

CRISPR/Cas9-Mediated Gene Therapy for Glioblastoma: A Scoping Review

 , , ,
, , , 

Abstract
1. Introduction
2. CRISPR/Cas9 Gene Editing
2.1. Brief Historical Overview of CRISPR/Cas9 Technology
2.2. CRISPR/Cas9 Technology
2.3. Applications in Gene Therapy
2.4. Principles of CRISPR/Cas9 Gene Editing Technology
3. CRISPR/Cas9-Mediated GBM Therapy
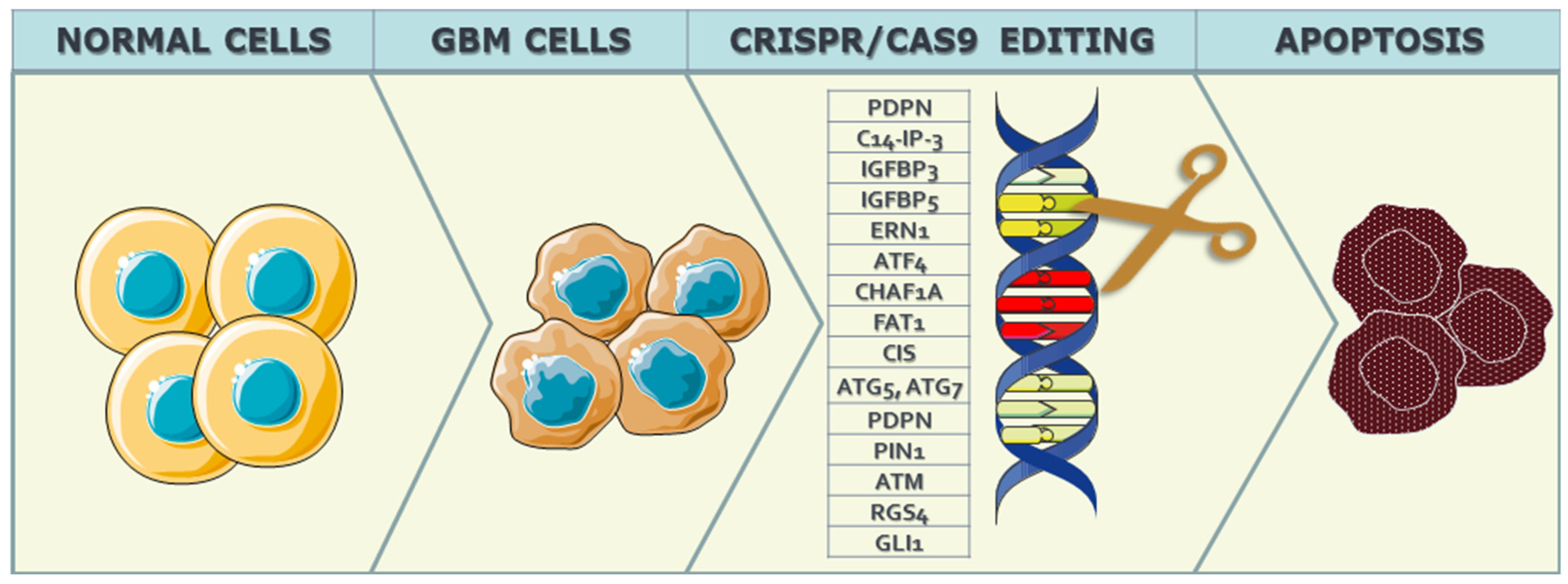
3.1. Targeting Specific Genetic Mutations in GBM
3.1.1. Cell Cycle Regulation
3.1.2. Cell-Interphase-Related Targets
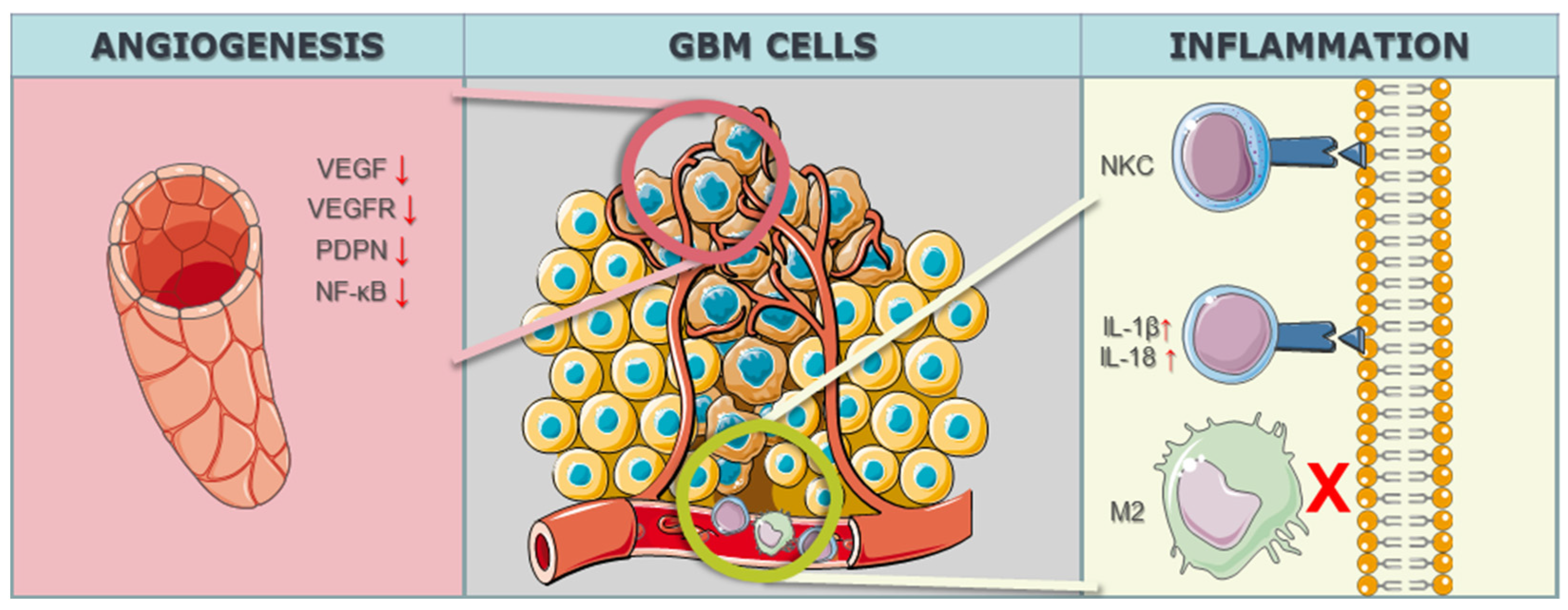
3.1.3. Microenvironmental CRISPR/Cas9 Targets in GBM Cells
3.2. Contribution of CRISPR/Cas9 Technology in Alleviating Therapy Resistance of GBM
4. Efficacy and Safety Considerations
5. Future Directions and Considerations
5.1. Ethical and Regulatory Considerations
5.2. Possibilities for Personalized Gene Editing in GBM
5.3. Limitations and Challenges of the CRISPR/Ca9 Therapy
5.4. Toxicology of CRISPR/Cas9 in GBM Therapy
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Begagić, E.; Pugonja, R.; Bečulić, H.; Čeliković, A.; Tandir Lihić, L.; Kadić Vukas, S.; Čejvan, L.; Skomorac, R.; Selimović, E.; Jaganjac, B.; et al. Molecular Targeted Therapies in Glioblastoma Multiforme: A Systematic Overview of Global Trends and Findings. Brain Sci. 2023, 13, 1602. [Google Scholar] [CrossRef] [PubMed]
- Stoyanov, G.S.; Lyutfi, E.; Georgieva, R.; Georgiev, R.; Dzhenkov, D.L.; Petkova, L.; Ivanov, B.D.; Kaprelyan, A.; Ghenev, P. Reclassification of Glioblastoma Multiforme According to the 2021 World Health Organization Classification of Central Nervous System Tumors: A Single Institution Report and Practical Significance. Cureus 2022, 14, e21822. [Google Scholar] [CrossRef] [PubMed]
- Jain, K.K. A Critical Overview of Targeted Therapies for Glioblastoma. Front. Oncol. 2018, 8, 419. [Google Scholar] [CrossRef] [PubMed]
- Tatebayashi, K.; Nakayama, N.; Sakamoto, D.; Iida, T.; Ono, S.; Matsuda, I.; Enomoto, Y.; Tanaka, M.; Fujita, M.; Hirota, S.; et al. Clinical Significance of Early Venous Filling Detected via Preoperative Angiography in Glioblastoma. Cancers 2023, 15, 3800. [Google Scholar] [CrossRef] [PubMed]
- Angom, R.S.; Nakka, N.M.R.; Bhattacharya, S. Advances in Glioblastoma Therapy: An Update on Current Approaches. Brain Sci. 2023, 13, 1536. [Google Scholar] [CrossRef] [PubMed]
- Agosti, E.; Zeppieri, M.; De Maria, L.; Tedeschi, C.; Fontanella, M.M.; Panciani, P.P.; Ius, T. Glioblastoma Immunotherapy: A Systematic Review of the Present Strategies and Prospects for Advancements. Int. J. Mol. Sci. 2023, 24, 15037. [Google Scholar] [CrossRef] [PubMed]
- Riemenschneider, M.J.; Jeuken, J.W.; Wesseling, P.; Reifenberger, G. Molecular diagnostics of gliomas: State of the art. Acta Neuropathol. 2010, 120, 567–584. [Google Scholar] [CrossRef] [PubMed]
- Isachesku, E.; Braicu, C.; Pirlog, R.; Kocijancic, A.; Busuioc, C.; Pruteanu, L.-L.; Pandey, D.P.; Berindan-Neagoe, I. The Role of Non-Coding RNAs in Epigenetic Dysregulation in Glioblastoma Development. Int. J. Mol. Sci. 2023, 24, 16320. [Google Scholar] [CrossRef]
- Senhaji, N.; Squalli Houssaini, A.; Lamrabet, S.; Louati, S.; Bennis, S. Molecular and Circulating Biomarkers in Patients with Glioblastoma. Int. J. Mol. Sci. 2022, 23, 7474. [Google Scholar] [CrossRef]
- Khlidj, Y. What did CRISPR-Cas9 accomplish in its first 10 years? Biochem. Med. 2023, 33, 030601. [Google Scholar] [CrossRef]
- Peixoto, J.; Príncipe, C.; Pestana, A.; Osório, H.; Pinto, M.T.; Prazeres, H.; Soares, P.; Lima, R.T. Using a Dual CRISPR/Cas9 Approach to Gain Insight into the Role of LRP1B in Glioblastoma. Int. J. Mol. Sci. 2023, 24, 11285. [Google Scholar] [CrossRef]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Gallo, K.; Srinageshwar, B.; Ward, A.; Diola, C.; Dunbar, G.; Rossignol, J.; Bakke, J. Inducible Knockout of 14-3-3β Attenuates Proliferation and Spheroid Formation in a Human Glioblastoma Cell Line U87MG. Brain Sci. 2023, 13, 868. [Google Scholar] [CrossRef] [PubMed]
- Motoche-Monar, C.; Ordoñez, J.E.; Chang, O.; Gonzales-Zubiate, F.A. gRNA Design: How Its Evolution Impacted on CRISPR/Cas9 Systems Refinement. Biomolecules 2023, 13, 1698. [Google Scholar] [CrossRef]
- Ding, S.; Liu, J.; Han, X.; Tang, M. CRISPR/Cas9-Mediated Genome Editing in Cancer Therapy. Int. J. Mol. Sci. 2023, 24, 16325. [Google Scholar] [CrossRef] [PubMed]
- Begagić, E.; Pugonja, R.; Bečulić, H.; Selimović, E.; Skomorac, R.; Saß, B.; Pojskić, M. The new era of spinal surgery: Exploring the utilization of exoscopes as a viable alternative to operative microscopes—A systematic review and meta-analysis. World Neurosurg. 2023, in press. [Google Scholar] [CrossRef] [PubMed]
