
The Impact of Microglia on Neurodevelopment and Brain Function in Autism


Abstract
:1. Introduction
2. The Source of Microglia
3. Neurodevelopmental Abnormalities on ASD Caused by Microglia
3.1. Microglia Affect Neurogenesis in ASD
3.2. Microglia Affect the Neural Circuits on ASD
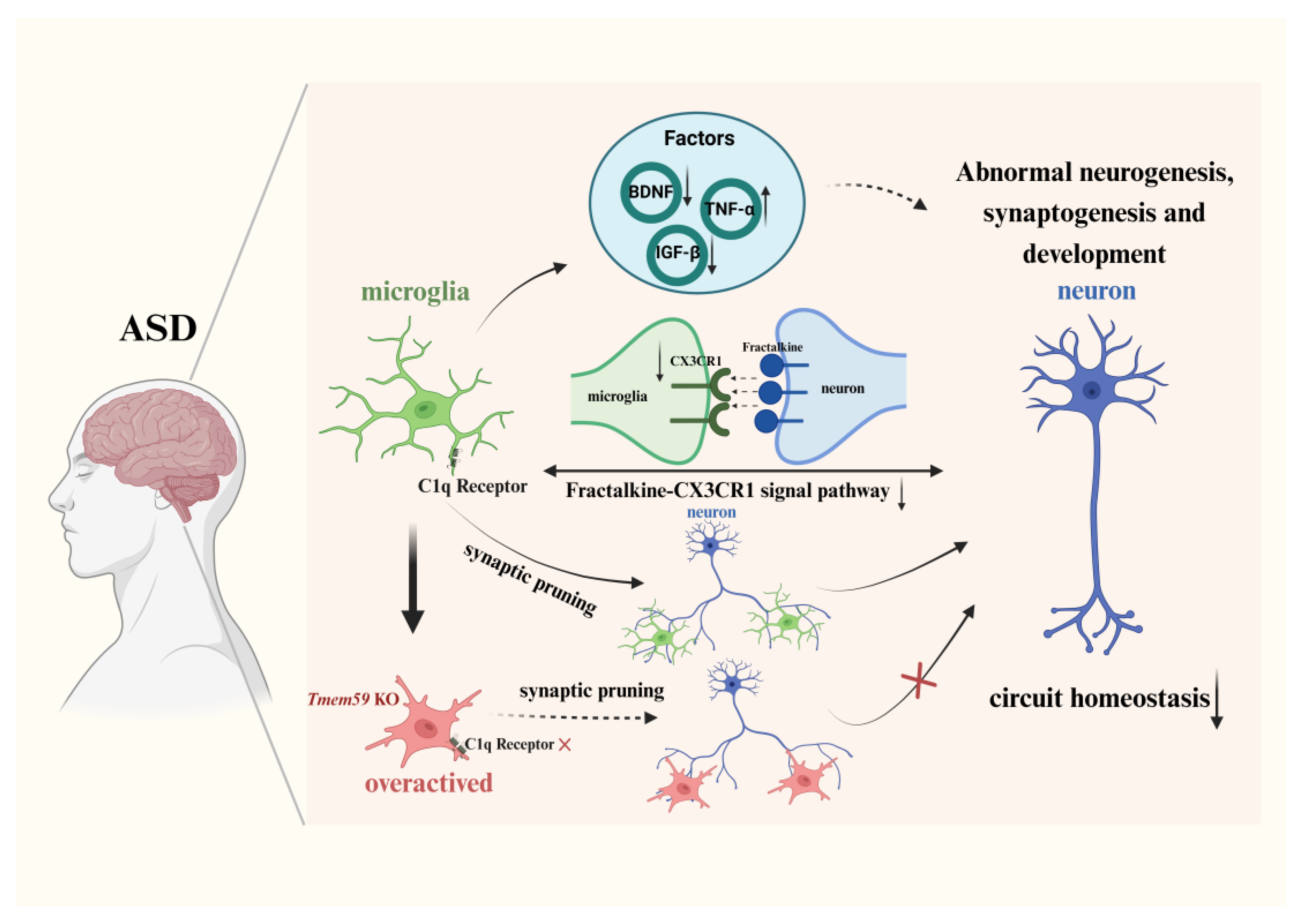
3.2.1. Synapse Formation and Pruning via the Complement Pathway
3.2.2. Neural Circuit Formation and Signal Pathway
4. Factors Released by Microglia in ASD
4.1. Deficient BDNF Associated with Microglia Causes Abnormal ASD
4.2. Reduced IGF-1 Associated with Microglia Causes Abnormal ASD
4.3. Increased TNF-α Associated with Microglia Causes Abnormal ASD
5. The Immune Function of Microglia on ASD
5.1. Migration and Monitor of Microglia
5.2. Different Status of Microglia
6. Interaction between Microglia and Other Cells in the CNS
6.1. Interaction between Microglia and Neurons
6.2. Interaction between Microglia and Astrocyte
6.3. Interaction between Microglia and Oligodendrocyte
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Malwane, M.I.; Nguyen, E.B.; Trejo, S.; Kim, E.Y.; Cucalón-Calderón, J.R. A Delayed Diagnosis of Autism Spectrum Disorder in the Setting of Complex Attention Deficit Hyperactivity Disorder. Cureus 2022, 14, e25825. [Google Scholar] [CrossRef] [PubMed]
- Lai, M.-C.; Lombardo, M.V.; Baron-Cohen, S. Autism. Lancet 2014, 383, 896–910. [Google Scholar] [CrossRef] [PubMed]
- Longo, F.; Klann, E. Reciprocal Control of Translation and Transcription in Autism Spectrum Disorder. EMBO Rep. 2021, 22, e52110. [Google Scholar] [CrossRef] [PubMed]
- Park, H.R.; Lee, J.M.; Moon, H.E.; Lee, D.S.; Kim, B.-N.; Kim, J.; Kim, D.G.; Paek, S.H. A Short Review on the Current Understanding of Autism Spectrum Disorders. Exp. Neurobiol. 2016, 25, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Quesnel-Vallières, M.; Weatheritt, R.J.; Cordes, S.P.; Blencowe, B.J. Autism Spectrum Disorder: Insights into Convergent Mechanisms from Transcriptomics. Nat. Rev. Genet. 2019, 20, 51–63. [Google Scholar] [CrossRef]
- Gandal, M.J.; Haney, J.R.; Wamsley, B.; Yap, C.X.; Parhami, S.; Emani, P.S.; Chang, N.; Chen, G.T.; Hoftman, G.D.; de Alba, D.; et al. Broad Transcriptomic Dysregulation Occurs across the Cerebral Cortex in ASD. Nature 2022, 611, 532–539. [Google Scholar] [CrossRef]
- Salter, M.W.; Stevens, B. Microglia Emerge as Central Players in Brain Disease. Nat. Med. 2017, 23, 1018–1027. [Google Scholar] [CrossRef]
- Kracht, L.; Borggrewe, M.; Eskandar, S.; Brouwer, N.; Chuva De Sousa Lopes, S.M.; Laman, J.D.; Scherjon, S.A.; Prins, J.R.; Kooistra, S.M.; Eggen, B.J.L. Human Fetal Microglia Acquire Homeostatic Immune-Sensing Properties Early in Development. Science 2020, 369, 530–537. [Google Scholar] [CrossRef]
- Prinz, M.; Jung, S.; Priller, J. Microglia Biology: One Century of Evolving Concepts. Cell 2019, 179, 292–311. [Google Scholar] [CrossRef]
- Ronaldson, P.T.; Davis, T.P. Regulation of Blood-Brain Barrier Integrity by Microglia in Health and Disease: A Therapeutic Opportunity. J. Cereb. Blood Flow Metab. 2020, 40, S6–S24. [Google Scholar] [CrossRef]
- Nayak, D.; Zinselmeyer, B.H.; Corps, K.N.; McGavern, D.B. In Vivo Dynamics of Innate Immune Sentinels in the CNS. IntraVital 2012, 1, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Davalos, D.; Grutzendler, J.; Yang, G.; Kim, J.V.; Zuo, Y.; Jung, S.; Littman, D.R.; Dustin, M.L.; Gan, W.-B. ATP Mediates Rapid Microglial Response to Local Brain Injury in Vivo. Nat. Neurosci. 2005, 8, 752–758. [Google Scholar] [CrossRef] [PubMed]
- Stence, N.; Waite, M.; Dailey, M.E. Dynamics of Microglial Activation: A Confocal Time-Lapse Analysis in Hippocampal Slices. Glia 2001, 33, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Eugenín, E.A.; Eckardt, D.; Theis, M.; Willecke, K.; Bennett, M.V.L.; Sáez, J.C. Microglia at Brain Stab Wounds Express Connexin 43 and in Vitro Form Functional Gap Junctions after Treatment with Interferon-γ and Tumor Necrosis Factor-α. Proc. Natl. Acad. Sci. USA 2001, 98, 4190–4195. [Google Scholar] [CrossRef]
- Xu, Z.-X.; Kim, G.H.; Tan, J.-W.; Riso, A.E.; Sun, Y.; Xu, E.Y.; Liao, G.-Y.; Xu, H.; Lee, S.-H.; Do, N.-Y.; et al. Elevated Protein Synthesis in Microglia Causes Autism-like Synaptic and Behavioral Aberrations. Nat. Commun. 2020, 11, 1797. [Google Scholar] [CrossRef]
- Kitamura, T.; Miyake, T.; Fujita, S. Genesis of Resting Microglia in the Gray Matter of Mouse Hippocampus. J. Comp. Neurol. 1984, 226, 421–433. [Google Scholar] [CrossRef]
- Beers, D.R.; Henkel, J.S.; Xiao, Q.; Zhao, W.; Wang, J.; Yen, A.A.; Siklos, L.; McKercher, S.R.; Appel, S.H. Wild-Type Microglia Extend Survival in PU.1 Knockout Mice with Familial Amyotrophic Lateral Sclerosis. Proc. Natl. Acad. Sci. USA 2006, 103, 16021–16026. [Google Scholar] [CrossRef]
- Murabe, Y.; Sano, Y. Morphological Studies on Neuroglia. VI. Postnatal Development of Microglial Cells. Cell Tissue Res. 1982, 225, 469–485. [Google Scholar] [CrossRef]
- Rio-Hortega, P. THE MICROGLIA. Lancet 1939, 233, 1023–1026. [Google Scholar] [CrossRef]
