
Sarcoidosis Associated Pulmonary Hypertension


Abstract
1. Introduction
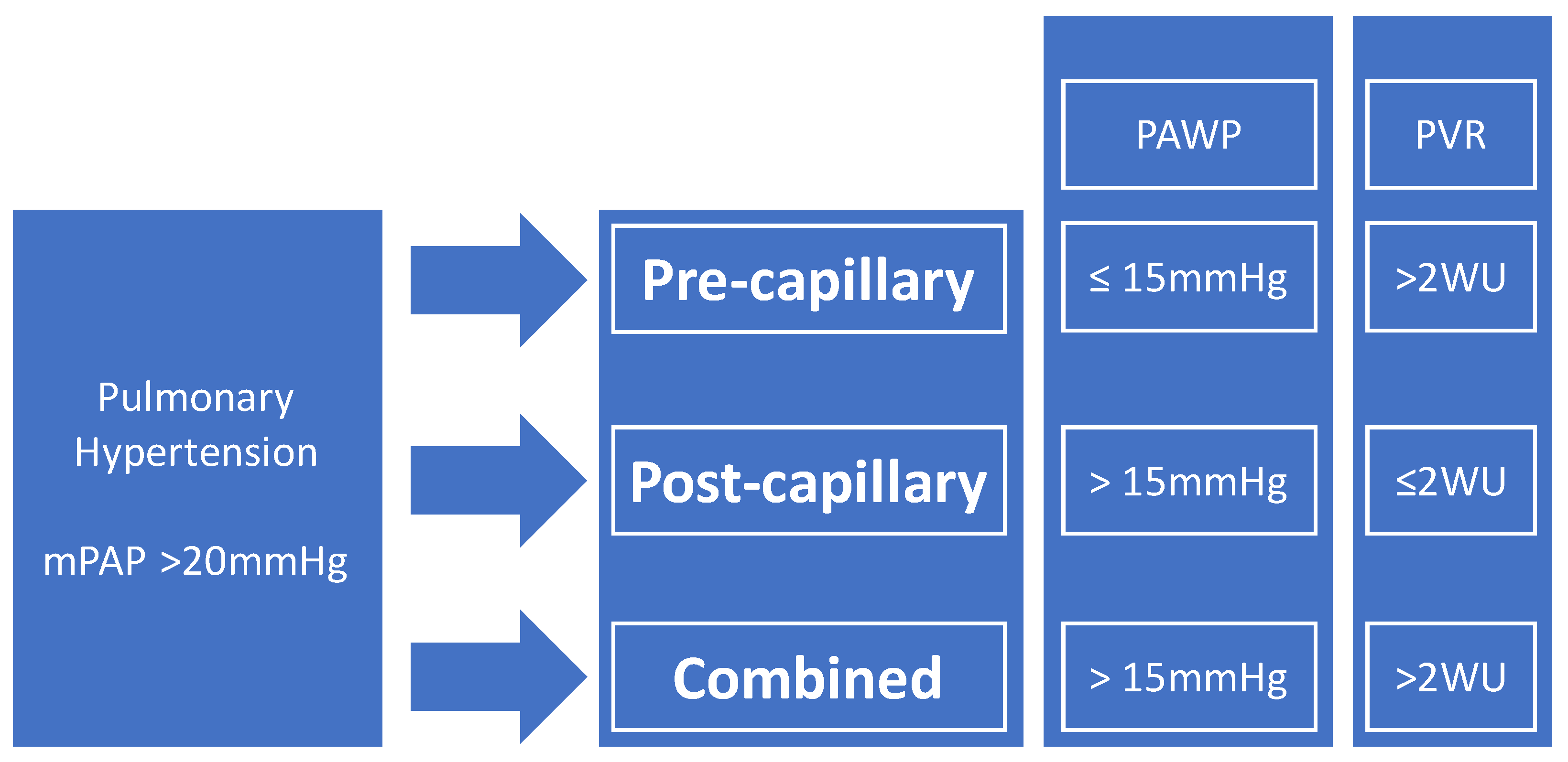
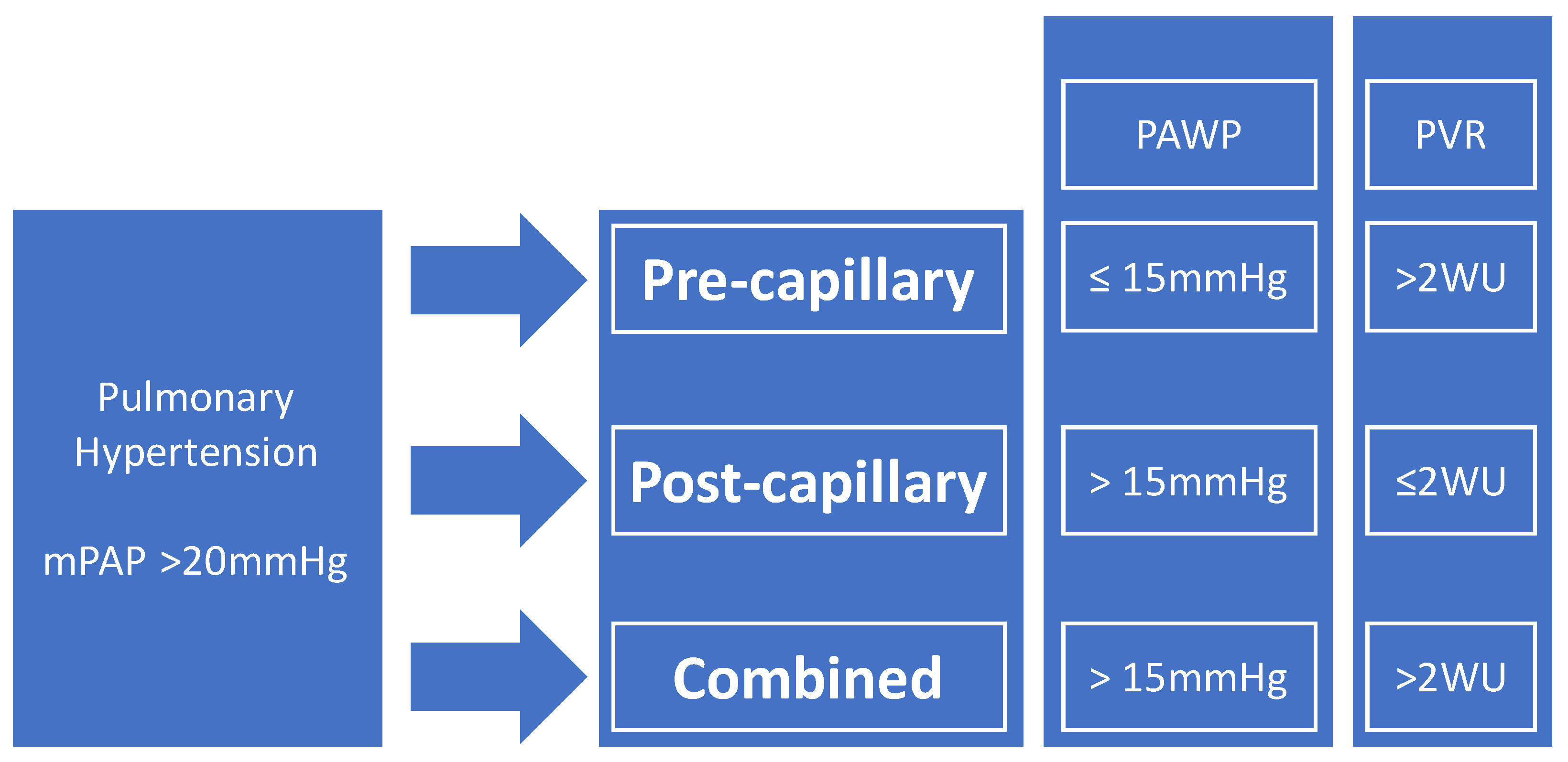
2. Definitions of PH in Sarcoidosis
3. Epidemiology
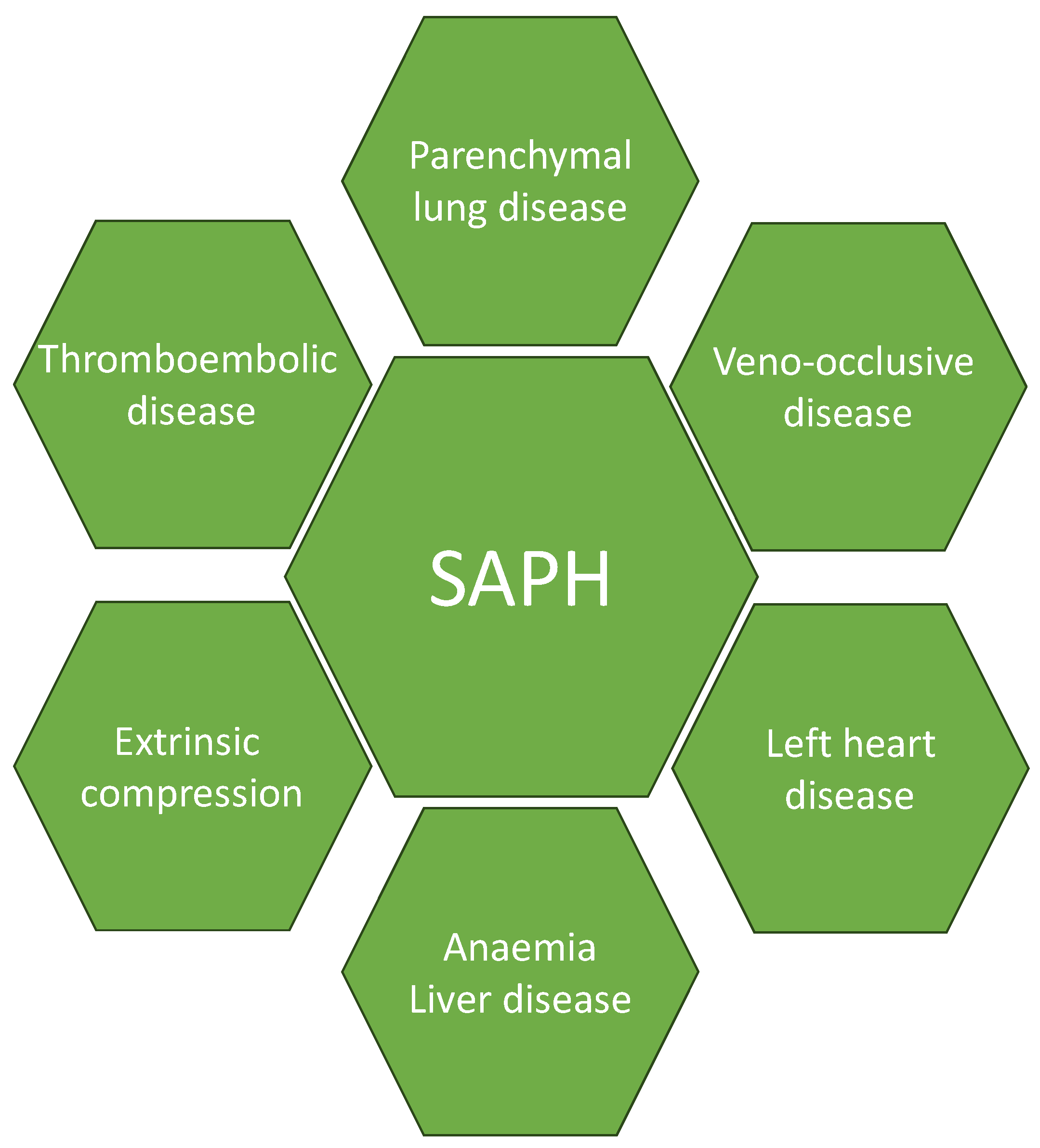
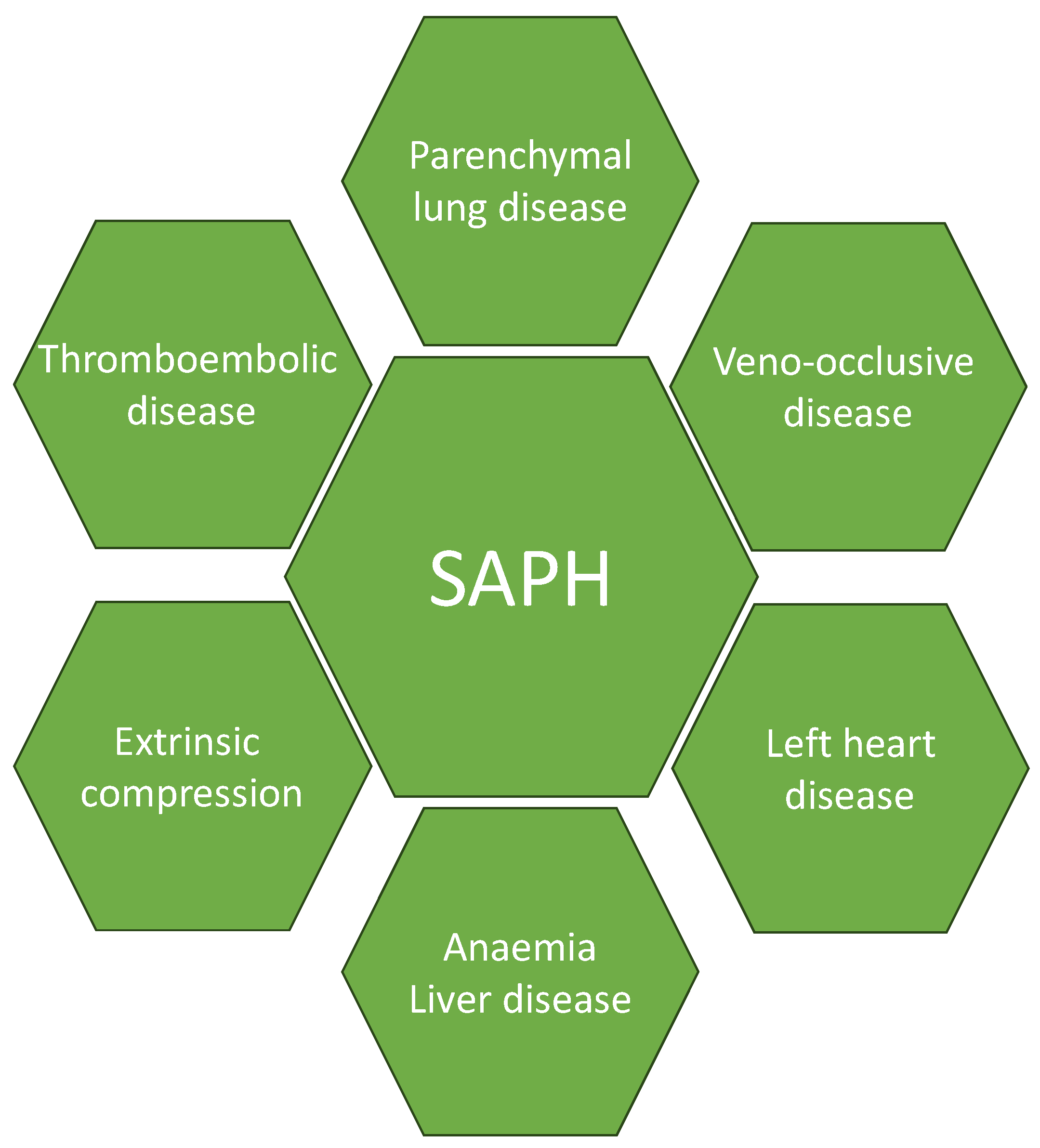
4. Pathophysiology
4.1. Parenchymal Lung Disease
4.2. Cardiac Diseases
4.3. Veno-Occlusive Disease
4.4. Thromboembolic Disease
4.5. Extrinsic Compressors and Other Factors
5. Screening and Diagnosis
5.1. Clinical Symptoms and Signs
5.2. Electrocardiogram (ECG)
5.3. Chest X-ray (CXR) and CT
5.4. Functional Tests
5.5. Ventilation/Perfusion (V/Q) Scan
5.6. Transthoracic Echocardiography (TTE)
5.7. Cardiovascular Magnetic Resonance (CMR) and Positron Emission Tomography (PET)
5.8. Invasive Assessment
5.9. Multidisciplinary Approach in the Diagnosis and Management
6. Clinical Management
7. Drug Treatments of SAPH
7.1. Endothelin Receptor Antagonists
7.2. Phosphodiesterase 5 Inhibitors (PDE5i)
7.3. Soluble Guanylate Cyclase Stimulator
7.4. Prostacyclin-Based Therapy
7.5. Combination Therapy
7.6. Immunosuppressive Therapy
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Churg, A.; Carrington, C.B.; Gupta, R. Necrotizing Sarcoid Granulomatosis. Chest 1979, 76, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Baughman, R.P.; Teirstein, A.S.; Judson, M.A.; Rossman, M.D.; Yeager, H.; Bresnitz, E.A.; DePalo, L.; Hunninghake, G.; Iannuzzi, M.C.; Johns, C.J.; et al. Clinical Characteristics of Patients in a Case Control Study of Sarcoidosis. Am. J. Respir. Crit. Care Med. 2001, 164, 1885–1889. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.; Yadav, D.; Puranik, N.; Guleria, R.; Jin, J.O. Sarcoidosis: Causes, Diagnosis, Clinical Features, and Treatments. J. Clin. Med. 2020, 9, 1081. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Guzman, E.; Farver, C.; Parambil, J.; Culver, D.A. Pulmonary hypertension caused by sarcoidosis. Clin. Chest Med. 2008, 29, 549–563. [Google Scholar] [CrossRef] [PubMed]
- Samaranayake, C.B.; McCabe, C.; Wort, S.J.; Price, L.C. Sarcoidosis associated pulmonary hypertension: An update. Curr. Opin. Pulm. Med. 2021, 27, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Kovacs, G.; Hoeper, M.M.; Badagliacca, R.; Berger, R.M.F.; Brida, M.; Carlsen, J.; Coats, A.J.S.; Escribano-Subias, P.; Ferrari, P.; et al. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: Developed by the task force for the diagnosis and treatment of pulmonary hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS). Endorsed by the International Society for Heart and Lung Transplantation (ISHLT) and the European Reference Network on rare respiratory diseases (ERN-LUNG). Eur. Heart J. 2022, 43, 3618–3731. [Google Scholar] [CrossRef] [PubMed]