- Begagić, E.; Bečulić, H.; Skomorac, R.; Pojskić, M. Accessible Spinal Surgery: Transformation Through the Implementation of Exoscopes As Substitutes for Conventional Microsurgery in Low- and Middle-Income Settings. Cureus 2023, 15, e45350. [Google Scholar] [CrossRef]
- Li, C.; Brant, E.; Budak, H.; Zhang, B. CRISPR/Cas: A Nobel Prize award-winning precise genome editing technology for gene therapy and crop improvement. J. Zhejiang Univ. Sci. B 2021, 22, 253–284. [Google Scholar] [CrossRef]
- Mohammadzadeh, I.; Qujeq, D.; Yousefi, T.; Ferns, G.A.; Maniati, M.; Vaghari-Tabari, M. CRISPR/Cas9 gene editing: A new therapeutic approach in the treatment of infection and autoimmunity. IUBMB Life 2020, 72, 1603–1621. [Google Scholar] [CrossRef]
- Gostimskaya, I. CRISPR-Cas9: A History of Its Discovery and Ethical Considerations of Its Use in Genome Editing. Biochemistry 2022, 87, 777–788. [Google Scholar] [CrossRef]
- Ishino, Y.; Krupovic, M.; Forterre, P. History of CRISPR-Cas from Encounter with a Mysterious Repeated Sequence to Genome Editing Technology. J. Bacteriol. 2018, 200, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Kozovska, Z.; Rajcaniova, S.; Munteanu, P.; Dzacovska, S.; Demkova, L. CRISPR: History and perspectives to the future. Biomed. Pharmacother. 2021, 141, 111917. [Google Scholar] [CrossRef] [PubMed]
- Janik, E.; Niemcewicz, M.; Ceremuga, M.; Krzowski, L.; Saluk-Bijak, J.; Bijak, M. Various Aspects of a Gene Editing System—CRISPR–Cas9. Int. J. Mol. Sci. 2020, 21, 9604. [Google Scholar] [CrossRef] [PubMed]
- Xing, H.; Meng, L.H. CRISPR-cas9: A powerful tool towards precision medicine in cancer treatment. Acta Pharmacol. Sin. 2020, 41, 583–587. [Google Scholar] [CrossRef]
- Jiang, C.; Meng, L.; Yang, B.; Luo, X. Application of CRISPR/Cas9 gene editing technique in the study of cancer treatment. Clin. Genet. 2020, 97, 73–88. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-W.; Gao, C.; Zheng, Y.-M.; Yi, L.; Lu, J.-C.; Huang, X.-Y.; Cai, J.-B.; Zhang, P.-F.; Cui, Y.-H.; Ke, A.-W. Current applications and future perspective of CRISPR/Cas9 gene editing in cancer. Mol. Cancer 2022, 21, 57. [Google Scholar] [CrossRef]
- Hazafa, A.; Mumtaz, M.; Farooq, M.F.; Bilal, S.; Chaudhry, S.N.; Firdous, M.; Naeem, H.; Ullah, M.O.; Yameen, M.; Mukhtiar, M.S.; et al. CRISPR/Cas9: A powerful genome editing technique for the treatment of cancer cells with present challenges and future directions. Life Sci. 2020, 263, 118525. [Google Scholar] [CrossRef]
- Hsu, M.N.; Chang, Y.H.; Truong, V.A.; Lai, P.L.; Nguyen, T.K.N.; Hu, Y.C. CRISPR technologies for stem cell engineering and regenerative medicine. Biotechnol. Adv. 2019, 37, 107447. [Google Scholar] [CrossRef]
- Jhu, M.-Y.; Ellison, E.E.; Sinha, N.R. CRISPR gene editing to improve crop resistance to parasitic plants. Front. Genome Ed. 2023, 5, 1289416. [Google Scholar] [CrossRef]
- Hirsch, F.; Iphofen, R.; Koporc, Z. Ethics assessment in research proposals adopting CRISPR technology. Biochem. Med. 2019, 29, 020202. [Google Scholar] [CrossRef]
- Gumer, J.M. The Wisdom of Germline Editing: An Ethical Analysis of the Use of CRISPR-Cas9 to Edit Human Embryos. New. Bioeth. 2019, 25, 137–152. [Google Scholar] [CrossRef] [PubMed]
- Sauvagère, S.; Siatka, C. CRISPR-Cas: ‘The Multipurpose Molecular Tool’ for Gene Therapy and Diagnosis. Genes 2023, 14, 1542. [Google Scholar] [PubMed]
- Gupta, S.; Kumar, A.; Patel, R.; Kumar, V. Genetically modified crop regulations: Scope and opportunity using the CRISPR-Cas9 genome editing approach. Mol. Biol. Rep. 2021, 48, 4851–4863. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B. CRISPR/Cas gene therapy. J. Cell. Physiol. 2021, 236, 2459–2481. [Google Scholar] [CrossRef]
- Yu, W.; Wu, Z. Use of AAV Vectors for CRISPR-Mediated In Vivo Genome Editing in the Retina. Methods Mol. Biol. 2019, 1950, 123–139. [Google Scholar] [CrossRef]
- Asmamaw Mengstie, M. Viral Vectors for the in Vivo Delivery of CRISPR Components: Advances and Challenges. Front. Bioeng. Biotechnol. 2022, 10, 895713. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zheng, W.; Liu, S.; Li, B.; Jiang, X. Delivery of CRISPR/Cas9 by Novel Strategies for Gene Therapy. Chembiochem 2019, 20, 634–643. [Google Scholar] [CrossRef]
- Uddin, F.; Rudin, C.M.; Sen, T. CRISPR Gene Therapy: Applications, Limitations, and Implications for the Future. Front. Oncol. 2020, 10, 1387. [Google Scholar] [CrossRef]
- Lim, J.M.; Kim, H.H. Basic Principles and Clinical Applications of CRISPR-Based Genome Editing. Yonsei Med. J. 2022, 63, 105–113. [Google Scholar] [CrossRef]
- Horodecka, K.; Düchler, M. CRISPR/Cas9: Principle, Applications, and Delivery through Extracellular Vesicles. Int. J. Mol. Sci. 2021, 22, 6072. [Google Scholar] [CrossRef]
- Tabassum, T.; Pietrogrande, G.; Healy, M.; Wolvetang, E.J. CRISPR-Cas9 Direct Fusions for Improved Genome Editing via Enhanced Homologous Recombination. Int. J. Mol. Sci. 2023, 24, 14701. [Google Scholar] [CrossRef]