- Lawson, L.J.; Perry, V.H.; Dri, P.; Gordon, S. Heterogeneity in the Distribution and Morphology of Microglia in the Normal Adult Mouse Brain. Neuroscience 1990, 39, 151–170. [Google Scholar] [CrossRef]
- Prinz, M.; Masuda, T.; Wheeler, M.A.; Quintana, F.J. Microglia and Central Nervous System–Associated Macrophages—From Origin to Disease Modulation. Annu. Rev. Immunol. 2021, 39, 251–277. [Google Scholar] [CrossRef] [PubMed]
- Squarzoni, P.; Thion, M.S.; Garel, S. Neuronal and Microglial Regulators of Cortical Wiring: Usual and Novel Guideposts. Front. Neurosci. 2015, 9, 248. [Google Scholar] [CrossRef] [PubMed]
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef]
- Thion, M.S.; Ginhoux, F.; Garel, S. Microglia and Early Brain Development: An Intimate Journey. Science 2018, 362, 185–189. [Google Scholar] [CrossRef]
- Vandenbark, A.A.; Offner, H.; Matejuk, S.; Matejuk, A. Microglia and Astrocyte Involvement in Neurodegeneration and Brain Cancer. J. Neuroinflammation 2021, 18, 298. [Google Scholar] [CrossRef]
- Cunningham, C.L.; Martínez-Cerdeño, V.; Noctor, S.C. Microglia Regulate the Number of Neural Precursor Cells in the Developing Cerebral Cortex. J. Neurosci. 2013, 33, 4216. [Google Scholar] [CrossRef] [PubMed]
- Wong, F.K.; Marín, O. Developmental Cell Death in the Cerebral Cortex. Annu. Rev. Cell Dev. Biol. 2019, 35, 523–542. [Google Scholar] [CrossRef]
- Diaz-Aparicio, I.; Paris, I.; Sierra-Torre, V.; Plaza-Zabala, A.; Rodríguez-Iglesias, N.; Márquez-Ropero, M.; Beccari, S.; Huguet, P.; Abiega, O.; Alberdi, E.; et al. Microglia Actively Remodel Adult Hippocampal Neurogenesis through the Phagocytosis Secretome. J. Neurosci. 2020, 40, 1453–1482. [Google Scholar] [CrossRef]
- Ziv, Y.; Ron, N.; Butovsky, O.; Landa, G.; Sudai, E.; Greenberg, N.; Cohen, H.; Kipnis, J.; Schwartz, M. Immune Cells Contribute to the Maintenance of Neurogenesis and Spatial Learning Abilities in Adulthood. Nat. Neurosci. 2006, 9, 268–275. [Google Scholar] [CrossRef]
- Morgan, S.C.; Taylor, D.L.; Pocock, J.M. Microglia Release Activators of Neuronal Proliferation Mediated by Activation of Mitogen-Activated Protein Kinase, Phosphatidylinositol-3-Kinase/Akt and Delta-Notch Signalling Cascades. J. Neurochem. 2004, 90, 89–101. [Google Scholar] [CrossRef]
- Antony, J.M.; Paquin, A.; Nutt, S.L.; Kaplan, D.R.; Miller, F.D. Endogenous Microglia Regulate Development of Embryonic Cortical Precursor Cells. J. Neurosci. Res. 2011, 89, 286–298. [Google Scholar] [CrossRef] [PubMed]
- Comer, A.L.; Carrier, M.; Tremblay, M.-È.; Cruz-Martín, A. The Inflamed Brain in Schizophrenia: The Convergence of Genetic and Environmental Risk Factors That Lead to Uncontrolled Neuroinflammation. Front. Cell. Neurosci. 2020, 14, 274. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, K.; He, X.; Zhou, J.; Jin, C.; Shen, L.; Gao, Y.; Tian, M.; Zhang, H. Structural, Functional, and Molecular Imaging of Autism Spectrum Disorder. Neurosci. Bull. 2021, 37, 1051–1071. [Google Scholar] [CrossRef] [PubMed]
- Maldonado-Ruiz, R.; Garza-Ocañas, L.; Camacho, A. Inflammatory Domains Modulate Autism Spectrum Disorder Susceptibility during Maternal Nutritional Programming. Neurochem. Int. 2019, 126, 109–117. [Google Scholar] [CrossRef]
- Dutra, M.L.; Dias, P.; Freiberger, V.; Ventura, L.; Comim, C.M.; Martins, D.F.; Bobinski, F. Maternal Immune Activation Induces Autism-like Behavior and Reduces Brain-Derived Neurotrophic Factor Levels in the Hippocampus and Offspring Cortex of C57BL/6 Mice. Neurosci. Lett. 2023, 793, 136974. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Lv, K.; Du, Z.; Zhang, D.; Chen, M.; Luo, J.; Wang, L.; Liu, T.; Gong, H.; Fan, X. Minocycline Improves Autism-Related Behaviors by Modulating Microglia Polarization in a Mouse Model of Autism. Int. Immunopharmacol. 2023, 122, 110594. [Google Scholar] [CrossRef]
- Walton, N.M.; Sutter, B.M.; Laywell, E.D.; Levkoff, L.H.; Kearns, S.M.; Marshall, G.P.; Scheffler, B.; Steindler, D.A. Microglia Instruct Subventricular Zone Neurogenesis. Glia 2006, 54, 815–825. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, M.-È.; Lowery, R.L.; Majewska, A.K. Microglial Interactions with Synapses Are Modulated by Visual Experience. PLoS Biol. 2010, 8, e1000527. [Google Scholar] [CrossRef]
- Wake, H.; Moorhouse, A.J.; Jinno, S.; Kohsaka, S.; Nabekura, J. Resting Microglia Directly Monitor the Functional State of Synapses In Vivo and Determine the Fate of Ischemic Terminals. J. Neurosci. 2009, 29, 3974–3980. [Google Scholar] [CrossRef]
- Parkhurst, C.N.; Yang, G.; Ninan, I.; Savas, J.N.; Yates, J.R.; Lafaille, J.J.; Hempstead, B.L.; Littman, D.R.; Gan, W.-B. Microglia Promote Learning-Dependent Synapse Formation through BDNF. Cell 2013, 155, 1596–1609. [Google Scholar] [CrossRef]
- Miyamoto, A.; Wake, H.; Ishikawa, A.W.; Eto, K.; Shibata, K.; Murakoshi, H.; Koizumi, S.; Moorhouse, A.J.; Yoshimura, Y.; Nabekura, J. Microglia Contact Induces Synapse Formation in Developing Somatosensory Cortex. Nat. Commun. 2016, 7, 12540. [Google Scholar] [CrossRef]
- Roumier, A.; Béchade, C.; Poncer, J.-C.; Smalla, K.-H.; Tomasello, E.; Vivier, E.; Gundelfinger, E.D.; Triller, A.; Bessis, A. Impaired Synaptic Function in the Microglial KARAP/DAP12-Deficient Mouse. J. Neurosci. 2004, 24, 11421–11428. [Google Scholar] [CrossRef] [PubMed]