- Duong, H.; Bonham, C.A. Sarcoidosis-associated Pulmonary Hypertension: Pathophysiology, Diagnosis, and Treatment. Clin. Pulm. Med. 2018, 25, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.; Berghold, A.; Scheidl, S.; Olschewski, H. Pulmonary arterial pressure during rest and exercise in healthy subjects: A systematic review. Eur. Respir. J. 2009, 34, 888–894. [Google Scholar] [CrossRef]
- Kovacs, G.; Olschewski, A.; Berghold, A.; Olschewski, H. Pulmonary vascular resistances during exercise in normal subjects: A systematic review. Eur. Respir. J. 2012, 39, 319–328. [Google Scholar] [CrossRef]
- Maron, B.A.; Hess, E.; Maddox, T.M.; Opotowsky, A.R.; Tedford, R.J.; Lahm, T.; Joynt, K.E.; Kass, D.J.; Stephens, T.; Stanislawski, M.A.; et al. Association of Borderline Pulmonary Hypertension With Mortality and Hospitalization in a Large Patient Cohort: Insights From the Veterans Affairs Clinical Assessment, Reporting, and Tracking Program. Circulation 2016, 133, 1240–1248. [Google Scholar] [CrossRef]
- Douschan, P.; Kovacs, G.; Avian, A.; Foris, V.; Gruber, F.; Olschewski, A.; Olschewski, H. Mild Elevation of Pulmonary Arterial Pressure as a Predictor of Mortality. Am. J. Respir. Crit. Care Med. 2018, 197, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Kolte, D.; Lakshmanan, S.; Jankowich, M.D.; Brittain, E.L.; Maron, B.A.; Choudhary, G. Mild Pulmonary Hypertension Is Associated With Increased Mortality: A Systematic Review and Meta-Analysis. J. Am. Heart Assoc. 2018, 7, e009729. [Google Scholar] [CrossRef] [PubMed]
- Pabst, S.; Hammerstingl, C.; Grau, N.; Kreuz, J.; Grohe, C.; Juergens, U.R.; Nickenig, G.; Skowasch, D. Pulmonary arterial hypertension in patients with sarcoidosis: The Pulsar single center experience. Adv. Exp. Med. Biol. 2013, 755, 299–305. [Google Scholar] [CrossRef] [PubMed]
- Alhamad, E.H.; Idrees, M.M.; Alanezi, M.O.; Alboukai, A.A.; Shaik, S.A. Sarcoidosis-associated pulmonary hypertension: Clinical features and outcomes in Arab patients. Ann. Thorac. Med. 2010, 5, 86–91. [Google Scholar] [CrossRef]
- Huitema, M.P.; Bakker, A.L.M.; Mager, J.J.; Rensing, B.; Smits, F.; Snijder, R.J.; Grutters, J.C.; Post, M.C. Prevalence of pulmonary hypertension in pulmonary sarcoidosis: The first large European prospective study. Eur. Respir. J. 2019, 54, 1900897. [Google Scholar] [CrossRef]
- Zhang, S.; Tong, X.; Zhang, T.; Wang, D.; Liu, S.; Wang, L.; Fan, H. Prevalence of Sarcoidosis-Associated Pulmonary Hypertension: A Systematic Review and Meta-Analysis. Front. Cardiovasc. Med. 2021, 8, 809594. [Google Scholar] [CrossRef]
- Shorr, A.F.; Helman, D.L.; Davies, D.B.; Nathan, S.D. Pulmonary hypertension in advanced sarcoidosis: Epidemiology and clinical characteristics. Eur. Respir. J. 2005, 25, 783–788. [Google Scholar] [CrossRef]