- Yip, B.H. Recent Advances in CRISPR/Cas9 Delivery Strategies. Biomolecules 2020, 10, 839. [Google Scholar] [CrossRef] [PubMed]
- Reuven, N.; Adler, J.; Myers, N.; Shaul, Y. CRISPR Co-Editing Strategy for Scarless Homology-Directed Genome Editing. Int. J. Mol. Sci. 2021, 22, 3741. [Google Scholar] [CrossRef] [PubMed]
- Loureiro, A.; da Silva, G.J. CRISPR-Cas: Converting A Bacterial Defence Mechanism into A State-of-the-Art Genetic Manipulation Tool. Antibiotics 2019, 8, 18. [Google Scholar] [CrossRef]
- Karginov, A.V.; Tarutina, M.G.; Lapteva, A.R.; Pakhomova, M.D.; Galliamov, A.A.; Filkin, S.Y.; Fedorov, A.N.; Agaphonov, M.O. A Split-Marker System for CRISPR-Cas9 Genome Editing in Methylotrophic Yeasts. Int. J. Mol. Sci. 2023, 24, 8173. [Google Scholar] [CrossRef] [PubMed]
- Kang, X.; Wang, Y.; Liu, P.; Huang, B.; Zhou, B.; Lu, S.; Geng, W.; Tang, H. Progresses, Challenges, and Prospects of CRISPR/Cas9 Gene-Editing in Glioma Studies. Cancers 2023, 15, 396. [Google Scholar] [CrossRef]
- Qazi, M.A.; Vora, P.; Venugopal, C.; Sidhu, S.S.; Moffat, J.; Swanton, C.; Singh, S.K. Intratumoral heterogeneity: Pathways to treatment resistance and relapse in human glioblastoma. Ann. Oncol. 2017, 28, 1448–1456. [Google Scholar] [CrossRef] [PubMed]
- Lake, J.A.; Donson, A.M.; Prince, E.; Davies, K.D.; Nellan, A.; Green, A.L.; Mulcahy Levy, J.; Dorris, K.; Vibhakar, R.; Hankinson, T.C.; et al. Targeted fusion analysis can aid in the classification and treatment of pediatric glioma, ependymoma, and glioneuronal tumors. Pediatr. Blood Cancer 2020, 67, e28028. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, X.; Li, J.; Liang, J.; Ren, X.; Yun, D.; Liu, J.; Fan, J.; Zhang, Y.; Zhang, J.; et al. PDPN contributes to constructing immunosuppressive microenvironment in IDH wildtype glioma. Cancer Gene Therapy 2023, 30, 345–357. [Google Scholar] [CrossRef]
- Nayak, S.; Aich, M.; Kumar, A.; Sengupta, S.; Bajad, P.; Dhapola, P.; Paul, D.; Narta, K.; Purkrait, S.; Mehani, B.; et al. Novel internal regulators and candidate miRNAs within miR-379/miR-656 miRNA cluster can alter cellular phenotype of human glioblastoma. Sci. Rep. 2018, 8, 7673. [Google Scholar] [CrossRef]
- Rodvold, J.J.; Xian, S.; Nussbacher, J.; Tsui, B.; Cameron Waller, T.; Searles, S.C.; Lew, A.; Jiang, P.; Babic, I.; Nomura, N.; et al. IRE1α and IGF signaling predict resistance to an endoplasmic reticulum stress-inducing drug in glioblastoma cells. Sci. Rep. 2020, 10, 8348. [Google Scholar] [CrossRef] [PubMed]
- Vu, H.T.; Kobayashi, M.; Hegazy, A.M.; Tadokoro, Y.; Ueno, M.; Kasahara, A.; Takase, Y.; Nomura, N.; Peng, H.; Ito, C.; et al. Autophagy inhibition synergizes with calcium mobilization to achieve efficient therapy of malignant gliomas. Cancer Sci. 2018, 109, 2497–2508. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Du, B.; Jiang, H.; Gao, J. Over-expression of CHAF1A promotes cell proliferation and apoptosis resistance in glioblastoma cells via AKT/FOXO3a/Bim pathway. Biochem. Biophys. Res. Commun. 2016, 469, 1111–1116. [Google Scholar] [CrossRef]
- Kranz, D.; Boutros, M. A synthetic lethal screen identifies FAT1 as an antagonist of caspase-8 in extrinsic apoptosis. Embo J. 2014, 33, 181–197. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, T.; Morimoto, T.; Maeoka, R.; Matsuda, R.; Nakamura, M.; Nishimura, F.; Ouji, N.; Yamada, S.; Nakagawa, I.; Park, Y.S.; et al. CIS deletion by CRISPR/Cas9 enhances human primary natural killer cell functions against allogeneic glioblastoma. J. Exp. Clin. Cancer Res. 2023, 42, 205. [Google Scholar] [CrossRef] [PubMed]
- Zielke, S.; Meyer, N.; Mari, M.; Abou-El-Ardat, K.; Reggiori, F.; van Wijk, S.J.L.; Kögel, D.; Fulda, S. Loperamide, pimozide, and STF-62247 trigger autophagy-dependent cell death in glioblastoma cells. Cell. Death Dis. 2018, 9, 994. [Google Scholar] [CrossRef] [PubMed]
- Maggio, J.; Cardama, G.A.; Armando, R.G.; Balcone, L.; Sobol, N.T.; Gomez, D.E.; Mengual Gómez, D.L. Key role of PIN1 in telomere maintenance and oncogenic behavior in a human glioblastoma model. Oncol. Rep. 2023, 49, 91. [Google Scholar] [CrossRef]
- Ali, R.; Alabdullah, M.; Miligy, I.; Normatova, M.; Babaei-Jadidi, R.; Nateri, A.S.; Rakha, E.A.; Madhusudan, S. ATM Regulated PTEN Degradation Is XIAP E3 Ubiquitin Ligase Mediated in p85α Deficient Cancer Cells and Influence Platinum Sensitivity. Cells 2019, 8, 1271. [Google Scholar] [CrossRef]
- Guda, M.R.; Velpula, K.K.; Asuthkar, S.; Cain, C.P.; Tsung, A.J. Targeting RGS4 Ablates Glioblastoma Proliferation. Int. J. Mol. Sci. 2020, 21, 3300. [Google Scholar] [CrossRef]
- Ranjan, A.; Srivastava, S.K. Penfluridol suppresses glioblastoma tumor growth by Akt-mediated inhibition of GLI1. Oncotarget 2017, 8, 32960–32976. [Google Scholar] [CrossRef]