- Hammond, T.R.; Robinton, D.; Stevens, B. Microglia and the Brain: Complementary Partners in Development and Disease. Annu. Rev. Cell Dev. Biol. 2018, 34, 523–544. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Chiou, B.; Gilman, C.K.; Luo, R.; Koshi, T.; Yu, D.; Oak, H.C.; Giera, S.; Johnson-Venkatesh, E.; Muthukumar, A.K.; et al. A Splicing Isoform of GPR56 Mediates Microglial Synaptic Refinement via Phosphatidylserine Binding. EMBO J. 2020, 39, e104136. [Google Scholar] [CrossRef] [PubMed]
- Sarn, N.; Jaini, R.; Thacker, S.; Lee, H.; Dutta, R.; Eng, C. Cytoplasmic-Predominant Pten Increases Microglial Activation and Synaptic Pruning in a Murine Model with Autism-like Phenotype. Mol. Psychiatry 2021, 26, 1458–1471. [Google Scholar] [CrossRef]
- Meng, J.; Han, L.; Zheng, N.; Wang, T.; Xu, H.; Jiang, Y.; Wang, Z.; Liu, Z.; Zheng, Q.; Zhang, X.; et al. Microglial Tmem59 Deficiency Impairs Phagocytosis of Synapse and Leads to Autism-Like Behaviors in Mice. J. Neurosci. 2022, 42, 4958–4979. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.; Nicholls, L.; Assareh, H.; Fang, Z.; Amos, T.G.; Edwards, R.J.; Assareh, A.A.; Voineagu, I. Transcriptome Analysis of Human Brain Tissue Identifies Reduced Expression of Complement Complex C1Q Genes in Rett Syndrome. BMC Genom. 2016, 17, 427. [Google Scholar] [CrossRef]
- Wadhera, T. Brain Network Topology Unraveling Epilepsy and ASD Association: Automated EEG-Based Diagnostic Model. Expert Syst. Appl. 2021, 186, 115762. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, L.; Fang, H.; Wang, F.; Gao, T.; Zhu, Q.; Jiao, G.; Ke, X. Social Brain Network of Children with Autism Spectrum Disorder: Characterization of Functional Connectivity and Potential Association with Stereotyped Behavior. Brain Sci. 2023, 13, 280. [Google Scholar] [CrossRef]
- Li, Q.; Barres, B.A. Microglia and Macrophages in Brain Homeostasis and Disease. Nat. Rev. Immunol. 2018, 18, 225–242. [Google Scholar] [CrossRef]
- Tatavarty, V.; Torrado Pacheco, A.; Groves Kuhnle, C.; Lin, H.; Koundinya, P.; Miska, N.J.; Hengen, K.B.; Wagner, F.F.; Van Hooser, S.D.; Turrigiano, G.G. Autism-Associated Shank3 Is Essential for Homeostatic Compensation in Rodent V1. Neuron 2020, 106, 769–777. [Google Scholar] [CrossRef]
- Dawson, M.S.; Gordon-Fleet, K.; Yan, L.; Tardos, V.; He, H.; Mui, K.; Nawani, S.; Asgarian, Z.; Catani, M.; Fernandes, C.; et al. Sexual Dimorphism in the Social Behaviour of Cntnap2-Null Mice Correlates with Disrupted Synaptic Connectivity and Increased Microglial Activity in the Anterior Cingulate Cortex. Commun. Biol. 2023, 6, 846. [Google Scholar] [CrossRef] [PubMed]
- Choe, K.Y.; Bethlehem, R.A.I.; Safrin, M.; Dong, H.; Salman, E.; Li, Y.; Grinevich, V.; Golshani, P.; DeNardo, L.A.; Peñagarikano, O.; et al. Oxytocin Normalizes Altered Circuit Connectivity for Social Rescue of the Cntnap2 Knockout Mouse. Neuron 2022, 110, 795–808.e6. [Google Scholar] [CrossRef] [PubMed]
- Maezawa, I.; Jin, L.-W. Rett Syndrome Microglia Damage Dendrites and Synapses by the Elevated Release of Glutamate. J. Neurosci. 2010, 30, 5346–5356. [Google Scholar] [CrossRef]
- Schafer, D.P.; Heller, C.T.; Gunner, G.; Heller, M.; Gordon, C.; Hammond, T.; Wolf, Y.; Jung, S.; Stevens, B. Microglia Contribute to Circuit Defects in Mecp2 Null Mice Independent of Microglia-Specific Loss of Mecp2 Expression. eLife 2016, 5, e15224. [Google Scholar] [CrossRef] [PubMed]
- Jafari, M.; Ghadami, E.; Dadkhah, T.; Akhavan-Niaki, H. PI3k/AKT Signaling Pathway: Erythropoiesis and Beyond. J. Cell. Physiol. 2019, 234, 2373–2385. [Google Scholar] [CrossRef]
- Bao, B.; Zahiri, J.; Gazestani, V.H.; Lopez, L.; Xiao, Y.; Kim, R.; Wen, T.H.; Chiang, A.W.T.; Nalabolu, S.; Pierce, K.; et al. A Predictive Ensemble Classifier for the Gene Expression Diagnosis of ASD at Ages 1 to 4 Years. Mol. Psychiatry 2023, 28, 822–833. [Google Scholar] [CrossRef]
- Sharma, A.; Bhalla, S.; Mehan, S. PI3K/AKT/mTOR Signalling Inhibitor Chrysophanol Ameliorates Neurobehavioural and Neurochemical Defects in Propionic Acid-Induced Experimental Model of Autism in Adult Rats. Metab. Brain Dis. 2022, 37, 1909–1929. [Google Scholar] [CrossRef]
- Wang, L.; Chen, J.; Hu, Y.; Liao, A.; Zheng, W.; Wang, X.; Lan, J.; Shen, J.; Wang, S.; Yang, F.; et al. Progranulin Improves Neural Development via the PI3K/Akt/GSK-3β Pathway in the Cerebellum of a VPA-Induced Rat Model of ASD. Transl. Psychiatry 2022, 12, 114. [Google Scholar] [CrossRef]
- Ueno, M.; Fujita, Y.; Tanaka, T.; Nakamura, Y.; Kikuta, J.; Ishii, M.; Yamashita, T. Layer V Cortical Neurons Require Microglial Support for Survival during Postnatal Development. Nat. Neurosci. 2013, 16, 543–551. [Google Scholar] [CrossRef]
- Li, Y.; Du, X.-F.; Liu, C.-S.; Wen, Z.-L.; Du, J.-L. Reciprocal Regulation between Resting Microglial Dynamics and Neuronal Activity in Vivo. Dev. Cell 2012, 23, 1189–1202. [Google Scholar] [CrossRef] [PubMed]
- Bernardino, L.; Agasse, F.; Silva, B.; Ferreira, R.; Grade, S.; Malva, J.O. Tumor Necrosis Factor-Alpha Modulates Survival, Proliferation, and Neuronal Differentiation in Neonatal Subventricular Zone Cell Cultures. Stem Cells 2008, 26, 2361–2371. [Google Scholar] [CrossRef] [PubMed]