- Baughman, R.P.; Shlobin, O.A.; Wells, A.U.; Alhamad, E.H.; Culver, D.A.; Barney, J.; Cordova, F.C.; Carmona, E.M.; Scholand, M.B.; Wijsenbeek, M.; et al. Clinical features of sarcoidosis associated pulmonary hypertension: Results of a multi-national registry. Respir. Med. 2018, 139, 72–78. [Google Scholar] [CrossRef]
- Islam, M.I.; Eggert, M.; Bernens, M.; Sill, J. Pulmonary veno-occlusive disease in sarcoidosis patient: A rare entity causing pulmonary hypertension. Chest 2020, 158, A2117–A2118. [Google Scholar] [CrossRef]
- Nunes, H.; Humbert, M.; Capron, F.; Brauner, M.; Sitbon, O.; Battesti, J.P.; Simonneau, G.; Valeyre, D. Pulmonary hypertension associated with sarcoidosis: Mechanisms, haemodynamics and prognosis. Thorax 2006, 61, 68. [Google Scholar] [CrossRef]
- Ungprasert, P.; Srivali, N.; Wijarnpreecha, K.; Thongprayoon, C. Sarcoidosis and risk of venous thromboembolism: A systematic review and meta-analysis. Sarcoidosis Vasc. Diffus. Lung Dis. 2015, 32, 182–187. [Google Scholar]
- Condado, J.F.; Babaliaros, V.; Henry, T.S.; Kaebnick, B.; Kim, D.; Staton, G.W., Jr. Pulmonary stenting for the treatment of sarcoid induced pulmonary vascular stenosis. Sarcoidosis Vasc. Diffus. Lung Dis. 2016, 33, 281–287. [Google Scholar]
- Damuth, T.E.; Bower, J.S.; Cho, K.; Dantzker, D.R. Major pulmonary artery stenosis causing pulmonary hypertension in sarcoidosis. Chest 1980, 78, 888–891. [Google Scholar] [CrossRef] [PubMed]
- Hamilton-Craig, C.R.; Slaughter, R.; McNeil, K.; Kermeen, F.; Walters, D.L. Improvement after angioplasty and stenting of pulmonary arteries due to sarcoid mediastinal fibrosis. Heart Lung Circ. 2009, 18, 222–225. [Google Scholar] [CrossRef] [PubMed]
- Hoffstein, V.; Ranganathan, N.; Mullen, J.B. Sarcoidosis simulating pulmonary veno-occlusive disease. Am. Rev. Respir. Dis. 1986, 134, 809–811. [Google Scholar] [CrossRef] [PubMed]
- Takemura, T.; Matsui, Y.; Saiki, S.; Mikami, R. Pulmonary vascular involvement in sarcoidosis: A report of 40 autopsy cases. Hum. Pathol. 1992, 23, 1216–1223. [Google Scholar] [CrossRef]
- Lal, C.; Medarov, B.I.; Judson, M.A. Interrelationship between sleep-disordered breathing and sarcoidosis. Chest 2015, 148, 1105–1114. [Google Scholar] [CrossRef]
- Sonnweber, T.; Pizzini, A.; Tancevski, I.; Löffler-Ragg, J.; Weiss, G. Anaemia, iron homeostasis and pulmonary hypertension: A review. Intern. Emerg. Med. 2020, 15, 573–585. [Google Scholar] [CrossRef]
- Nickel, N.P.; Galura, G.M.; Zuckerman, M.J.; Hakim, M.N.; Alkhateeb, H.; Mukherjee, D.; Austin, E.D.; Heresi, G.A. Liver abnormalities in pulmonary arterial hypertension. Pulm. Circ. 2021, 11, 20458940211054304. [Google Scholar] [CrossRef]
- Shlobin, O.A.; Kouranos, V.; Barnett, S.D.; Alhamad, E.H.; Culver, D.A.; Barney, J.; Cordova, F.C.; Carmona, E.M.; Scholand, M.B.; Wijsenbeek, M.; et al. Physiological predictors of survival in patients with sarcoidosis-associated pulmonary hypertension: Results from an international registry. Eur. Respir. J. 2020, 55, 1901747. [Google Scholar] [CrossRef]
