss in older-old persons. Neurology 2020, 94, e142–e152. [Google Scholar] [CrossRef]
- Hasegawa, M.; Arai, T.; Akiyama, H.; Nonaka, T.; Mori, H.; Hashimoto, T.; Yamazaki, M.; Oyanagi, K. TDP-43 is deposited in the Guam Parkinsonism-dementia complex brains. Brain 2007, 130, 1386–1394. [Google Scholar] [CrossRef]
- Miklossy, J.; Steele, J.C.; Yu, S.; McCall, S.; Sandberg, G.; McGeer, E.G.; McGeer, P.L. Enduring involvement of tau, beta-amyloid, alpha-synuclein, ubiquitin and TDP-43 pathology in the amyotrophic lateral sclerosis/parkinsonism-dementia complex of Guam (ALS/PDC). Acta Neuropathol. 2008, 116, 625–637. [Google Scholar] [CrossRef] [PubMed]
- Geser, F.; Winton, M.J.; Kwong, L.K.; Xu, Y.; Xie, S.X.; Igaz, L.M.; Garruto, R.M.; Perl, D.P.; Galasko, D.; Lee, V.M.; et al. Pathological TDP-43 in parkinsonism–dementia complex and amyotrophic lateral sclerosis of Guam. Acta Neuropathol. 2008, 115, 133–145. [Google Scholar] [CrossRef]
- Mimuro, M.; Yoshida, M.; Kuzuhara, S.; Kokubo, Y. Amyotrophic lateral sclerosis and Parkinsonism-dementia complex of the Hohara focus of the Kii Peninsula: A multiple proteinopathy? Neuropathology 2018, 38, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Condello, C.; Ayers, J.I.; Dalgard, C.L.; Garcia Garcia, M.M.; Rivera, B.M.; Seeley, W.W.; Perl, D.P.; Prusiner, S.B. Guam ALS-PDC is a distinct double-prion disorder featuring both tau and aβ prions. Proc. Natl. Acad. Sci. USA 2023, 120, e2220984120. [Google Scholar] [CrossRef] [PubMed]
- Honda, H.; Sasagasako, N.; Shen, C.; Shijo, M.; Hamasaki, H.; Suzuki, S.O.; Tsuboi, Y.; Fujii, N.; Iwaki, T. DCTN1 F52L mutation case of Perry syndrome with progressive supranuclear palsy-like tauopathy. Parkinsonism Relat. Disord. 2018, 51, 105–110. [Google Scholar] [CrossRef]
- Chung, E.J.; Kim, S.J.; Kim, E.J.; Ahn, J.W.; Huh, G.Y.; Cho, H.J.; Cairns, N.J. Neuropathological findings in a south Korean patient with Perry syndrome. Clin. Neuropathol. 2020, 39, 80–85. [Google Scholar] [CrossRef]
- Tafur, L.; Hinterndorfer, K.; Gabus, C.; Lamanna, C.; Bergmann, A.; Sadian, Y.; Hamdi, F.; Kyrilis, F.L.; Kastritis, P.L.; Loewith, R. Cryo-EM structure of the SEA complex. Nature 2022, 611, 399–404. [Google Scholar] [CrossRef]
- Schweighauser, M.; Arseni, D.; Bacioglu, M.; Huang, M.; Lövestam, S.; Shi, Y.; Yang, Y.; Zhang, W.; Kotecha, A.; Garringer, H.J.; et al. Age-dependent formation of TMEM106B amyloid filaments in human brains. Nature 2022, 605, 310–314. [Google Scholar] [CrossRef]
- Chang, A.; Xiang, X.; Wang, J.; Lee, C.; Arakhamia, T.; Simjanoska, M.; Wang, C.; Carlomagno, Y.; Zhang, G.; Dhingra, S.; et al. Homotypic fibrillization of TMEM106B across diverse neurodegenerative diseases. Cell 2022, 185, 1346–1355.e15. [Google Scholar] [CrossRef]
- Jiang, Y.X.; Cao, Q.; Sawaya, M.R.; Abskharon, R.; Ge, P.; DeTure, M.; Dickson, D.W.; Fu, J.Y.; Ogorzalek Loo, R.R.; Loo, J.A.; et al. Amyloid fibrils in FTLD-TDP are composed of TMEM106B and not TDP-43. Nature 2022, 605, 304–309. [Google Scholar] [CrossRef]
- Lee, Y.C.; Chung, C.P.; Chang, M.H.; Wang, S.J.; Liao, Y.C. NOTCH3 Cysteine-altering variant is an important risk factor for stroke in the Taiwanese population. Neurology 2020, 94, e87–e96. [Google Scholar] [CrossRef] [PubMed]