- Robinson-Agramonte, M.d.l.A.; Michalski, B.; Vidal-Martinez, B.; Hernández, L.R.; Santiesteban, M.W.; Fahnestock, M. BDNF, proBDNF and IGF-1 Serum Levels in Naïve and Medicated Subjects with Autism. Sci. Rep. 2022, 12, 13768. [Google Scholar] [CrossRef] [PubMed]
- Tamayo, J.M.; Rose, D.; Church, J.S.; Schwartzer, J.J.; Ashwood, P. Maternal Allergic Asthma Induces Prenatal Neuroinflammation. Brain Sci. 2022, 12, 1041. [Google Scholar] [CrossRef]
- Lee, J.H.; Espinera, A.R.; Chen, D.; Choi, K.-E.; Caslin, A.Y.; Won, S.; Pecoraro, V.; Xu, G.-Y.; Wei, L.; Yu, S.P. Neonatal Inflammatory Pain and Systemic Inflammatory Responses as Possible Environmental Factors in the Development of Autism Spectrum Disorder of Juvenile Rats. J. Neuroinflamm. 2016, 13, 109. [Google Scholar] [CrossRef] [PubMed]
- Sciara, A.N.; Beasley, B.; Crawford, J.D.; Anderson, E.P.; Carrasco, T.; Zheng, S.; Ordway, G.A.; Chandley, M.J. Neuroinflammatory Gene Expression Alterations in Anterior Cingulate Cortical White and Gray Matter of Males with Autism Spectrum Disorder. Autism Res. 2020, 13, 870–884. [Google Scholar] [CrossRef]
- Zahedi, E.; Sadr, S.-S.; Sanaeierad, A.; Roghani, M. Chronic Acetyl-L-Carnitine Treatment Alleviates Behavioral Deficits and Neuroinflammation through Enhancing Microbiota Derived-SCFA in Valproate Model of Autism. Biomed. Pharmacother. 2023, 163, 114848. [Google Scholar] [CrossRef]
- Abruzzo, P.M.; Matté, A.; Bolotta, A.; Federti, E.; Ghezzo, A.; Guarnieri, T.; Marini, M.; Posar, A.; Siciliano, A.; De Franceschi, L.; et al. Plasma Peroxiredoxin Changes and Inflammatory Cytokines Support the Involvement of Neuro-Inflammation and Oxidative Stress in Autism Spectrum Disorder. J. Transl. Med. 2019, 17, 332. [Google Scholar] [CrossRef]
- Tsilioni, I.; Patel, A.B.; Pantazopoulos, H.; Berretta, S.; Conti, P.; Leeman, S.E.; Theoharides, T.C. IL-37 Is Increased in Brains of Children with Autism Spectrum Disorder and Inhibits Human Microglia Stimulated by Neurotensin. Proc. Natl. Acad. Sci. USA 2019, 116, 21659–21665. [Google Scholar] [CrossRef]
- Tsilioni, I.; Pantazopoulos, H.; Conti, P.; Leeman, S.E.; Theoharides, T.C. IL-38 Inhibits Microglial Inflammatory Mediators and Is Decreased in Amygdala of Children with Autism Spectrum Disorder. Proc. Natl. Acad. Sci. USA 2020, 117, 16475–16480. [Google Scholar] [CrossRef]
- Chen, H.-R.; Chen, C.-W.; Mandhani, N.; Short-Miller, J.C.; Smucker, M.R.; Sun, Y.-Y.; Kuan, C.-Y. Monocytic Infiltrates Contribute to Autistic-like Behaviors in a Two-Hit Model of Neurodevelopmental Defects. J. Neurosci. 2020, 40, 9386–9400. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Huang, L.; Li, X.; Li, H.; Zhou, Y.; Zhu, H.; Pan, T.; Kendrick, K.M.; Xu, W. Immunological Cytokine Profiling Identifies TNF-α as a Key Molecule Dysregulated in Autistic Children. Oncotarget 2017, 8, 82390–82398. [Google Scholar] [CrossRef]
- Van de los Angeles Robinson-Agramonte, M.; Michalski, B.; Fernández, L.G.; Vidal-Martinez, B.; Cuesta, H.V.; Rizo, C.M.; Fahnestock, M. Effect of NON-INVASIVE Brain Stimulation on Behavior and Serum Brain-derived Neurotrophic Factor and Insulin-like Growth Factor-1 Levels in Autistic Patients. Drug Dev. Res. 2021, 82, 716–723. [Google Scholar] [CrossRef]
- Qi, C.; Chen, A.; Mao, H.; Hu, E.; Ge, J.; Ma, G.; Ren, K.; Xue, Q.; Wang, W.; Wu, S. Excitatory and Inhibitory Synaptic Imbalance Caused by Brain-Derived Neurotrophic Factor Deficits During Development in a Valproic Acid Mouse Model of Autism. Front. Mol. Neurosci. 2022, 15, 860275. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.M.Y.; Yau, S.-Y.; Chan, M.M.Y.; Wong, C.-K.; Chan, A.S. Altered Cytokine and BDNF Levels in Individuals with Autism Spectrum Disorders. Brain Sci. 2022, 12, 460. [Google Scholar] [CrossRef] [PubMed]
- Yan, Q.; Rosenfeld, R.D.; Matheson, C.R.; Hawkins, N.; Lopez, O.T.; Bennett, L.; Welcher, A.A. Expression of Brain-Derived Neurotrophic Factor Protein in the Adult Rat Central Nervous System. Neuroscience 1997, 78, 431–448. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, K.D.; Dreyfus, C.F.; Black, I.B. Brain-Derived Neurotrophic Factor in Astrocytes, Oligodendrocytes, and Microglia/Macrophages after Spinal Cord Injury. Neurobiol. Dis. 2000, 7, 574–585. [Google Scholar] [CrossRef]
- Kowiański, P.; Lietzau, G.; Czuba, E.; Waśkow, M.; Steliga, A.; Moryś, J. BDNF: A Key Factor with Multipotent Impact on Brain Signaling and Synaptic Plasticity. Cell. Mol. Neurobiol. 2018, 38, 579–593. [Google Scholar] [CrossRef]
- Huang, L.; Jin, J.; Chen, K.; You, S.; Zhang, H.; Sideris, A.; Norcini, M.; Recio-Pinto, E.; Wang, J.; Gan, W.-B.; et al. BDNF Produced by Cerebral Microglia Promotes Cortical Plasticity and Pain Hypersensitivity after Peripheral Nerve Injury. PLoS Biol. 2021, 19, e3001337. [Google Scholar] [CrossRef]