- Kouranos, V.; Sharma, R. Cardiac sarcoidosis: State-of-the-art review. Heart 2021, 107, 1591–1599. [Google Scholar] [CrossRef] [PubMed]
- Guazzi, M.; Borlaug, B.A. Pulmonary hypertension due to left heart disease. Circulation 2012, 126, 975–990. [Google Scholar] [CrossRef]
- Baughman, R.P.; Engel, P.J.; Taylor, L.; Lower, E.E. Survival in sarcoidosis-associated pulmonary hypertension: The importance of hemodynamic evaluation. Chest 2010, 138, 1078–1085. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.B.; Mor-Avi, V.; Murtagh, G.; Bonham, C.A.; Laffin, L.J.; Hogarth, D.K.; Medvedofsky, D.; Lang, R.M.; Patel, A.R. Right Heart Involvement in Patients with Sarcoidosis. Echocardiography 2016, 33, 734–741. [Google Scholar] [CrossRef]
- Swigris, J.J.; Olson, A.L.; Huie, T.J.; Fernandez-Perez, E.R.; Solomon, J.J.; Sprunger, D.; Brown, K.K. Increased risk of pulmonary embolism among US decedents with sarcoidosis from 1988 to 2007. Chest 2011, 140, 1261–1266. [Google Scholar] [CrossRef] [PubMed]
- Ungprasert, P.; Crowson, C.S.; Matteson, E.L. Association of Sarcoidosis With Increased Risk of VTE: A Population-Based Study, 1976 to 2013. Chest 2017, 151, 425–430. [Google Scholar] [CrossRef]
- Crawshaw, A.P.; Wotton, C.J.; Yeates, D.G.; Goldacre, M.J.; Ho, L.P. Evidence for association between sarcoidosis and pulmonary embolism from 35-year record linkage study. Thorax 2011, 66, 447–448. [Google Scholar] [CrossRef]
- Boucly, A.; Cottin, V.; Nunes, H.; Jaïs, X.; Tazi, A.; Prévôt, G.; Reynaud-Gaubert, M.; Dromer, C.; Viacroze, C.; Horeau-Langlard, D.; et al. Management and long-term outcomes of sarcoidosis-associated pulmonary hypertension. Eur. Respir. J. 2017, 50, 1700465. [Google Scholar] [CrossRef]
- Savale, L.; Huitema, M.; Shlobin, O.; Kouranos, V.; Nathan, S.D.; Nunes, H.; Gupta, R.; Grutters, J.C.; Culver, D.A.; Post, M.C.; et al. WASOG statement on the diagnosis and management of sarcoidosis-associated pulmonary hypertension. Eur. Respir. Rev. 2022, 31, 210165. [Google Scholar] [CrossRef]
- Ng, C.S.; Wells, A.U.; Padley, S.P. A CT sign of chronic pulmonary arterial hypertension: The ratio of main pulmonary artery to aortic diameter. J. Thorac. Imaging 1999, 14, 270–278. [Google Scholar] [CrossRef]
- Huitema, M.P.; Spee, M.; Vorselaars, V.M.; Boerman, S.; Snijder, R.J.; van Es, H.W.; Reesink, H.J.; Grutters, J.C.; Post, M.C. Pulmonary artery diameter to predict pulmonary hypertension in pulmonary sarcoidosis. Eur. Respir. J. 2016, 47, 673–676. [Google Scholar] [CrossRef] [PubMed]
- Stein, P.D.; Fowler, S.E.; Goodman, L.R.; Gottschalk, A.; Hales, C.A.; Hull, R.D.; Leeper, K.V., Jr.; Popovich, J., Jr.; Quinn, D.A.; Sos, T.A.; et al. Multidetector computed tomography for acute pulmonary embolism. N. Engl. J. Med. 2006, 354, 2317–2327. [Google Scholar] [CrossRef] [PubMed]