- Cho, B.P.H.; Nannoni, S.; Harshfield, E.L.; Tozer, D.; Gräf, S.; Bell, S.; Markus, H.S. NOTCH3 variants are more common than expected in the general population and associated with stroke and vascular dementia: An analysis of 200 000 participants. J. Neurol. Neurosurg. Psychiatry 2021, 92, 694–701. [Google Scholar] [CrossRef]
- Kim, Y.; Bae, J.S.; Lee, J.Y.; Song, H.K.; Lee, J.H.; Lee, M.; Kim, C.; Lee, S.H. Genotype and phenotype differences in CADASIL from an Asian perspective. Int. J. Mol. Sci. 2022, 23, 11506. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, S.; Yoshimoto, T.; Ohara, M.; Takagaki, M.; Nakamura, H.; Watanabe, K.; Gon, Y.; Todo, K.; Sasaki, T.; Araki, H.; et al. Effect of the RNF213 p.R4810K variant on the progression of intracranial artery stenosis: A 15-year follow-up study. Neurol. Genet. 2022, 8, e200029. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, T.; Tanaka, K.; Koga, J.; Saito, S.; Yamagami, H.; Nakaoku, Y.; Ogata, S.; Nishimura, K.; Yamaguchi, E.; Chiba, T.; et al. Impact of the RNF213 p.R4810K variant on endovascular therapy for large-vessel occlusion stroke. Stroke Vasc. Interv. Neurol. 2022, 2, e000396. [Google Scholar] [CrossRef]
- Ihara, M.; Yamamoto, Y.; Hattori, Y.; Liu, W.; Kobayashi, H.; Ishiyama, H.; Yoshimoto, T.; Miyawaki, S.; Clausen, T.; Bang, O.Y.; et al. Moyamoya disease: Diagnosis and interventions. Lancet Neurol. 2022, 21, 747–758. [Google Scholar] [CrossRef] [PubMed]
- Yabe, I.; Yaguchi, H.; Kato, Y.; Miki, Y.; Takahashi, H.; Tanikawa, S.; Shirai, S.; Takahashi, I.; Kimura, M.; Hama, Y.; et al. Mutations in bassoon in individuals with familial and sporadic progressive supranuclear palsy-like syndrome. Sci. Rep. 2018, 8, 819. [Google Scholar] [CrossRef]
- Schattling, B.; Engler, J.B.; Volkmann, C.; Rothammer, N.; Woo, M.S.; Petersen, M.; Winkler, I.; Kaufmann, M.; Rosenkranz, S.C.; Fejtova, A.; et al. Bassoon proteinopathy drives neurodegeneration in multiple sclerosis. Nat. Neurosci. 2019, 22, 887–896. [Google Scholar] [CrossRef]
- Schroer, T.A. Dynactin. Annu. Rev. Cell Dev. Biol. 2004, 20, 759–779. [Google Scholar] [CrossRef]
- Holzbaur, E.L.F.; Hammarback, J.A.; Paschal, B.M.; Kravit, N.G.; Pfister, K.K.; Vallee, R.B. Homology of a 150 K cytoplasmic dynein-associated polypeptide with the drosophila gene glued. Nature 1991, 351, 579–583. [Google Scholar] [CrossRef]
- Gill, S.R.; Schroer, T.A.; Szilak, I.; Steuer, E.R.; Sheetz, M.P.; Cleveland, D.W. Dynactin, a conserved, ubiquitously expressed component of an activator of vesicle motility mediated by cytoplasmic dynein. J. Cell Biol. 1991, 115, 1639–1650. [Google Scholar] [CrossRef]
- Karki, S.; Holzbaur, E.L.F. Affinity chromatography demonstrates a direct binding between cytoplasmic dynein and the dynactin complex. J. Biol. Chem. 1995, 270, 28806–28811. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, K.T.; Vallee, R.B. Cytoplasmic dynein binds dynactin through a direct interaction between the intermediate chains and p150 Glued. J. Cell Biol. 1995, 131, 1507–1516. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Foster, H.E.; Rondelet, A.; Lacey, S.E.; Bahi-Buisson, N.; Bird, A.W.; Carter, A.P. Cryo-EM reveals how human cytoplasmic dynein is auto-inhibited and activated. Cell 2017, 169, 1303–1314.e18. [Google Scholar] [CrossRef]