- Esemen, Y.; Awan, M.; Parwez, R.; Baig, A.; Rahman, S.; Masala, I.; Franchini, S.; Giakoumettis, D. Molecular Pathogenesis of Glioblastoma in Adults and Future Perspectives: A Systematic Review. Int. J. Mol. Sci. 2022, 23, 2607. [Google Scholar] [CrossRef] [PubMed]
- Fierro, J., Jr.; DiPasquale, J.; Perez, J.; Chin, B.; Chokpapone, Y.; Tran, A.M.; Holden, A.; Factoriza, C.; Sivagnanakumar, N.; Aguilar, R.; et al. Dual-sgRNA CRISPR/Cas9 knockout of PD-L1 in human U87 glioblastoma tumor cells inhibits proliferation, invasion, and tumor-associated macrophage polarization. Sci. Rep. 2022, 12, 2417. [Google Scholar] [CrossRef] [PubMed]
- Lumibao, J.C.; Haak, P.L.; Kolossov, V.L.; Chen, J.E.; Stutchman, J.; Ruiz, A.; Sivaguru, M.; Sarkaria, J.N.; Harley, B.A.C.; Steelman, A.J.; et al. CHCHD2 mediates glioblastoma cell proliferation, mitochondrial metabolism, hypoxia-induced invasion and therapeutic resistance. Int. J. Oncol. 2023, 63, 117. [Google Scholar] [CrossRef]
- Toledano, S.; Sabag, A.D.; Ilan, N.; Liburkin-Dan, T.; Kessler, O.; Neufeld, G. Plexin-A2 enables the proliferation and the development of tumors from glioblastoma derived cells. Cell Death Dis. 2023, 14, 41. [Google Scholar] [CrossRef] [PubMed]
- Harutyunyan, A.S.; Krug, B.; Chen, H.; Papillon-Cavanagh, S.; Zeinieh, M.; De Jay, N.; Deshmukh, S.; Chen, C.C.L.; Belle, J.; Mikael, L.G.; et al. H3K27M induces defective chromatin spread of PRC2-mediated repressive H3K27me2/me3 and is essential for glioma tumorigenesis. Nat. Commun. 2019, 10, 1262. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Liu, R.; Liu, C.; Zhang, H.; Lu, Y. Nanos3, a cancer-germline gene, promotes cell proliferation, migration, chemoresistance, and invasion of human glioblastoma. Cancer Cell. Int. 2020, 20, 197. [Google Scholar] [CrossRef] [PubMed]
- Godoy, P.R.D.V.; Pour Khavari, A.; Rizzo, M.; Sakamoto-Hojo, E.T.; Haghdoost, S. Targeting NRF2, Regulator of Antioxidant System, to Sensitize Glioblastoma Neurosphere Cells to Radiation-Induced Oxidative Stress. Oxidative Med. Cell. Longev. 2020, 2020, 2534643. [Google Scholar] [CrossRef]
- Zhang, F.; Liu, R.; Zhang, H.; Liu, C.; Liu, C.; Lu, Y. Suppressing Dazl modulates tumorigenicity and stemness in human glioblastoma cells. BMC Cancer 2020, 20, 673. [Google Scholar] [CrossRef]
- Liu, J.; Sareddy, G.R.; Zhou, M.; Viswanadhapalli, S.; Li, X.; Lai, Z.; Tekmal, R.R.; Brenner, A.; Vadlamudi, R.K. Differential Effects of Estrogen Receptor β Isoforms on Glioblastoma Progression. Cancer Res. 2018, 78, 3176–3189. [Google Scholar] [CrossRef]
- Bulstrode, H.; Johnstone, E.; Marques-Torrejon, M.A.; Ferguson, K.M.; Bressan, R.B.; Blin, C.; Grant, V.; Gogolok, S.; Gangoso, E.; Gagrica, S.; et al. Elevated FOXG1 and SOX2 in glioblastoma enforces neural stem cell identity through transcriptional control of cell cycle and epigenetic regulators. Genes. Dev. 2017, 31, 757–773. [Google Scholar] [CrossRef]
- Saenz-Antoñanzas, A.; Moncho-Amor, V.; Auzmendi-Iriarte, J.; Elua-Pinin, A.; Rizzoti, K.; Lovell-Badge, R.; Matheu, A. CRISPR/Cas9 Deletion of SOX2 Regulatory Region 2 (SRR2) Decreases SOX2 Malignant Activity in Glioblastoma. Cancers 2021, 13, 1574. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Wan, X.; Huang, T.; Zeng, C.; Sastry, N.; Wu, B.; James, C.D.; Horbinski, C.; Nakano, I.; Zhang, W.; et al. SRSF3-Regulated RNA Alternative Splicing Promotes Glioblastoma Tumorigenicity by Affecting Multiple Cellular Processes. Cancer Res. 2019, 79, 5288–5301. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, J.; Pao, G.M.; Shokhirev, M.N.; Verma, I.M. Glioblastoma Model Using Human Cerebral Organoids. Cell. Rep. 2018, 23, 1220–1229. [Google Scholar] [CrossRef] [PubMed]
- Smolkin, T.; Nir-Zvi, I.; Duvshani, N.; Mumblat, Y.; Kessler, O.; Neufeld, G. Complexes of plexin-A4 and plexin-D1 convey semaphorin-3C signals to induce cytoskeletal collapse in the absence of neuropilins. J. Cell. Sci. 2018, 131, jcs208298. [Google Scholar] [CrossRef] [PubMed]
- Prolo, L.M.; Li, A.; Owen, S.F.; Parker, J.J.; Foshay, K.; Nitta, R.T.; Morgens, D.W.; Bolin, S.; Wilson, C.M.; Vega, L.J.; et al. Targeted genomic CRISPR-Cas9 screen identifies MAP4K4 as essential for glioblastoma invasion. Sci. Rep. 2019, 9, 14020. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yang, C.H.; Schultz, A.P.; Sims, M.M.; Miller, D.D.; Pfeffer, L.M. Brahma-Related Gene-1 (BRG1) promotes the malignant phenotype of glioblastoma cells. J. Cell. Mol. Med. 2021, 25, 2956–2966. [Google Scholar] [CrossRef] [PubMed]
- Shao, W.; Azam, Z.; Guo, J.; To, S.S.T. Oncogenic potential of PIK3CD in glioblastoma is exerted through cytoskeletal proteins PAK3 and PLEK2. Lab. Investig. 2022, 102, 1314–1322. [Google Scholar] [CrossRef]