- Cristino, L.M.F.; Chaves Filho, A.J.M.; Custódio, C.S.; Vasconcelos, S.M.M.; de Sousa, F.C.F.; Sanders, L.L.O.; de Lucena, D.F.; Macedo, D.S. Animal Model of Neonatal Immune Challenge by Lipopolysaccharide: A Study of Sex Influence in Behavioral and Immune/Neurotrophic Alterations in Juvenile Mice. Neuroimmunomodulation 2022, 29, 391–401. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, Q.; Su, D.; Li, L.; Xiao, C.; He, H.; You, Z.; Zhou, T. Akebia Saponin D Acts via the PPAR-Gamma Pathway to Reprogramme a pro-Neurogenic Microglia That Can Restore Hippocampal Neurogenesis in Mice Exposed to Chronic Mild Stress. CNS Neurosci. Ther. 2023, 29, 2555–2571. [Google Scholar] [CrossRef] [PubMed]
- Arsenijevic, Y.; Weiss, S. Insulin-Like Growth Factor-I Is a Differentiation Factor for Postmitotic CNS Stem Cell-Derived Neuronal Precursors: Distinct Actions from Those of Brain-Derived Neurotrophic Factor. J. Neurosci. 1998, 18, 2118–2128. [Google Scholar] [CrossRef] [PubMed]
- Popken, G.J.; Hodge, R.D.; Ye, P.; Zhang, J.; Ng, W.; O’Kusky, J.R.; D’Ercole, A.J. In Vivo Effects of Insulin-like Growth Factor-I (IGF-I) on Prenatal and Early Postnatal Development of the Central Nervous System. Eur. J. Neurosci. 2004, 19, 2056–2068. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.M.; Mervis, R.F.; Niu, S.-L.; Salem, N.; Witters, L.A.; Tseng, V.; Reinhardt, R.; Bondy, C.A. Insulin-like Growth Factor 1 Is Essential for Normal Dendritic Growth. J. Neurosci. Res. 2003, 73, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Torres-Aleman, I.; Naftolin, F.; Robbins, R.J. Trophic Effects of Insulin-like Growth Factor-I on Fetal Rat Hypothalamic Cells in Culture. Neuroscience 1990, 35, 601–608. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Perez, A.I.; Borrajo, A.; Diaz-Ruiz, C.; Garrido-Gil, P.; Labandeira-Garcia, J.L. Crosstalk between Insulin-like Growth Factor-1 and Angiotensin-II in Dopaminergic Neurons and Glial Cells: Role in Neuroinflammation and Aging. Oncotarget 2016, 7, 30049–30067. [Google Scholar] [CrossRef] [PubMed]
- Quesada, A.; Romeo, H.E.; Micevych, P. Distribution and Localization Patterns of Estrogen Receptor-Beta and Insulin-like Growth Factor-1 Receptors in Neurons and Glial Cells of the Female Rat Substantia Nigra: Localization of ERbeta and IGF-1R in Substantia Nigra. J. Comp. Neurol. 2007, 503, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Suh, H.-S.; Zhao, M.-L.; Derico, L.; Choi, N.; Lee, S.C. Insulin-like Growth Factor 1 and 2 (IGF1, IGF2) Expression in Human Microglia: Differential Regulation by Inflammatory Mediators. J. Neuroinflamm. 2013, 10, 37. [Google Scholar] [CrossRef]
- Bennett, M.L.; Barres, B.A. A Genetically Distinct Microglial Subset Promotes Myelination. EMBO J. 2017, 36, 3269–3271. [Google Scholar] [CrossRef]
- Reinhardt, R.R.; Bondy, C.A. Insulin-like Growth Factors Cross the Blood-Brain Barrier. Endocrinology 1994, 135, 1753–1761. [Google Scholar] [CrossRef]
- Costales, J.; Kolevzon, A. The Therapeutic Potential of Insulin-Like Growth Factor-1 in Central Nervous System Disorders. Neurosci. Biobehav. Rev. 2016, 63, 207–222. [Google Scholar] [CrossRef]
- Abedini, M.; Mashayekhi, F.; Salehi, Z. Analysis of Insulin-like Growth Factor-1 Serum Levels and Promoter (Rs12579108) Polymorphism in the Children with Autism Spectrum Disorders. J. Clin. Neurosci. 2022, 99, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Vithayathil, J.; Pucilowska, J.; Landreth, G.E. ERK/MAPK signaling and autism spectrum disorders. Prog. Brain Res. 2018, 241, 63–112. [Google Scholar] [CrossRef] [PubMed]
- Kolevzon, A.; Breen, M.S.; Siper, P.M.; Halpern, D.; Frank, Y.; Rieger, H.; Weismann, J.; Trelles, M.P.; Lerman, B.; Rapaport, R.; et al. Clinical Trial of Insulin-like Growth Factor-1 in Phelan-McDermid Syndrome. Mol. Autism 2022, 13, 17. [Google Scholar] [CrossRef]
- Fischer, I.; Shohat, S.; Levy, G.; Bar, E.; Trangle, S.S.; Efrati, S.; Barak, B. Hyperbaric Oxygen Therapy Alleviates Social Behavior Dysfunction and Neuroinflammation in a Mouse Model for Autism Spectrum Disorders. Int. J. Mol. Sci. 2022, 23, 11077. [Google Scholar] [CrossRef] [PubMed]
- Welser-Alves, J.V.; Milner, R. Microglia Are the Major Source of TNF-α and TGF-β in Postnatal Glial Cultures; Regulation by Cytokines, Lipopolysaccharide, and Vitronectin. Neurochem. Int. 2013, 63, 47–53. [Google Scholar] [CrossRef]
- Chen, Z.; Palmer, T.D. Differential Roles of TNFR1 and TNFR2 Signaling in Adult Hippocampal Neurogenesis. Brain Behav. Immun. 2013, 30, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Prinz, M.; Priller, J. The Role of Peripheral Immune Cells in the CNS in Steady State and Disease. Nat. Neurosci. 2017, 20, 136–144. [Google Scholar] [CrossRef]
- Swinnen, N.; Smolders, S.; Avila, A.; Notelaers, K.; Paesen, R.; Ameloot, M.; Brône, B.; Legendre, P.; Rigo, J.-M. Complex Invasion Pattern of the Cerebral Cortex Bymicroglial Cells during Development of the Mouse Embryo. Glia 2013, 61, 150–163. [Google Scholar] [CrossRef]
- Low, D.; Ginhoux, F. Recent Advances in the Understanding of Microglial Development and Homeostasis. Cell. Immunol. 2018, 330, 68–78. [Google Scholar] [CrossRef]
- Hattori, Y. The Microglia-Blood Vessel Interactions in the Developing Brain. Neurosci. Res. 2023, 187, 58–66. [Google Scholar] [CrossRef] [PubMed]
- West, A.P.; Shadel, G.S.; Ghosh, S. Mitochondria in Innate Immune Responses. Nat. Rev. Immunol. 2011, 11, 389–402. [Google Scholar] [CrossRef] [PubMed]