- Urnavicius, L.; Lau, C.K.; Elshenawy, M.M.; Morales-Rios, E.; Motz, C.; Yildiz, A.; Carter, A.P. Cryo-EM shows how dynactin recruits two dyneins for faster movement. Nature 2018, 554, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Olenick, M.A.; Holzbaur, E.L.F. Dynein activators and adaptors at a glance. J. Cell Sci. 2019, 132, jcs227132. [Google Scholar] [CrossRef] [PubMed]
- Reck-Peterson, S.L.; Redwine, W.B.; Vale, R.D.; Carter, A.P. The cytoplasmic dynein transport machinery and its many cargoes. Nat. Rev. Mol. Cell Biol. 2018, 19, 382–398. [Google Scholar] [CrossRef]
- Canty, J.T.; Yildiz, A. Activation and regulation of cytoplasmic dynein. Trends Biochem. Sci. 2020, 45, 440–453. [Google Scholar] [CrossRef]
- Waterman-Storer, C.M.; Karki, S.; Holzbaur, E.F.L. The p150Glued component of the dynactin complex binds to both microtubules and the actin-related protein centractin (Arp-1). Proc. Natl. Acad. Sci. USA 1995, 92, 1634–1638. [Google Scholar] [CrossRef]
- Vaughan, K.T.; Tynan, S.H.; Faulkner, N.E.; Echeverri, C.J.; Vallee, R.B. Colocalization of cytoplasmic dynein with dynactin and CLIP-170 at microtubule distal ends. J. Cell Sci. 1999, 112, 1437–1447. [Google Scholar] [CrossRef]
- Culver–Hanlon, T.L.; Lex, S.A.; Stephens, A.D.; Quintyne, N.J.; King, S.J. A microtubule-binding domain in dynactin increases dynein processivity by skating along microtubules. Nat. Cell Biol. 2006, 8, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Moughamian, A.J.; Holzbaur, E.L. Dynactin is required for transport initiation from the distal axon. Neuron 2012, 74, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, T.E.; Machamer, J.; O’Hara, K.; Kim, J.H.; Collins, S.E.; Wong, M.Y.; Sahin, B.; Imlach, W.; Yang, Y.; Levitan, E.S.; et al. The p150Glued CAP-Gly domain regulates initiation of retrograde transport at synaptic termini. Neuron 2012, 74, 344–360. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, H.; Liu, W.; Wang, J.; Zhang, J.; Chang, X.; Huang, S.; Pang, X.; Guo, J.; Wang, Q.; et al. A novel Q93H missense mutation in DCTN1 caused distal hereditary motor neuropathy type 7B and Perry syndrome from a Chinese family. Neurol. Sci. 2021, 42, 3695–3705. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, P.; Sarma, G.R.K.; Murgod, U.; Srinivas, M.; Roy, A.K. Perry syndrome with a novel mutation and a rare presentation: First report from India. Ann. Indian Acad. Neurol. 2022, 25, 703–706. [Google Scholar] [CrossRef] [PubMed]
- Boardman, J.; Mascareno Ponte, M.; Chaouch, A.; Kobylecki, C. Perry syndrome with intrafamilial heterogeneity in presentation and survival including acute respiratory failure: Case series. Mov. Disord. Clin. Pract. 2022, 9, 816–820. [Google Scholar] [CrossRef] [PubMed]
- Dulski, J.; Cerquera-Cleves, C.; Milanowski, L.; Kwiatek-Majkusiak, J.; Koziorowski, D.; Ross, O.A.; Pentela-Nowicka, J.; Sławek, J.; Wszolek, Z.K. L-Dopa response, choreic dyskinesia, and dystonia in Perry syndrome. Parkinsonism Relat. Disord. 2022, 100, 19–23. [Google Scholar] [CrossRef]