- Chen, L.; Fang, W.; Chen, W.; Wei, Y.; Ding, J.; Li, J.; Lin, J.; Wu, Q. Deciphering the molecular mechanism of the THBS1 gene in the TNF signaling axis in glioma stem cells. Cell. Signal. 2023, 106, 110656. [Google Scholar] [CrossRef]
- Ezgi, O.-G.; Ezgi Yagmur, K.; Ali Cenk, A.; Ipek, B.; Ahmet, C.; Sheikh, N.; Martin, B.; Fidan, S.-P.; Tunc, M.; Can, A.; et al. Epigenetic-focused CRISPR/Cas9 screen identifies ASH2L as a regulator of glioblastoma cell survival. bioRxiv 2022. peer review. [Google Scholar] [CrossRef]
- Nieland, L.; van Solinge, T.S.; Cheah, P.S.; Morsett, L.M.; El Khoury, J.; Rissman, J.I.; Kleinstiver, B.P.; Broekman, M.L.D.; Breakefield, X.O.; Abels, E.R. CRISPR-Cas knockout of miR21 reduces glioma growth. Mol. Ther. Oncolytics 2022, 25, 121–136. [Google Scholar] [CrossRef]
- Uceda-Castro, R.; van Asperen, J.V.; Vennin, C.; Sluijs, J.A.; van Bodegraven, E.J.; Margarido, A.S.; Robe, P.A.J.; van Rheenen, J.; Hol, E.M. GFAP splice variants fine-tune glioma cell invasion and tumour dynamics by modulating migration persistence. Sci. Rep. 2022, 12, 424. [Google Scholar] [CrossRef] [PubMed]
- Han, N.; Hu, G.; Shi, L.; Long, G.; Yang, L.; Xi, Q.; Guo, Q.; Wang, J.; Dong, Z.; Zhang, M. Notch1 ablation radiosensitizes glioblastoma cells. Oncotarget 2017, 8, 88059–88068. [Google Scholar] [CrossRef] [PubMed]
- Eisemann, T.; Costa, B.; Harter, P.N.; Wick, W.; Mittelbronn, M.; Angel, P.; Peterziel, H. Podoplanin expression is a prognostic biomarker but may be dispensable for the malignancy of glioblastoma. Neuro-Oncol. 2019, 21, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Szymura, S.J.; Bernal, G.M.; Wu, L.; Zhang, Z.; Crawley, C.D.; Voce, D.J.; Campbell, P.A.; Ranoa, D.E.; Weichselbaum, R.R.; Yamini, B. DDX39B interacts with the pattern recognition receptor pathway to inhibit NF-κB and sensitize to alkylating chemotherapy. BMC Biol. 2020, 18, 32. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.I.; Wang, Y.T.; Wang, Y.S.; Wu, C.Y.; Li, C.C. Involvement of BIG1 and BIG2 in regulating VEGF expression and angiogenesis. FASEB J. 2019, 33, 9959–9973. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Lee, E.A.; Kong, E.; Chon, H.; Llaiqui-Condori, M.; Park, C.H.; Park, B.Y.; Kang, N.R.; Yoo, J.S.; Lee, H.S.; et al. An agonistic anti-Tie2 antibody suppresses the normal-to-tumor vascular transition in the glioblastoma invasion zone. Exp. Mol. Med. 2023, 55, 470–484. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Marisetty, A.; Schrand, B.; Gabrusiewicz, K.; Hashimoto, Y.; Ott, M.; Grami, Z.; Kong, L.Y.; Ling, X.; Caruso, H.; et al. Osteopontin mediates glioblastoma-associated macrophage infiltration and is a potential therapeutic target. J. Clin. Investig. 2019, 129, 137–149. [Google Scholar] [CrossRef]
- Chen, P.A.; Shrivastava, G.; Balcom, E.F.; McKenzie, B.A.; Fernandes, J.; Branton, W.G.; Wheatley, B.M.; Petruk, K.; van Landeghem, F.K.H.; Power, C. Absent in melanoma 2 regulates tumor cell proliferation in glioblastoma multiforme. J. Neurooncol. 2019, 144, 265–273. [Google Scholar] [CrossRef]
- Wu, W.; Wu, Y.; Mayer, K.; von Rosenstiel, C.; Schecker, J.; Baur, S.; Würstle, S.; Liesche-Starnecker, F.; Gempt, J.; Schlegel, J. Lipid Peroxidation Plays an Important Role in Chemotherapeutic Effects of Temozolomide and the Development of Therapy Resistance in Human Glioblastoma. Transl. Oncol. 2020, 13, 100748. [Google Scholar] [CrossRef]
- Han, X.; Abdallah, M.O.E.; Breuer, P.; Stahl, F.; Bakhit, Y.; Potthoff, A.L.; Pregler, B.E.F.; Schneider, M.; Waha, A.; Wüllner, U.; et al. Downregulation of MGMT expression by targeted editing of DNA methylation enhances temozolomide sensitivity in glioblastoma. Neoplasia 2023, 44, 100929. [Google Scholar] [CrossRef]
- Tong, F.; Zhao, J.X.; Fang, Z.Y.; Cui, X.T.; Su, D.Y.; Liu, X.; Zhou, J.H.; Wang, G.X.; Qiu, Z.J.; Liu, S.Z.; et al. MUC1 promotes glioblastoma progression and TMZ resistance by stabilizing EGFRvIII. Pharmacol. Res. 2023, 187, 106606. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Cao, Z.; Wang, W.; Zou, C.; Wang, Y.; Pan, L.; Jia, B.; Zhang, K.; Zhang, W.; Li, W.; et al. Engineered Extracellular Vesicle-Delivered CRISPR/Cas9 for Radiotherapy Sensitization of Glioblastoma. ACS Nano 2023, 17, 16432–16447. [Google Scholar] [CrossRef]
- Rocha, C.R.R.; Rocha, A.R.; Silva, M.M.; Gomes, L.R.; Latancia, M.T.; Andrade-Tomaz, M.; de Souza, I.; Monteiro, L.K.S.; Menck, C.F.M. Revealing Temozolomide Resistance Mechanisms via Genome-Wide CRISPR Libraries. Cells 2020, 9, 2573. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Wang, X.; Ge, X.; Ding, F.; Shi, Z.; Ge, Z.; Huang, G.; Zhao, N.; Chen, D.; Zhang, J.; et al. Hypoxanthine phosphoribosyl transferase 1 metabolizes temozolomide to activate AMPK for driving chemoresistance of glioblastomas. Nat. Commun. 2023, 14, 5913. [Google Scholar] [CrossRef] [PubMed]