- Bourbonnais, J.M.; Samavati, L. Clinical predictors of pulmonary hypertension in sarcoidosis. Eur. Respir. J. 2008, 32, 296–302. [Google Scholar] [CrossRef]
- Mirsaeidi, M.; Omar, H.R.; Baughman, R.; Machado, R.; Sweiss, N. The association between BNP, 6MWD test, DLCO% and pulmonary hypertension in sarcoidosis. Sarcoidosis Vasc. Diffus. Lung Dis. 2016, 33, 317–320. [Google Scholar]
- Gupta, R.; Baughman, R.P.; Nathan, S.D.; Wells, A.U.; Kouranos, V.; Alhamad, E.H.; Culver, D.A.; Barney, J.; Carmona, E.M.; Cordova, F.C.; et al. The six-minute walk test in sarcoidosis associated pulmonary hypertension: Results from an international registry. Respir. Med. 2022, 196, 106801. [Google Scholar] [CrossRef]
- Tandon, R.; Baughman, R.P.; Stanley, J.; Khan, A.A. The link between chronic thromboembolic pulmonary hypertension and sarcoidosis: Association or visual masquerade? Sarcoidosis Vasc. Diffus. Lung Dis. 2017, 34, 352–355. [Google Scholar] [CrossRef]
- Kelle, S.; Bucciarelli-Ducci, C.; Judd, R.M.; Kwong, R.Y.; Simonetti, O.; Plein, S.; Raimondi, F.; Weinsaft, J.W.; Wong, T.C.; Carr, J. Society for Cardiovascular Magnetic Resonance (SCMR) recommended CMR protocols for scanning patients with active or convalescent phase COVID-19 infection. J. Cardiovasc. Magn. Reson. 2020, 22, 61. [Google Scholar] [CrossRef]
- Ahmadi, A.; Ohira, H.; Mielniczuk, L.M. FDG PET imaging for identifying pulmonary hypertension and right heart failure. Curr. Cardiol. Rep. 2015, 17, 555. [Google Scholar] [CrossRef]
- Hagan, G.; Southwood, M.; Treacy, C.; Ross, R.M.; Soon, E.; Coulson, J.; Sheares, K.; Screaton, N.; Pepke-Zaba, J.; Morrell, N.W.; et al. 18FDG PET imaging can quantify increased cellular metabolism in pulmonary arterial hypertension: A proof-of-principle study. Pulm. Circ. 2011, 1, 448–455. [Google Scholar] [CrossRef]
- Marsboom, G.; Wietholt, C.; Haney, C.R.; Toth, P.T.; Ryan, J.J.; Morrow, E.; Thenappan, T.; Bache-Wiig, P.; Piao, L.; Paul, J.; et al. Lung 18F-fluorodeoxyglucose positron emission tomography for diagnosis and monitoring of pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2012, 185, 670–679. [Google Scholar] [CrossRef]
- Maier, A.; Liao, S.L.; Lescure, T.; Robson, P.M.; Hirata, N.; Sartori, S.; Narula, N.; Vergani, V.; Soultanidis, G.; Morgenthau, A.; et al. Pulmonary Artery 18F-Fluorodeoxyglucose Uptake by PET/CMR as a Marker of Pulmonary Hypertension in Sarcoidosis. JACC Cardiovasc. Imaging 2022, 15, 108–120. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Ashek, A.; Wang, L.; Fang, W.; Dabral, S.; Dubois, O.; Cupitt, J.; Pullamsetti, S.S.; Cotroneo, E.; Jones, H.; et al. Heterogeneity in lung 18FDG uptake in pulmonary arterial hypertension: Potential of dynamic 18FDG positron emission tomography with kinetic analysis as a bridging biomarker for pulmonary vascular remodeling targeted treatments. Circulation 2013, 128, 1214–1224. [Google Scholar] [CrossRef] [PubMed]