- Stoker, T.B.; Dostal, V.; Cochius, J.; Williams-Gray, C.H.; Scherzer, C.R.; Wang, J.; Liu, G.; Coyle-Gilchrist, I. DCTN1 mutation associated Parkinsonism: Case series of three new families with Perry syndrome. J. Neurol. 2022, 269, 6667–6672. [Google Scholar] [CrossRef]
- Silva, E.; Itzcovich, T.; Niikado, M.; Caride, A.; Fernández, E.; Vázquez, J.C.; Romorini, L.; Marazita, M.; Sevlever, G.; Martinetto, H.; et al. Perry disease in an Argentine family due to the DCTN1 p.G67D variant. Parkinsonism Relat. Disord. 2022, 97, 63–64. [Google Scholar] [CrossRef]
- Pan, X.; Hong, Q.; Lu, X.; Li, Z.; Wang, L.; Chen, W.; Pan, S. A Chinese pedigree with Perry disease caused by the p.Y78H mutation in DCTN1: A 6-year clinical follow-up. Behav. Brain Res. 2023, 441, 114284. [Google Scholar] [CrossRef]
- Hwang, S.H.; Kim, E.J.; Hong, Y.B.; Joo, J.; Kim, S.M.; Nam, S.H.; Hong, H.D.; Kim, S.H.; Oh, K.; Lim, J.G.; et al. Distal hereditary motor neuropathy type 7B with dynactin 1 mutation. Mol. Med. Rep. 2016, 14, 3362–3368. [Google Scholar] [CrossRef] [PubMed]
- Münch, C.; Sedlmeier, R.; Meyer, T.; Homberg, V.; Sperfeld, A.D.; Kurt, A.; Prudlo, J.; Peraus, G.; Hanemann, C.O.; Stumm, G.; et al. Point mutations of the P150 subunit of dynactin (DCTN1) gene in ALS. Neurology 2004, 63, 724–726. [Google Scholar] [CrossRef] [PubMed]
- Konno, T.; Ross, O.A.; Teive, H.A.G.; Sławek, J.; Dickson, D.W.; Wszolek, Z.K. DCTN1-related neurodegeneration: Perry syndrome and beyond. Parkinsonism Relat. Disord. 2017, 41, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Dulski, J.; Konno, T.; Wszolek, Z. DCTN1-related neurodegeneration. In GeneReviews® [Internet]; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2010. Available online: https://www.ncbi.nlm.nih.gov/books/NBK47027/ (accessed on 17 December 2023).
- He, J.; Yu, W.; Liu, X.; Fan, D. An identical DCTN1 mutation in two Chinese siblings manifest as dHMN and ALS respectively: A case report. Amyotroph Lateral Scler Front. Degener. 2022, 23, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Goldman, J.S. Predictive genetic counseling for neurodegenerative diseases: Past, present, and future. Cold Spring Harb. Perspect. Med. 2020, 10, a036525. [Google Scholar] [CrossRef]
- Wider, C.; Dachsel, J.C.; Farrer, M.J.; Dickson, D.W.; Tsuboi, Y.; Wszolek, Z.K. Elucidating the genetics and pathology of Perry syndrome. J. Neurol. Sci. 2010, 289, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, K.; Saiki, S.; Furuya, N.; Yamada, D.; Imamichi, Y.; Li, Y.; Kawajiri, S.; Sasaki, H.; Koike, M.; Tsuboi, Y.; et al. P150glued-associated disorders are caused by activation of intrinsic apoptotic pathway. PLoS ONE 2014, 9, e94645. [Google Scholar] [CrossRef] [PubMed]
- Mishima, T.; Ishikawa, T.; Imamura, K.; Kondo, T.; Koshiba, Y.; Takahashi, R.; Takahashi, J.; Watanabe, A.; Fujii, N.; Tsuboi, Y.; et al. Cytoplasmic aggregates of dynactin in iPSC-derived tyrosine hydroxylase-positive neurons from a patient with Perry syndrome. Parkinsonism Relat. Disord. 2016, 30, 67–72. [Google Scholar] [CrossRef]
- Ishikawa, K.I.; Saiki, S.; Furuya, N.; Imamichi, Y.; Tsuboi, Y.; Hattori, N. P150glued deficiency impairs effective fusion between autophagosomes and lysosomes due to their redistribution to the cell periphery. Neurosci. Lett. 2019, 690, 181–187. [Google Scholar] [CrossRef]