- Pangrazzi, L.; Balasco, L.; Bozzi, Y. Oxidative Stress and Immune System Dysfunction in Autism Spectrum Disorders. Int. J. Mol. Sci. 2020, 21, 3293. [Google Scholar] [CrossRef] [PubMed]
- Lenaz, G. The Mitochondrial Production of Reactive Oxygen Species: Mechanisms and Implications in Human Pathology. IUBMB Life 2001, 52, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Rose, S.; Niyazov, D.M.; Rossignol, D.A.; Goldenthal, M.; Kahler, S.G.; Frye, R.E. Clinical and Molecular Characteristics of Mitochondrial Dysfunction in Autism Spectrum Disorder. Mol. Diagn. Ther. 2018, 22, 571–593. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Lin, J.; Zhang, H.; Khan, N.U.; Zhang, J.; Tang, X.; Cao, X.; Shen, L. Oxidative Stress in Autism Spectrum Disorder—Current Progress of Mechanisms and Biomarkers. Front. Psychiatry 2022, 13, 813304. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Fröhlich, H.; Torres, F.B.; Silva, R.L.; Poschet, G.; Agarwal, A.; Rappold, G.A. Mitochondrial Dysfunction and Oxidative Stress Contribute to Cognitive and Motor Impairment in FOXP1 Syndrome. Proc. Natl. Acad. Sci. USA 2022, 119, e2112852119. [Google Scholar] [CrossRef]
- Xiong, Y.; Chen, J.; Li, Y. Microglia and Astrocytes Underlie Neuroinflammation and Synaptic Susceptibility in Autism Spectrum Disorder. Front. Neurosci. 2023, 17, 1125428. [Google Scholar] [CrossRef]
- Carta, A.R.; Pisanu, A. Modulating Microglia Activity with PPAR-γ Agonists: A Promising Therapy for Parkinson’s Disease? Neurotox. Res. 2013, 23, 112–123. [Google Scholar] [CrossRef]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting Microglial Cells Are Highly Dynamic Surveillants of Brain Parenchyma in Vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef]
- Choi, S.-H.; Aid, S.; Kim, H.-W.; Jackson, S.H.; Bosetti, F. Inhibition of NADPH Oxidase Promotes Alternative and Anti–Inflammatory Microglial Activation during Neuroinflammation. J. Neurochem. 2012, 120, 292–301. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Leak, R.K.; Shi, Y.; Suenaga, J.; Gao, Y.; Zheng, P.; Chen, J. Microglial and Macrophage Polarization-New Prospects for Brain Repair. Nat. Rev. Neurol. 2015, 11, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Mosser, D.M.; Edwards, J.P. Exploring the Full Spectrum of Macrophage Activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef]
- Frank-Cannon, T.C.; Alto, L.T.; McAlpine, F.E.; Tansey, M.G. Does Neuroinflammation Fan the Flame in Neurodegenerative Diseases? Mol. Neurodegener. 2009, 4, 47. [Google Scholar] [CrossRef] [PubMed]
- Jha, M.K.; Lee, W.-H.; Suk, K. Functional Polarization of Neuroglia: Implications in Neuroinflammation and Neurological Disorders. Biochem. Pharmacol. 2016, 103, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Ren, C.; Li, D.; Zhou, Q.; Hu, X. Mitochondria-Targeted TPP-MoS2 with Dual Enzyme Activity Provides Efficient Neuroprotection through M1/M2 Microglial Polarization in an Alzheimer’s Disease Model. Biomaterials 2020, 232, 119752. [Google Scholar] [CrossRef]
- Song, G.J.; Nam, Y.; Jo, M.; Jung, M.; Koo, J.Y.; Cho, W.; Koh, M.; Park, S.B.; Suk, K. A Novel Small-Molecule Agonist of PPAR-γ Potentiates an Anti-Inflammatory M2 Glial Phenotype. Neuropharmacology 2016, 109, 159–169. [Google Scholar] [CrossRef]
- Yang, Z.; Liu, B.; Yang, L.; Zhang, C. Platycodigenin as Potential Drug Candidate for Alzheimer’s Disease via Modulating Microglial Polarization and Neurite Regeneration. Molecules 2019, 24, 3207. [Google Scholar] [CrossRef]
- Zhao, D.; Mokhtari, R.; Pedrosa, E.; Birnbaum, R.; Zheng, D.; Lachman, H.M. Transcriptome Analysis of Microglia in a Mouse Model of Rett Syndrome: Differential Expression of Genes Associated with Microglia/Macrophage Activation and Cellular Stress. Mol. Autism. 2017, 8, 17. [Google Scholar] [CrossRef]
- Pósfai, B.; Cserép, C.; Orsolits, B.; Dénes, Á. New Insights into Microglia-Neuron Interactions: A Neuron’s Perspective. Neuroscience 2019, 405, 103–117. [Google Scholar] [CrossRef]
- Virgone-Carlotta, A.; Uhlrich, J.; Akram, M.N.; Ressnikoff, D.; Chrétien, F.; Domenget, C.; Gherardi, R.; Despars, G.; Jurdic, P.; Honnorat, J.; et al. Mapping and Kinetics of Microglia/Neuron Cell-to-Cell Contacts in the 6-OHDA Murine Model of Parkinson’s Disease. Glia 2013, 61, 1645–1658. [Google Scholar] [CrossRef] [PubMed]
- Eyo, U.B.; Peng, J.; Murugan, M.; Mo, M.; Lalani, A.; Xie, P.; Xu, P.; Margolis, D.J.; Wu, L.-J. Regulation of Physical Microglia-Neuron Interactions by Fractalkine Signaling after Status Epilepticus. eNeuro 2016, 3, ENEURO.0209-16.2016. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Yao, Y.; Qi, H.; Yang, J.; Zhang, C.; Zhang, A.; Liu, X.; Zhang, C.; Gan, G.; Zhu, X. Microglia Sense and Suppress Epileptic Neuronal Hyperexcitability. Pharmacol. Res. 2023, 195, 106881. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic Pruning by Microglia Is Necessary for Normal Brain Development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef] [PubMed]