- Álvarez, M.M.; Biayna, J.; Supek, F. TP53-dependent toxicity of CRISPR/Cas9 cuts is differential across genomic loci and can confound genetic screening. Nat. Commun. 2022, 13, 4520. [Google Scholar] [CrossRef] [PubMed]
- Ihry, R.J.; Worringer, K.A.; Salick, M.R.; Frias, E.; Ho, D.; Theriault, K.; Kommineni, S.; Chen, J.; Sondey, M.; Ye, C.; et al. p53 inhibits CRISPR-Cas9 engineering in human pluripotent stem cells. Nat. Med. 2018, 24, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Barbosa, K.; Cheng, K.; Leiserson, M.D.M.; Jain, P.; Deshpande, A.; Wilson, D.M.; Ryan, B.M.; Luo, J.; Ronai, Z.e.A.; et al. A systematic genome-wide mapping of oncogenic mutation selection during CRISPR-Cas9 genome editing. Nat. Commun. 2021, 12, 6512. [Google Scholar] [CrossRef]
- Enache, O.M.; Rendo, V.; Abdusamad, M.; Lam, D.; Davison, D.; Pal, S.; Currimjee, N.; Hess, J.; Pantel, S.; Nag, A.; et al. Cas9 activates the p53 pathway and selects for p53-inactivating mutations. Nat. Genet. 2020, 52, 662–668. [Google Scholar] [CrossRef]
- Hwang, W.Y.; Fu, Y.; Reyon, D.; Maeder, M.L.; Tsai, S.Q.; Sander, J.D.; Peterson, R.T.; Yeh, J.R.; Joung, J.K. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat. Biotechnol. 2013, 31, 227–229. [Google Scholar] [CrossRef]
- Bečulić, H.; Begagić, E.; Skomorac, R.; Mašović, A.; Selimović, E.; Pojskić, M. ChatGPT’s contributions to the evolution of neurosurgical practice and education: A systematic review of benefits, concerns and limitations. Med. Glas. 2024, 21. [Google Scholar] [CrossRef]
- Ayanoğlu, F.B.; Elçin, A.E.; Elçin, Y.M. Bioethical issues in genome editing by CRISPR-Cas9 technology. Turk. J. Biol. 2020, 44, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Zuo, E.; Sun, Y.; Yuan, T.; He, B.; Zhou, C.; Ying, W.; Liu, J.; Wei, W.; Zeng, R.; Li, Y.; et al. A rationally engineered cytosine base editor retains high on-target activity while reducing both DNA and RNA off-target effects. Nat. Methods 2020, 17, 600–604. [Google Scholar] [CrossRef] [PubMed]
- Yunta, E. Ethical Issues in Genome Editing using Crispr/Cas9 System. J. Clin. Res. Bioeth. 2016, 7, 1000266. [Google Scholar] [CrossRef]
- Ray, U.; Raghavan, S.C. Modulation of DNA double-strand break repair as a strategy to improve precise genome editing. Oncogene 2020, 39, 6393–6405. [Google Scholar] [CrossRef] [PubMed]
- Stoltz, K.; Sinyuk, M.; Hale, J.S.; Wu, Q.; Otvos, B.; Walker, K.; Vasanji, A.; Rich, J.N.; Hjelmeland, A.B.; Lathia, J.D. Development of a Sox2 reporter system modeling cellular heterogeneity in glioma. Neuro-Oncol. 2015, 17, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.F.; Li, C.; Xi, S.Y.; Chen, F.R.; Wang, J.; Zhang, Z.Q.; Liu, Y.; Li, X.; Chen, Z.P. Whole exome sequencing reveals the genetic heterogeneity and evolutionary history of primary gliomas and matched recurrences. Comput. Struct. Biotechnol. J. 2022, 20, 2235–2246. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Sage, J.C.; Miller, M.R.; Verhaak, R.G.; Hippenmeyer, S.; Vogel, H.; Foreman, O.; Bronson, R.T.; Nishiyama, A.; Luo, L.; et al. Mosaic analysis with double markers reveals tumor cell of origin in glioma. Cell 2011, 146, 209–221. [Google Scholar] [CrossRef]
- Bhat, K.P.L.; Balasubramaniyan, V.; Vaillant, B.; Ezhilarasan, R.; Hummelink, K.; Hollingsworth, F.; Wani, K.; Heathcock, L.; James, J.D.; Goodman, L.D.; et al. Mesenchymal differentiation mediated by NF-κB promotes radiation resistance in glioblastoma. Cancer Cell 2013, 24, 331–346. [Google Scholar] [CrossRef]
- Ene, C.I.; Holland, E.C. Personalized medicine for gliomas. Surg. Neurol. Int. 2015, 6, S89–S95. [Google Scholar] [CrossRef]
- Rosenblum, D.; Gutkin, A.; Kedmi, R.; Ramishetti, S.; Veiga, N.; Jacobi, A.M.; Schubert, M.S.; Friedmann-Morvinski, D.; Cohen, Z.R.; Behlke, M.A.; et al. CRISPR-Cas9 genome editing using targeted lipid nanoparticles for cancer therapy. Sci. Adv. 2020, 6, eabc9450. [Google Scholar] [CrossRef]
- Luo, Y.L.; Xu, C.F.; Li, H.J.; Cao, Z.T.; Liu, J.; Wang, J.L.; Du, X.J.; Yang, X.Z.; Gu, Z.; Wang, J. Macrophage-Specific in Vivo Gene Editing Using Cationic Lipid-Assisted Polymeric Nanoparticles. ACS Nano 2018, 12, 994–1005. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, C.A.; Brandes, N.; Bueno, R.; Trinidad, M.; Mazumder, T.; Yu, B.; Hwang, B.; Chang, C.; Liu, J.; Sun, Y.; et al. Mitigation of chromosome loss in clinical CRISPR-Cas9-engineered T cells. Cell 2023, 186, 4567–4582.e20. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.; Merkel, O.M. Immunogenicity of Cas9 Protein. J. Pharm. Sci. 2020, 109, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Crudele, J.M.; Chamberlain, J.S. Cas9 immunity creates challenges for CRISPR gene editing therapies. Nat. Commun. 2018, 9, 3497. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Chang, R.; Yang, H.; Zhao, T.; Hong, Y.; Kong, H.E.; Sun, X.; Qin, Z.; Jin, P.; Li, S.; et al. CRISPR/Cas9-mediated gene editing ameliorates neurotoxicity in mouse model of Huntington’s disease. J. Clin. Investig. 2017, 127, 2719–2724. [Google Scholar] [CrossRef] [PubMed]