- Deshimaru, M.; Kinoshita-Kawada, M.; Kubota, K.; Watanabe, T.; Tanaka, Y.; Hirano, S.; Ishidate, F.; Hiramoto, M.; Ishikawa, M.; Uehara, Y.; et al. DCTN1 binds to TDP-43 and regulates TDP-43 aggregation. Int. J. Mol. Sci. 2021, 22, 3985. [Google Scholar] [CrossRef]
- Mishima, T.; Deshimaru, M.; Watanabe, T.; Kubota, K.; Kinoshita-Kawada, M.; Yuasa-Kawada, J.; Takasaki, K.; Uehara, Y.; Jinno, S.; Iwasaki, K.; et al. Behavioral defects in a DCTN1G71A transgenic mouse model of Perry syndrome. Neurosci. Lett. 2018, 666, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Deshimaru, M.; Mishima, T.; Watanabe, T.; Kubota, K.; Hosoi, M.; Kinoshita-Kawada, M.; Yuasa-Kawada, J.; Ikeda, M.; Mori, M.; Murata, Y.; et al. Behavioral profile in a Dctn1G71A knock-in mouse model of Perry disease. Neurosci. Lett. 2021, 764, 136234. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Lai, C.; Shim, H.; Xie, C.; Sun, L.; Long, C.X.; Ding, J.; Li, Y.; Cai, H. Genetic ablation of dynactin P150Glued in postnatal neurons causes preferential degeneration of spinal motor neurons in aged mice. Mol. Neurodegener. 2018, 13, 10. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Yang, X.; Zheng, J.; Sgobio, C.; Sun, L.; Cai, H. Deficiency of Perry syndrome-associated P150Glued in midbrain dopaminergic neurons leads to progressive neurodegeneration and endoplasmic reticulum abnormalities. NPJ Parkinsons Dis. 2023, 9, 35. [Google Scholar] [CrossRef] [PubMed]
- Levy, J.R.; Sumner, C.J.; Caviston, J.P.; Tokito, M.K.; Ranganathan, S.; Ligon, L.A.; Wallace, K.E.; LaMonte, B.H.; Harmison, G.G.; Puls, I.; et al. A motor neuron disease-associated mutation in p150Glued perturbs dynactin function and induces protein aggregation. J. Cell Biol. 2006, 172, 733–745. [Google Scholar] [CrossRef] [PubMed]
- Araki, E.; Tsuboi, Y.; Daechsel, J.; Milnerwood, A.; Vilarino-Guell, C.; Fujii, N.; Mishima, T.; Oka, T.; Hara, H.; Fukae, J.; et al. A novel DCTN1 mutation with late-onset Parkinsonism and frontotemporal atrophy. Mov. Disord. 2014, 29, 1201–1204. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.; Lin, X.; Chandran, J.; Shim, H.; Yang, W.J.; Cai, H. The G59S mutation in p150glued causes dysfunction of dynactin in mice. J. Neurosci. 2007, 27, 13982–13990. [Google Scholar] [CrossRef] [PubMed]
- Chevalier-Larsen, E.S.; Wallace, K.E.; Pennise, C.R.; Holzbaur, E.L. Lysosomal proliferation and distal degeneration in motor neurons expressing the G59S mutation in the P150Glued subunit of dynactin. Hum. Mol. Genet. 2008, 17, 1946–1955. [Google Scholar] [CrossRef]
- Laird, F.M.; Farah, M.H.; Ackerley, S.; Hoke, A.; Maragakis, N.; Rothstein, J.D.; Griffin, J.; Price, D.L.; Martin, L.J.; Wong, P.C. Motor neuron disease occurring in a mutant dynactin mouse model is characterized by defects in vesicular trafficking. J. Neurosci. 2008, 28, 1997–2005. [Google Scholar] [CrossRef]
- Lazarus, J.E.; Moughamian, A.J.; Tokito, M.K.; Holzbaur, E.L. Dynactin subunit P150Glued is a neuron-specific anti-catastrophe factor. PLoS Biol. 2013, 11, e1001611. [Google Scholar] [CrossRef]