- Hoshiko, M.; Arnoux, I.; Avignone, E.; Yamamoto, N.; Audinat, E. Deficiency of the Microglial Receptor CX3CR1 Impairs Postnatal Functional Development of Thalamocortical Synapses in the Barrel Cortex. J. Neurosci. 2012, 32, 15106–15111. [Google Scholar] [CrossRef] [PubMed]
- Harrison, J.K.; Jiang, Y.; Chen, S.; Xia, Y.; Maciejewski, D.; McNamara, R.K.; Streit, W.J.; Salafranca, M.N.; Adhikari, S.; Thompson, D.A.; et al. Role for Neuronally Derived Fractalkine in Mediating Interactions between Neurons and CX3CR1-Expressing Microglia. Proc. Natl. Acad. Sci. USA 1998, 95, 10896–10901. [Google Scholar] [CrossRef]
- Fernández De Cossío, L.; Guzmán, A.; Van Der Veldt, S.; Luheshi, G.N. Prenatal Infection Leads to ASD-like Behavior and Altered Synaptic Pruning in the Mouse Offspring. Brain Behav. Immun. 2017, 63, 88–98. [Google Scholar] [CrossRef]
- Ishizuka, K.; Fujita, Y.; Kawabata, T.; Kimura, H.; Iwayama, Y.; Inada, T.; Okahisa, Y.; Egawa, J.; Usami, M.; Kushima, I.; et al. Rare Genetic Variants in CX3CR1 and Their Contribution to the Increased Risk of Schizophrenia and Autism Spectrum Disorders. Transl. Psychiatry 2017, 7, e1184. [Google Scholar] [CrossRef]
- Horiuchi, M.; Smith, L.; Maezawa, I.; Jin, L.-W. CX3CR1 Ablation Ameliorates Motor and Respiratory Dysfunctions and Improves Survival of a Rett Syndrome Mouse Model. Brain Behav. Immun. 2017, 60, 106–116. [Google Scholar] [CrossRef]
- Endo, F.; Kasai, A.; Soto, J.S.; Yu, X.; Qu, Z.; Hashimoto, H.; Gradinaru, V.; Kawaguchi, R.; Khakh, B.S. Molecular Basis of Astrocyte Diversity and Morphology across the CNS in Health and Disease. Science 2022, 378, eadc9020. [Google Scholar] [CrossRef]
- Araki, T.; Ikegaya, Y.; Koyama, R. The Effects of Microglia- and Astrocyte-derived Factors on Neurogenesis in Health and Disease. Eur. J. Neurosci. 2021, 54, 5880–5901. [Google Scholar] [CrossRef] [PubMed]
- Allen, N.J.; Barres, B.A. Neuroscience: Glia—More than Just Brain Glue. Nature 2009, 457, 675–677. [Google Scholar] [CrossRef]
- Baxter, P.S.; Dando, O.; Emelianova, K.; He, X.; McKay, S.; Hardingham, G.E.; Qiu, J. Microglial Identity and Inflammatory Responses Are Controlled by the Combined Effects of Neurons and Astrocytes. Cell Rep. 2021, 34, 108882. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Brennan, F.H.; Popovich, P.G.; Zhou, M. Microglia Maintain the Normal Structure and Function of the Hippocampal Astrocyte Network. Glia 2022, 70, 1359–1379. [Google Scholar] [CrossRef] [PubMed]
- Kern, J.K.; Geier, D.A.; Sykes, L.K.; Geier, M.R. Relevance of Neuroinflammation and Encephalitis in Autism. Front. Cell. Neurosci. 2016, 9, 519. [Google Scholar] [CrossRef]
- Vakilzadeh, G.; Martinez-Cerdeño, V. Pathology and Astrocytes in Autism. Neuropsychiatr. Dis. Treat. 2023, 19, 841–850. [Google Scholar] [CrossRef]
- Vakilzadeh, G.; Falcone, C.; Dufour, B.; Hong, T.; Noctor, S.C.; Martínez-Cerdeño, V. Decreased Number and Increased Activation State of Astrocytes in Gray and White Matter of the Prefrontal Cortex in Autism. Cereb. Cortex 2022, 32, 4902–4912. [Google Scholar] [CrossRef]
- Jha, M.K.; Jo, M.; Kim, J.-H.; Suk, K. Microglia-Astrocyte Crosstalk: An Intimate Molecular Conversation. Neuroscientist 2019, 25, 227–240. [Google Scholar] [CrossRef]
- Traetta, M.E.; Uccelli, N.A.; Zárate, S.C.; Gómez Cuautle, D.; Ramos, A.J.; Reinés, A. Long-Lasting Changes in Glial Cells Isolated from Rats Subjected to the Valproic Acid Model of Autism Spectrum Disorder. Front. Pharmacol. 2021, 12, 707859. [Google Scholar] [CrossRef]
- Allen, M.; Huang, B.S.; Notaras, M.J.; Lodhi, A.; Barrio-Alonso, E.; Lituma, P.J.; Wolujewicz, P.; Witztum, J.; Longo, F.; Chen, M.; et al. Astrocytes Derived from ASD Individuals Alter Behavior and Destabilize Neuronal Activity through Aberrant Ca2+ Signaling. Mol. Psychiatry 2022, 27, 2470–2484. [Google Scholar] [CrossRef]
- Wang, Q.; Kong, Y.; Wu, D.-Y.; Liu, J.-H.; Jie, W.; You, Q.-L.; Huang, L.; Hu, J.; Chu, H.-D.; Gao, F.; et al. Impaired Calcium Signaling in Astrocytes Modulates Autism Spectrum Disorder-like Behaviors in Mice. Nat. Commun. 2021, 12, 3321. [Google Scholar] [CrossRef] [PubMed]
- Moore, S.; Meschkat, M.; Ruhwedel, T.; Trevisiol, A.; Tzvetanova, I.D.; Battefeld, A.; Kusch, K.; Kole, M.H.P.; Strenzke, N.; Möbius, W.; et al. A Role of Oligodendrocytes in Information Processing. Nat. Commun. 2020, 11, 5497. [Google Scholar] [CrossRef]
- Miron, V.E.; Boyd, A.; Zhao, J.-W.; Yuen, T.J.; Ruckh, J.M.; Shadrach, J.L.; van Wijngaarden, P.; Wagers, A.J.; Williams, A.; Franklin, R.J.M.; et al. M2 Microglia/Macrophages Drive Oligodendrocyte Differentiation during CNS Remyelination. Nat. Neurosci. 2013, 16, 1211–1218. [Google Scholar] [CrossRef]
- Hagemeyer, N.; Hanft, K.-M.; Akriditou, M.-A.; Unger, N.; Park, E.S.; Stanley, E.R.; Staszewski, O.; Dimou, L.; Prinz, M. Microglia Contribute to Normal Myelinogenesis and to Oligodendrocyte Progenitor Maintenance during Adulthood. Acta Neuropathol. 2017, 134, 441–458. [Google Scholar] [CrossRef]
- Kigerl, K.A.; Gensel, J.C.; Ankeny, D.P.; Alexander, J.K.; Donnelly, D.J.; Popovich, P.G. Identification of Two Distinct Macrophage Subsets with Divergent Effects Causing Either Neurotoxicity or Regeneration in the Injured Mouse Spinal Cord. J. Neurosci. 2009, 29, 13435–13444. [Google Scholar] [CrossRef] [PubMed]