- Ovadia, M. Pulmonary Arterial 18F-FDG Uptake in Sarcoidosis: A Novel Biosignal for Subclinical Pulmonary Hypertension. JACC Cardiovasc. Imaging 2022, 15, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Milman, N.; Svendsen, C.B.; Iversen, M.; Videbaek, R.; Carlsen, J. Sarcoidosis-associated pulmonary hypertension: Acute vasoresponsiveness to inhaled nitric oxide and the relation to long-term effect of sildenafil. Clin. Respir. J. 2009, 3, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Preston, I.R.; Klinger, J.R.; Landzberg, M.J.; Houtchens, J.; Nelson, D.; Hill, N.S. Vasoresponsiveness of sarcoidosis-associated pulmonary hypertension. Chest 2001, 120, 866–872. [Google Scholar] [CrossRef] [PubMed]
- daSilva-deAbreu, A.; Mandras, S.A. Sarcoidosis-Associated Pulmonary Hypertension: An Updated Review and Discussion of the Clinical Conundrum. Curr. Probl. Cardiol. 2021, 46, 100506. [Google Scholar] [CrossRef]
- Huitema, M.P.; Mathijssen, H.; Mager, J.J.; Snijder, R.J.; Grutters, J.C.; Post, M.C. Sarcoidosis-Associated Pulmonary Hypertension. Semin. Respir. Crit. Care Med. 2020, 41, 659–672. [Google Scholar] [CrossRef]
- Cullivan, S.; Gaine, S.; Sitbon, O. New trends in pulmonary hypertension. Eur. Respir. Rev. 2023, 32, 220211. [Google Scholar] [CrossRef]
- Macera, F.; Vachiéry, J.L. Management of Pulmonary Hypertension in Left Heart Disease. Methodist. DeBakey Cardiovasc. J. 2021, 17, 115–123. [Google Scholar] [CrossRef]
- Le Pavec, J.; Valeyre, D.; Gazengel, P.; Holm, A.M.; Schultz, H.H.; Perch, M.; Le Borgne, A.; Reynaud-Gaubert, M.; Knoop, C.; Godinas, L.; et al. Lung transplantation for sarcoidosis: Outcome and prognostic factors. Eur. Respir. J. 2021, 58, 2003358. [Google Scholar] [CrossRef]
- Weill, D.; Benden, C.; Corris, P.A.; Dark, J.H.; Davis, R.D.; Keshavjee, S.; Lederer, D.J.; Mulligan, M.J.; Patterson, G.A.; Singer, L.G.; et al. A consensus document for the selection of lung transplant candidates: 2014--an update from the Pulmonary Transplantation Council of the International Society for Heart and Lung Transplantation. J. Heart Lung Transplant. 2015, 34, 1–15. [Google Scholar] [CrossRef]
- Fadel, E.; Mercier, O.; Mussot, S.; Leroy-Ladurie, F.; Cerrina, J.; Chapelier, A.; Simonneau, G.; Dartevelle, P. Long-term outcome of double-lung and heart-lung transplantation for pulmonary hypertension: A comparative retrospective study of 219 patients. Eur. J. Cardio-Thorac. Surg. 2010, 38, 277–284. [Google Scholar] [CrossRef]
- Shah, L. Lung Transplantation in Sarcoidosis. Semin. Respir. Crit. Care Med. 2007, 28, 134–140. [Google Scholar] [CrossRef]