- Hosaka, Y.; Inoshita, T.; Shiba-Fukushima, K.; Cui, C.; Arano, T.; Imai, Y.; Hattori, N. Reduced TDP-43 expression improves neuronal activities in a drosophila model of Perry syndrome. eBioMedicine 2017, 21, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Gaj, T.; Sirk, S.J.; Shui, S.L.; Liu, J. Genome-editing technologies: Principles and applications. Cold Spring Harb. Perspect. Biol. 2016, 8, a023754. [Google Scholar] [CrossRef] [PubMed]
- Bulaklak, K.; Gersbach, C.A. The once and future gene therapy. Nat. Commun. 2020, 11, 5820. [Google Scholar] [CrossRef] [PubMed]
- Horton, R.H.; Saade, D.; Markati, T.; Harriss, E.; Bönnemann, C.G.; Muntoni, F.; Servais, L. A systematic review of adeno-associated virus gene therapies in neurology: The need for consistent safety monitoring of a promising treatment. J. Neurol. Neurosurg. Psychiatry 2022, 93, 1276–1288. [Google Scholar] [CrossRef] [PubMed]
- Tacik, P.; Fiesel, F.C.; Fujioka, S.; Ross, O.A.; Pretelt, F.; Castañeda Cardona, C.; Kidd, A.; Hlavac, M.; Raizis, A.; Okun, M.S.; et al. Three families with Perry syndrome from distinct parts of the world. Parkinsonism Relat. Disord. 2014, 20, 884–888. [Google Scholar] [CrossRef] [PubMed]
- Trang, H.; Samuels, M.; Ceccherini, I.; Frerick, M.; Garcia-Teresa, M.A.; Peters, J.; Schoeber, J.; Migdal, M.; Markstrom, A.; Ottonello, G.; et al. Guidelines for diagnosis and management of congenital central hypoventilation syndrome. Orphanet J. Rare Dis. 2020, 15, 252. [Google Scholar] [CrossRef] [PubMed]
- Mishima, T.; Fujioka, S.; Fukae, J.; Yuasa-Kawada, J.; Tsuboi, Y. Modeling Parkinson’s disease and atypical Parkinsonian syndromes using induced pluripotent stem cells. Int. J. Mol. Sci. 2018, 19, 3870. [Google Scholar] [CrossRef]
- Shad, M.U. Recent developments in pharmacotherapy of depression: Bench to bedside. J. Pers. Med. 2023, 13, 773. [Google Scholar] [CrossRef]
- Louis, E.D. Essential tremor: From bedside to bench and back to bedside. Curr. Opin. Neurol. 2014, 27, 461–467. [Google Scholar] [CrossRef]
- Hampton, T. Bench to bedside and back again may be key to clinical breakthroughs. JAMA 2017, 318, 16–17. [Google Scholar] [CrossRef]
- Manolio, T.A.; Fowler, D.M.; Starita, L.M.; Haendel, M.A.; MacArthur, D.G.; Biesecker, L.G.; Worthey, E.; Chisholm, R.L.; Green, E.D.; Jacob, H.J.; et al. Bedside back to bench: Building bridges between basic and clinical genomic research. Cell 2017, 169, 6–12. [Google Scholar] [CrossRef]
- Vollstedt, E.J.; Kasten, M.; Klein, C.; MJFF Global Genetic Parkinson’s Disease Study Group. Using global team science to identify genetic Parkinson’s disease worldwide. Ann. Neurol. 2019, 86, 153–157. [Google Scholar] [CrossRef]
- Lupușoru, G.; Ailincăi, I.; Frățilă, G.; Ungureanu, O.; Andronesi, A.; Lupușoru, M.; Banu, M.; Văcăroiu, I.; Dina, C.; Sinescu, I. Tumor lysis syndrome: An endless challenge in onco-nephrology. Biomedicines 2022, 10, 1012. [Google Scholar] [CrossRef]
- Boarescu, P.M.; Roşian, A.N.; Roşian, Ş.H. Transvenous lead extraction procedure-indications, methods, and complications. Biomedicines 2022, 10, 2780. [Google Scholar] [CrossRef]
- Mishima, T.; Fujioka, S.; Morishita, T.; Inoue, T.; Tsuboi, Y. Personalized medicine in Parkinson’s disease: New options for advanced treatments. J. Pers. Med. 2021, 11, 650. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Battaglia, S.; Nazzi, C.; Thayer, J.F. Heart’s tale of trauma: Fear-conditioned heart rate changes in post-traumatic stress disorder. Acta Psychiatr. Scand. 2023, 148, 463–466. [Google Scholar] [CrossRef]