- Wlodarczyk, A.; Holtman, I.R.; Krueger, M.; Yogev, N.; Bruttger, J.; Khorooshi, R.; Benmamar-Badel, A.; de Boer-Bergsma, J.J.; Martin, N.A.; Karram, K.; et al. A Novel Microglial Subset Plays a Key Role in Myelinogenesis in Developing Brain. EMBO J. 2017, 36, 3292–3308. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Linke, A.; Olson, L.; Kohli, J.; Kinnear, M.; Sereno, M.; Müller, R.; Carper, R.; Fishman, I. Cortical Myelination in Toddlers and Preschoolers with Autism Spectrum Disorder. Dev. Neurobiol. 2022, 82, 261–274. [Google Scholar] [CrossRef]
- Graciarena, M.; Seiffe, A.; Nait-Oumesmar, B.; Depino, A.M. Hypomyelination and Oligodendroglial Alterations in a Mouse Model of Autism Spectrum Disorder. Front. Cell. Neurosci. 2019, 12, 517. [Google Scholar] [CrossRef]
- Takanezawa, Y.; Tanabe, S.; Kato, D.; Ozeki, R.; Komoda, M.; Suzuki, T.; Baba, H.; Muramatsu, R. Microglial ASD-Related Genes Are Involved in Oligodendrocyte Differentiation. Sci. Rep. 2021, 11, 17825. [Google Scholar] [CrossRef]
- Gosselin, D.; Skola, D.; Coufal, N.G.; Holtman, I.R.; Schlachetzki, J.C.M.; Sajti, E.; Jaeger, B.N.; O’Connor, C.; Fitzpatrick, C.; Pasillas, M.P.; et al. An Environment-Dependent Transcriptional Network Specifies Human Microglia Identity. Science 2017, 356, eaal3222. [Google Scholar] [CrossRef]
- Fagerlund, I.; Dougalis, A.; Shakirzyanova, A.; Gómez-Budia, M.; Pelkonen, A.; Konttinen, H.; Ohtonen, S.; Fazaludeen, M.F.; Koskuvi, M.; Kuusisto, J.; et al. Microglia-like Cells Promote Neuronal Functions in Cerebral Organoids. Cells 2021, 11, 124. [Google Scholar] [CrossRef] [PubMed]
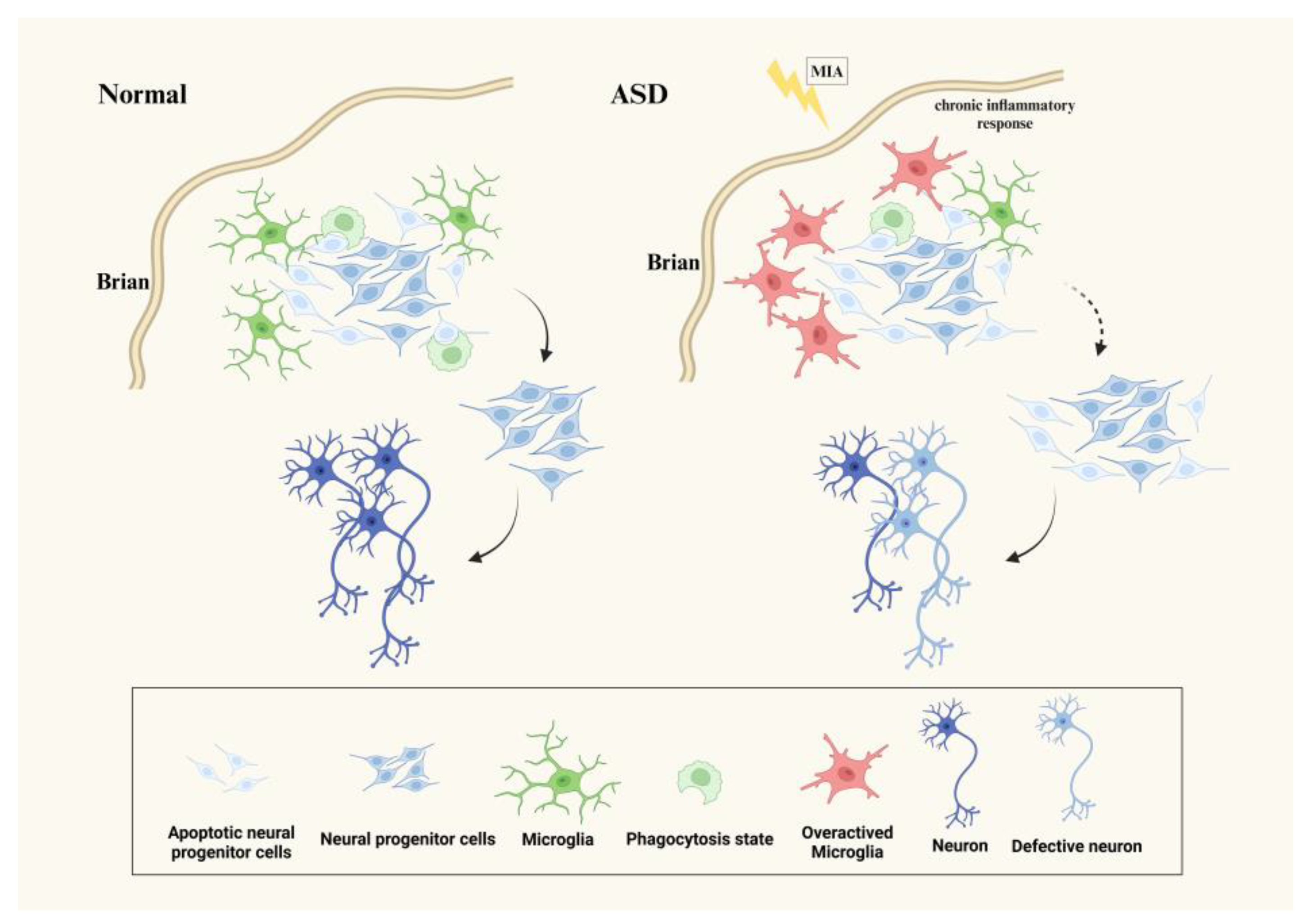

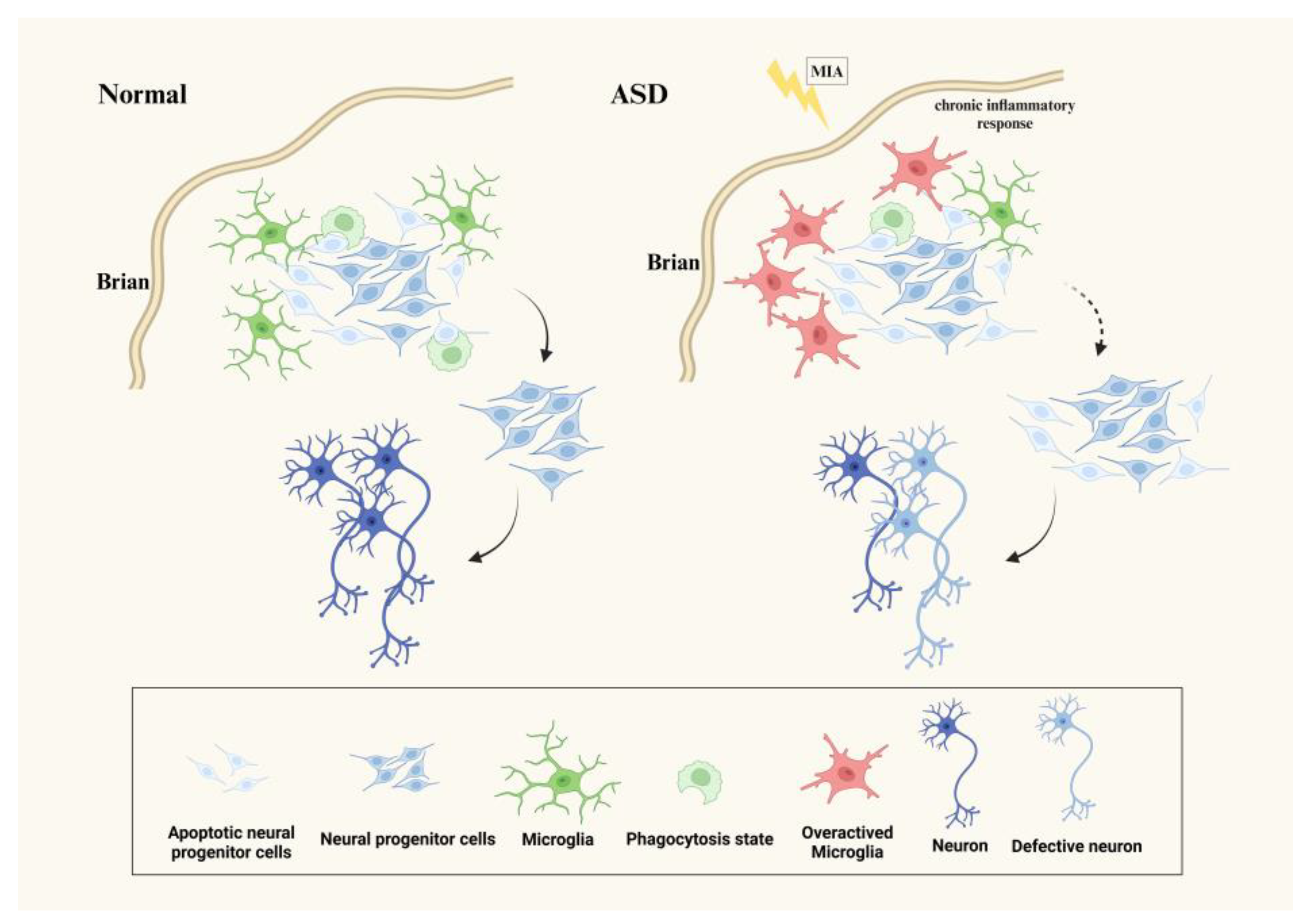{kind=link}
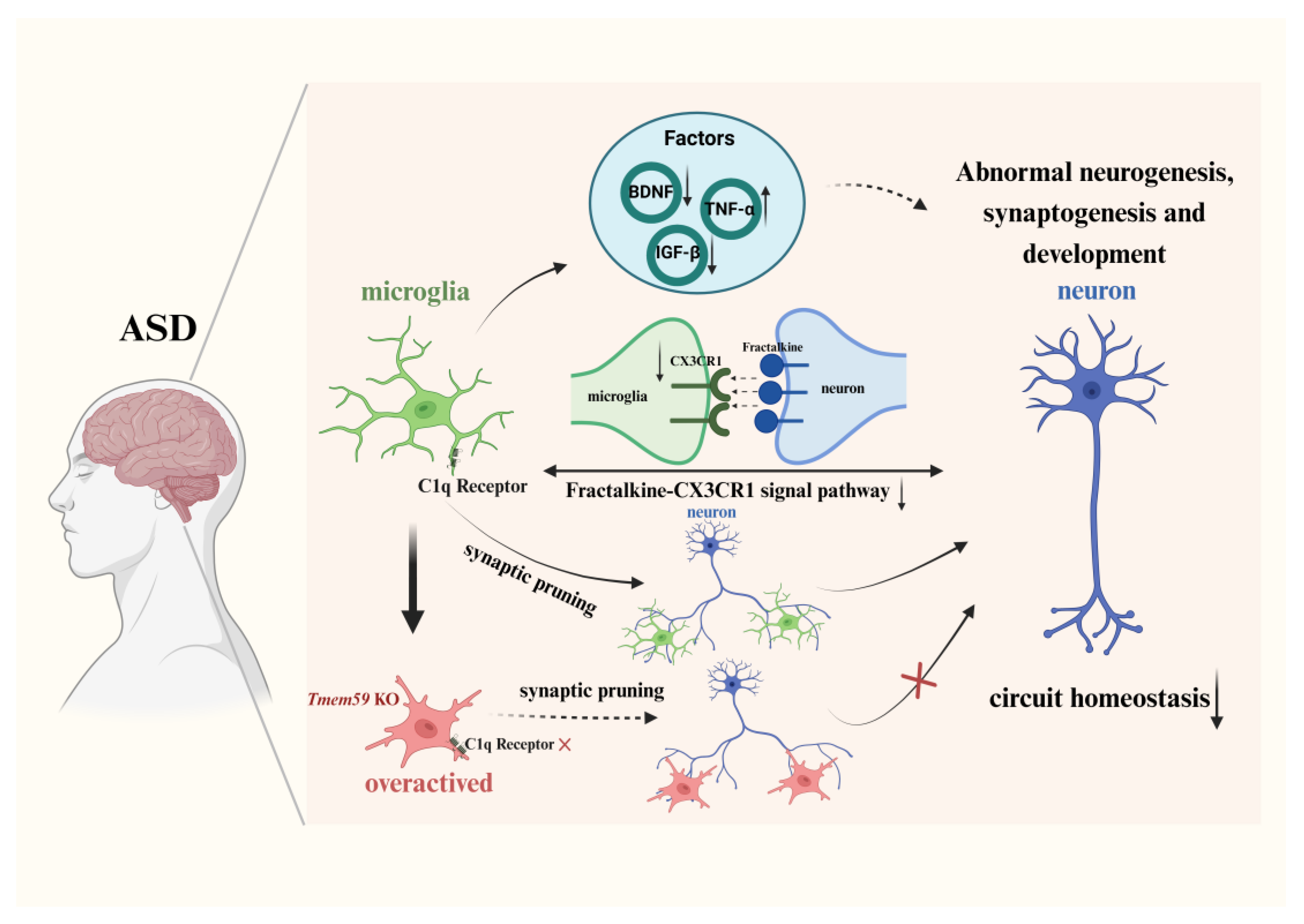{kind=link}
| Factor | Species | Area and Expression Level | Influence | Reference |
|---|---|---|---|---|
| IL-1α | Offspring of MAA mice | Brain of offspring ↑ | Passed through the placenta, induces ASD in offspring | [64] |
| IL-1β | ASD rat | Blood, brain ↑ | Hippocampal and cortical neuronal death, impairment of axonal integrity and myelination | [65] |
| ASD patient | ACC ↑ | Neurons decrease in size and number, and synaptic connections are damaged | [66] | |
| ASD rat | Cerebral cortex ↑ | Accompanied by microglial state transition inducing the ASD-like phenotype | [67] | |
| IL-6 | ASD patient | Plasma ↑ | Promote oxidative stress and neuroinflammation, leading to dysfunction in ASD | [68] |
| Offspring of MAA mice | Brain of offspring ↑ | Passed through the placenta, induces ASD in offspring | [64] | |
| IL-37 | ASD patient | Amygdala, prefrontal cortex ↑ | Inhibits the secretion of proinflammatory cytokines IL-1β and CXCL8 by cultured microglia in vitro | [69] |
| IL-38 | ASD patient | Amygdala ↓ | Inhibits the secretion of proinflammatory factors from cultured microglia in vitro | [70] |
| TNF-α | ASD Rat | Blood, brain ↑ | Hippocampal and cortical neuronal death, impairment of axonal integrity and myelination | [65] |
| Offspring of MAA mice | Brain of offspring ↑ | Passed through the placenta, induces ASD in offspring | [64] | |
| ASD Rat | Cerebral cortex ↑ | Accompanied by microglial state transition inducing the ASD-like phenotype | [67] | |
| HI Offspring of MIA mice | Brain ↑ | Impairing prominent network connections in offspring mice; induces ASD | [71] | |
| ASD patient | Serum ↑ | Expression levels are positively correlated with ASD severity | [72] | |
| IFN-γ | Offspring of MAA mice | Brain of offspring ↑ | Passed through the placenta; induces ASD in offspring | [64] |
| GM-CSF | Offspring of MAA mice | Brain of offspring ↑ | Passed through the placenta; induces ASD in offspring | [64] |
| IGF-1 | ASD patient | Anterior cingulate cortex↓ | Neurons decrease in size and number, and synaptic connections are damaged | [66] |
| ASD patient | Serum ↓ | Affects synaptic connections by activating MAPK and PI3K/AKt signaling pathways | [73] | |
| NF-κB | HI offspring of MIA mice | Brain ↑ | Impairing prominent network connections in offspring mice induces ASD | [71] |
| BDNF | ASD mice | ACC ↓ | Excitatory/inhibitory imbalance, impaired synaptic plasticity and function | [74] |
| ASD patient | Serum ↓ | Expression levels are negatively correlated with ASD severity | [75] | |
| ASD patient | Serum ↓ | Affects synaptic connections by activating MAPK and PI3K/Akt signaling pathways | [73] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, Y.; Wang, Z. The Impact of Microglia on Neurodevelopment and Brain Function in Autism. Biomedicines 2024, 12, 210. https://doi.org/10.3390/biomedicines12010210
Luo Y, Wang Z. The Impact of Microglia on Neurodevelopment and Brain Function in Autism. Biomedicines. 2024; 12(1):210. https://doi.org/10.3390/biomedicines12010210
Chicago/Turabian StyleLuo, Yuyi, and Zhengbo Wang. 2024. "The Impact of Microglia on Neurodevelopment and Brain Function in Autism" Biomedicines 12, no. 1: 210. https://doi.org/10.3390/biomedicines12010210
APA StyleLuo, Y., & Wang, Z. (2024). The Impact of Microglia on Neurodevelopment and Brain Function in Autism. Biomedicines, 12(1), 210. https://doi.org/10.3390/biomedicines12010210