- Wryobeck, J.M.; Lippo, G.; McLaughlin, V.; Riba, M.; Rubenfire, M. Psychosocial aspects of pulmonary hypertension: A review. Psychosomatics 2007, 48, 467–475. [Google Scholar] [CrossRef]
- Bussotti, M.; Sommaruga, M. Anxiety and depression in patients with pulmonary hypertension: Impact and management challenges. Vasc. Health Risk Manag. 2018, 14, 349–360. [Google Scholar] [CrossRef]
- Rawlings, G.H.; Novakova, B.; Armstrong, I.; Thompson, A.R. A systematic review of psychological interventions in adults with pulmonary hypertension: Is the evidence-base disproportionate to the problem? Clin. Respir. J. 2023, 17, 966–972. [Google Scholar] [CrossRef]
- Currie, B.M.; Davies, E.W.; Beaudet, A.; Stassek, L.; Kleinman, L.; Baughman, R.P. Symptoms, impacts, and suitability of the Pulmonary Arterial Hypertension-Symptoms and Impact (PAH-SYMPACT™) questionnaire in patients with sarcoidosis-associated pulmonary hypertension (SAPH): A qualitative interview study. BMC Pulm. Med. 2021, 21, 365. [Google Scholar] [CrossRef]
- Chin, K.M.; Gomberg-Maitland, M.; Channick, R.N.; Cuttica, M.J.; Fischer, A.; Frantz, R.P.; Hunsche, E.; Kleinman, L.; McConnell, J.W.; McLaughlin, V.V.; et al. Psychometric Validation of the Pulmonary Arterial Hypertension-Symptoms and Impact (PAH-SYMPACT) Questionnaire: Results of the SYMPHONY Trial. Chest 2018, 154, 848–861. [Google Scholar] [CrossRef]
- Baughman, R.P.; Culver, D.A.; Cordova, F.C.; Padilla, M.; Gibson, K.F.; Lower, E.E.; Engel, P.J. Bosentan for Sarcoidosis-Associated Pulmonary Hypertension: A Double-Blind Placebo Controlled Randomized Trial. Chest 2014, 145, 810–817. [Google Scholar] [CrossRef]
- Judson, M.A.; Highland, K.B.; Kwon, S.; Donohue, J.F.; Aris, R.; Craft, N.; Burt, S.; Ford, H.J. Ambrisentan for sarcoidosis associated pulmonary hypertension. Sarcoidosis Vasc. Diffus. Lung Dis. 2011, 28, 139–145. [Google Scholar]
- Mathijssen, H.; Huitema, M.P.; Bakker, A.L.M.; Mager, J.J.; Snijder, R.J.; Grutters, J.C.; Post, M.C. Safety of macitentan in sarcoidosis-associated pulmonary hypertension: A case-series. Sarcoidosis Vasc. Diffus. Lung Dis. 2020, 37, 74–78. [Google Scholar] [CrossRef]
- Milman, N.; Burton, C.M.; Iversen, M.; Videbaek, R.; Jensen, C.V.; Carlsen, J. Pulmonary hypertension in end-stage pulmonary sarcoidosis: Therapeutic effect of sildenafil? J. Heart Lung Transplant. 2008, 27, 329–334. [Google Scholar] [CrossRef]
- Ford, H.J.; Baughman, R.P.; Aris, R.; Engel, P.; Donohue, J.F. Tadalafil therapy for sarcoidosis-associated pulmonary hypertension. Pulm. Circ. 2016, 6, 557–562. [Google Scholar] [CrossRef] [PubMed]
- Keir, G.J.; Walsh, S.L.; Gatzoulis, M.A.; Marino, P.S.; Dimopoulos, K.; Alonso, R.; Raposeiras-Roubin, S.; Renzoni, E.A.; Maher, T.M.; Wells, A.U.; et al. Treatment of sarcoidosis-associated pulmonary hypertension: A single centre retrospective experience using targeted therapies. Sarcoidosis Vasc. Diffus. Lung Dis. 2014, 31, 82–90. [Google Scholar]