- Tran, K.N.; Nguyen, N.P.K.; Nguyen, L.T.H.; Shin, H.M.; Yang, I.J. Screening for neuroprotective and rapid antidepressant-like effects of 20 essential oils. Biomedicines 2023, 11, 1248. [Google Scholar] [CrossRef]
- Polyák, H.; Galla, Z.; Nánási, N.; Cseh, E.K.; Rajda, C.; Veres, G.; Spekker, E.; Szabó, Á.; Klivényi, P.; Tanaka, M.; et al. The tryptophan-kynurenine metabolic system is suppressed in cuprizone-induced model of demyelination simulating progressive multiple sclerosis. Biomedicines 2023, 11, 945. [Google Scholar] [CrossRef]
- Battaglia, S.; Schmidt, A.; Hassel, S.; Tanaka, M. Editorial: Case reports in neuroimaging and stimulation. Front. Psychiatry 2023, 14, 1264669. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Features | Laboratory Features | |
|---|---|---|
| Cardinal | Supportive | Cardinal |
| (A) Parkinsonism * | (a) Rapid disease progression within five years of onset | (1) Genetic test: mutation in the DCTN1 gene |
| (B) Apathy or depression | (b) Onset younger than 50 years | (2) Pathology: nigral neuronal loss and TDP-43 pathology in the brainstem and basal ganglia |
| (C) Respiratory symptoms ** | ||
| (D) Unexpected weight loss | ||
| (E) Positive family history of parkinsonism or respiratory symptoms | ||
| Definite: Presence of (A) and (E) plus cardinal laboratory features of (1) positive genetic test; Presence of (A), (B), (C), and (D) plus cardinal laboratory features of (1) positive genetic test; Presence of (A), (B), (C), and (D) plus cardinal laboratory features of (2) TDP-43 pathologies. If evidence of other mutations or neurodegenerative disease pathology is present, both cardinal laboratory features must also be observed. | ||
| Probable: Presence of (A), (B), (C), (D), and (E). | ||
| Possible: Presence of (A) and (E) plus supportive clinical features of (a) or (b). | ||
| Disease | Parkinsonism | Apathy/ Depression | Weight Loss | Respiratory Symptoms | Autonomic Dysfunction | Vertical Gaze Palsy |
|---|---|---|---|---|---|---|
| Perry disease | + | + | + | + | + | + |
| PD | + | + | + | - | + | - |
| MSA | + | + | ± | + | + | - |
| PSP | + | + | - | - | - | + |
| FTLD | + | + | + | + | - | + |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mishima, T.; Yuasa-Kawada, J.; Fujioka, S.; Tsuboi, Y. Perry Disease: Bench to Bedside Circulation and a Team Approach. Biomedicines 2024, 12, 113. https://doi.org/10.3390/biomedicines12010113
Mishima T, Yuasa-Kawada J, Fujioka S, Tsuboi Y. Perry Disease: Bench to Bedside Circulation and a Team Approach. Biomedicines. 2024; 12(1):113. https://doi.org/10.3390/biomedicines12010113
Chicago/Turabian StyleMishima, Takayasu, Junichi Yuasa-Kawada, Shinsuke Fujioka, and Yoshio Tsuboi. 2024. "Perry Disease: Bench to Bedside Circulation and a Team Approach" Biomedicines 12, no. 1: 113. https://doi.org/10.3390/biomedicines12010113
APA StyleMishima, T., Yuasa-Kawada, J., Fujioka, S., & Tsuboi, Y. (2024). Perry Disease: Bench to Bedside Circulation and a Team Approach. Biomedicines, 12(1), 113. https://doi.org/10.3390/biomedicines12010113