- Cribbs, A.P.; Perera, S.M.W. Science and Bioethics of CRISPR-Cas9 Gene Editing: An Analysis Towards Separating Facts and Fiction. Yale J. Biol. Med. 2017, 90, 625–634. [Google Scholar] [PubMed]
- Tao, J.; Bauer, D.E.; Chiarle, R. Assessing and advancing the safety of CRISPR-Cas tools: From DNA to RNA editing. Nat. Commun. 2023, 14, 212. [Google Scholar] [CrossRef]
- Zha, M.J.; Cai, C.E.; He, P.M. Outlook on the Security and Potential Improvements of CRISPR-Cas9. Mol. Biotechnol. 2023, 65, 1729–1736. [Google Scholar] [CrossRef]
- Lu, Y.; Xue, J.; Deng, T.; Zhou, X.; Yu, K.; Deng, L.; Huang, M.; Yi, X.; Liang, M.; Wang, Y.; et al. Safety and feasibility of CRISPR-edited T cells in patients with refractory non-small-cell lung cancer. Nat. Med. 2020, 26, 732–740. [Google Scholar] [CrossRef]
- Deng, H.X.; Zhai, H.; Shi, Y.; Liu, G.; Lowry, J.; Liu, B.; Ryan, É.B.; Yan, J.; Yang, Y.; Zhang, N.; et al. Efficacy and long-term safety of CRISPR/Cas9 genome editing in the SOD1-linked mouse models of ALS. Commun. Biol. 2021, 4, 396. [Google Scholar] [CrossRef]
- Sledzinski, P.; Dabrowska, M.; Nowaczyk, M.; Olejniczak, M. Paving the way towards precise and safe CRISPR genome editing. Biotechnol. Adv. 2021, 49, 107737. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference Year | Targeted Gene | Targeted Molecules or Focus | Targeted Function | CRISPR-Cas9 Gene Editing | Therapy Efficiency or Outcome |
|---|---|---|---|---|---|
| Wang et al. [49] 2023 | PDPN | PDPN | Apoptosis; Cell proliferation | Knockdown | PDPN may contribute to GBM immune microenvironment, with AUCs for 1-, 3-, and 5-year OS at 0.887, 0.916, and 0.870. |
| Nayak et al. [50] 2018 | C14-IP-3 | EGFR, AKT, TP53, RAF1 | Regulation of proliferation and invasion | CRISPRa | miR-134 targets EGFR and RAF1, confirmed with luciferase assay. |
| Rodvold et al. [51] 2019 | IGFBP3, IGFBP5, ERN1, ATF4. | IGFBP3, IGFBP5, IRE1α, ATF4. | Apoptosis via UPR | Knockout | Nonresponder phenotype is linked to UPR gene expression, particularly ERN1 and ATF4. CRISPR-deletion of ERN1, IGFBP3, and IGFBP5 in U251 cells enhances responsiveness to 12ADT. |
| Thi Vu et al. [52] 2018 | ATG5 | ATG5 | Apoptosis, autophagy | Knockout | Ca2+ mobilization compounds combined with autophagy inhibition may be a novel therapy for GBM. |
| Peng et al. [53] 2018 | CHAF1A | AKT, FOXO3a, Bim | Proliferation and DNA repair | Knockout | CRISPR/Cas9 knockout of CHAF1A inhibits FOXO3a transactivity, upregulating Bim and caspase cleavage. |
| Kranz et al. [54] 2014 | FAT1 | Caspase-8 | Apoptosis via Death-Inducing Signaling Complex (DISC) | Knockout | FAT1 knockout with CRISPR/Cas9 increases susceptibility to death receptor-mediated apoptosis. |
| Nakazawa et al. [55] 2023 | CIS (deleted NKCs) | IFNɤ TNF | NK cells activation; apoptosis | Knockout | CIS deletion enhances NKC-mediated anti-tumor effects in allogeneic GBM. |
| Zielke et al. [56] 2018 | ATG5 ATG7 | ATG5 ATG7 | Autophagosome membrane | Knockout | Loperamide, pimozide, and STF-62247 induce ATG5- and ATG7-dependent cell death in GBM, preceded by autophagy induction. |
| Wang et al. [49] 2023 | PDPN | PDPN | Apoptosis; Cell proliferation | Knockdown | |
| Maggio et al. [57] 2023 | PIN1 | PIN1 enzyme | Apoptosis, migration, cell cycle progression | Knockout | PIN1 deletion in GBM diminishes active NF-κB, reducing il-8 and htert gene transcription. |
| Reem et al. [58] 2019 | ATM, PTEN, p85α, XIAP | PI3K, PIKK, p110α | Tumor suppressors | Knockout | ATM’s novel role in autophagy regulation via XIAP interaction is speculated. |
| Guda et al. [59] | RGS4 | MMP2 | Apoptosis (G protein signaling) | Knockout | Silencing RGS4 in GSC20 and GSC28 cells demonstrates anticancer effects, establishing RGS4 as a promoter of invasive behavior in GSCs. |
| Ranjan et al. [60] 2017 | GLI1 | PI3K/Akt | Apoptosis | Knockout | Penfluridol treatment suppresses Akt phosphorylation, reduces GLI1, OCT4, Nanog, Sox2 expression, inhibiting tumor growth. |
| Reference Year | Targeted Gene | Targeted Molecules/Focus | Targeted Function | CRISPR-Cas9 Gene Editing | Therapy Efficiency/Outcome |
|---|---|---|---|---|---|
| Cell proliferation | |||||
| Fierro et al. [62] 2022 | PD-L1 | PD-1 | proliferation, invasion, and macrophage polarization | Knockout | Dual-sgRNAs with repair template caused a 64% reduction in PD-L1 protein levels in U87 cells. |
| Lumibao et al. [63] 2023 | CHCHD2 | EGFRvIII | mitochondrial respiration, glutathione status, and cell growth inhibition | Knockout | CRISPR-Cas9 knockout of CHCHD2 in EGFRvIII-expressing U87 cells altered mitochondrial respiration, glutathione status, and decreased cell growth and invasion under normoxic and hypoxic conditions. |