- Baughman, R.P.; Shlobin, O.A.; Gupta, R.; Engel, P.J.; Stewart, J.I.; Lower, E.E.; Rahaghi, F.F.; Zeigler, J.; Nathan, S.D. Riociguat for Sarcoidosis-Associated Pulmonary Hypertension: Results of a 1-Year Double-Blind, Placebo-Controlled Trial. Chest 2022, 161, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Fisher, K.A.; Serlin, D.M.; Wilson, K.C.; Walter, R.E.; Berman, J.S.; Farber, H.W. Sarcoidosis-associated pulmonary hypertension: Outcome with long-term epoprostenol treatment. Chest 2006, 130, 1481–1488. [Google Scholar] [CrossRef]
- Baughman, R.P.; Judson, M.A.; Lower, E.E.; Highland, K.; Kwon, S.; Craft, N.; Engel, P.J. Inhaled iloprost for sarcoidosis associated pulmonary hypertension. Sarcoidosis Vasc. Diffus. Lung Dis. 2009, 26, 110–120. [Google Scholar]
- Abston, E.; Hon, S.; Lawrence, R.; Berman, J.; Govender, P.; Farber, H.W. Treatment of newly diagnosed sarcoid-associated pulmonary hypertension with ambrisentan and tadalafil combination therapy. Sarcoidosis Vasc. Diffus. Lung Dis. 2020, 37, 234–238. [Google Scholar] [CrossRef]
- Sumimoto, K.; Taniguchi, Y.; Emoto, N.; Hirata, K.I. Combination therapy with pulmonary arterial hypertension targeted drugs and immunosuppression can be a useful strategy for sarcoidosis-associated pulmonary hypertension: A case report. Eur. Heart J. Case Rep. 2021, 5, ytaa454. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
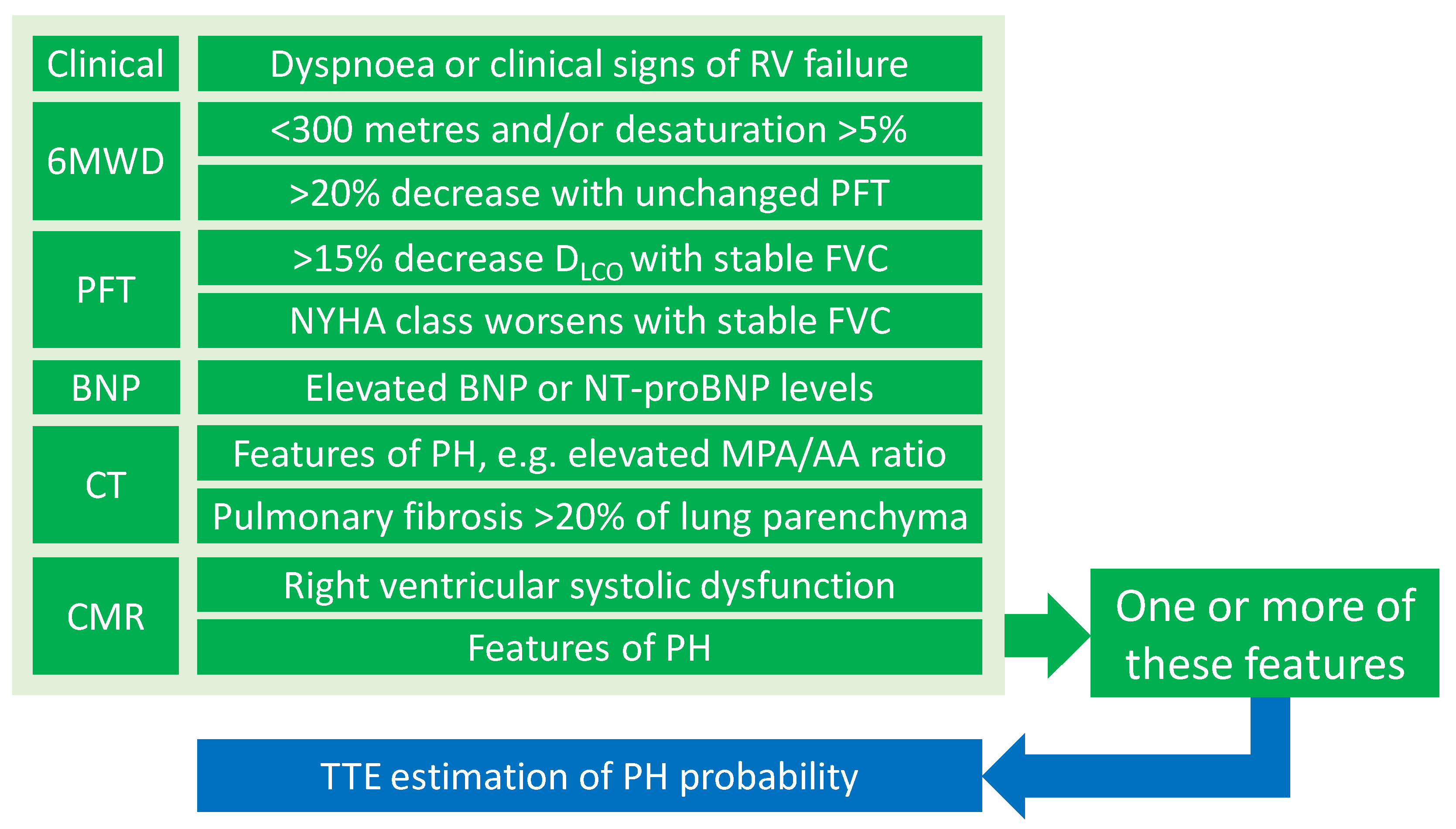{kind=link}
| Investigations | Salient Parameters |
|---|---|
| Electrocardiogram | Features of PH |
| Lung function test | Disproportionate reduction in DLCO; Kco |
| Six-minute walk test | Six-minute walk distance and desaturation |
| Arterial blood gas sample | Alveolar-arterial gradient |
| Blood test sample | BNP or NT-proBNP |
| Chest CT | MPA/AA diameter ratio, pulmonary fibrosis |
| CMR | RV dysfunction and PH features |
| Echocardiography | Probability of pulmonary hypertension |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, A.; Price, L.C.; Sharma, R.; Wells, A.U.; Kouranos, V. Sarcoidosis Associated Pulmonary Hypertension. Biomedicines 2024, 12, 177. https://doi.org/10.3390/biomedicines12010177
Liu A, Price LC, Sharma R, Wells AU, Kouranos V. Sarcoidosis Associated Pulmonary Hypertension. Biomedicines. 2024; 12(1):177. https://doi.org/10.3390/biomedicines12010177
Chicago/Turabian StyleLiu, Alexander, Laura C. Price, Rakesh Sharma, Athol U. Wells, and Vasileios Kouranos. 2024. "Sarcoidosis Associated Pulmonary Hypertension" Biomedicines 12, no. 1: 177. https://doi.org/10.3390/biomedicines12010177
APA StyleLiu, A., Price, L. C., Sharma, R., Wells, A. U., & Kouranos, V. (2024). Sarcoidosis Associated Pulmonary Hypertension. Biomedicines, 12(1), 177. https://doi.org/10.3390/biomedicines12010177