| Toledano et al. [64] 2023 | Plexin-A2 | β-galactosidase, MAPK, FARP2 | cytoskeletal organization, cell flattening, and cell cycle arrest | Knockout | Plexin-A2’s proproliferative effects are mediated via FARP2, FYN, and the GTPase activating (GAP) domain in its intracellular domain. |
| Gallo et al. [13] 2023 | 14-3-3β | Bad, FBI1, Raf-1, Cdc25b | proliferation and spheroid formation | Knockout | 14-3-3β knockout resulted in impaired proliferation and decreased cells within a 3D-spheroid of U87MG cells. |
| Meng et al. [65] 2018 | CDK7 | n/d | growth | Knockout | |
| Guda et al. [59] 2020 | RGS4 | MMP2 | proliferation | Knockout | |
| Zhang et al. [66] 2020 | Nanos3 | CD133, Oct4 | proliferation, migration, and chemoresistance | Knockdown | Nanos3 deletion reduced proliferation, migration, and invasion of GBM cells in vitro (p < 0.05), increased sensitivity to DOX and TMZ (p < 0.05), and inhibited subcutaneous xenograft tumor growth in vivo (p < 0.001). |
| Godoy et al. [67] 2020 | NRF2 | SOD | self-renewal and cell proliferation | Knockdown | NRF2 knockdown resulted in less self-renewal, more differentiated cells, and decreased proliferation after irradiation with low- and high-dose rate gamma rays. |
| Zhang et al. [68] 2020 | Dazl | CD133/Oct4/Nanog/Sox2 regulatory axis | proliferation | Knockout | Knocking down Dazl in A172, U251, and LN229 cell lines resulted in reduced proliferation rates and decreased migration of Dazl+/− cells compared to Dazl WT cells (p < 0.05) in both instances. |
| Liu et al. [69] 2018 | ERβ | ERβ1, ERβ2, ERβ3, ERβ4, ERβ5 (exon 8), mTOR and STAT-3 | proliferation and apoptosis | Knockout | ERβ KO cells exhibited high migratory and invasive potentials, while ERβ1 re-expression reduced this phenotype. |
| Cell renewal | |||||
| Bulstrode et al. [70] | Foxo3 | FOXG1, SOX2, EGFR, EGFRvIII | differentiation | Knockdown | FOXG1 deletion in patient-derived GBM stem cells increased astrocyte differentiation and up-regulated FOXO3 in vivo. |
| Saent—Antonanzas [71] 2021 | SRR2 | SOX2 | self-renowal capacity | Deletion | SOX2 ablation attenuated proliferation, and mutant cells could not be expanded in vitro. SRR2-deleted GBM cells displayed reduced SOX2 expression, decreased proliferative activity, and inhibited tumor initiation and growth in vivo. |
| Song et al. [72] 2019 | SRSF3 | SR proteins | glioma-associated alternative splicing | Knockout | ETV1 gene showed exon skipping at exon 7, and NDE1 gene showed replacement of terminal exon 9 with exon 9′, increasing their oncogenic activity in GSCs. |
| Cell migration | |||||
| Ogawa et al. [73] 2018 | TP53 | n/a | migration | Recombination | |
| Smolkin et al. [74] 2018 | NRP2 | Plexin-A4 Plexin-D1 Semaphorin-3C | migration | Knockout | Sema3D and Sema3G could not transduce signals without neuropilins. |
| Prolo et al. [75] 2019 | MAP4K4 | n/d | migration and invasion | Knockout | MAP4K4 knockout led to a 41% reduction in invasion compared to U138-Cas9 control. |
| Wang et al. [76] 2021 | BRG1 | STAT3 | migration, proliferation, and TMZ resistance | Knockout | BRG1-KO inhibited GBM cell migration and invasion, sensitizing cells to TMZ. |
| Shao et al. [77] 2022 | PIK3CD | PAK3 PLEK2 | migration and invasion | Knockout | SD2 and SD13 cells did not form any noticeable xenograft tumor even 26 days after implantation, whereas xenograft tumors could be clearly observed 7 days after implantation in the U87-MG |
| Chen et al. [78] 2023 | THBS1 | TNF | proliferation and migration | Knockout | THBS1 gene knockout promoted proliferation and migration in U251 cells and GSCs. |
| Fierro et al. [62] 2022 | PD-L1 | PD-L1 | proliferation, growth, invasion, and migration | Deletion | PD-L1 deletion reduced BrdU + proliferating U87 cells and prevented cell invasion. |
| Ozyerli-Gokna et al. [79] 2022 | ASH2L | SET1/MLL | proliferation and migration | Knockout | ASH2L knockout resulted in significant gene expression changes. |
| Nieland et al. [80] 2022 | miR21 | SOX2 | migration, invasion, and proliferation | Knockout | Proliferation significantly decreased in miR-21 KO in GL261, CT2A, and U87 cells. CT2A cells showed increased migration and invasion over GL261 cells. |
| Uceda-Castro et al. [81] 2022 | GFAP | GFAPα, GFAPδ | invasion | Knockout | GFAPδ and GFAPα isoforms differentially regulate glioma cell dynamics. Depletion of either isoform increases migratory capacity, with distinct invasion patterns into brain tissue. |
| Reference Year | Targeted Gene | Targeted Molecules | Targeted Function | CRISPR-Cas9 Gene Editing | Therapy Efficiency or Outcome |
|---|---|---|---|---|---|
| Angiogenesis | |||||
| Han et al. [82] 2017 | Notch1 | n/d | hypoxia, angiogenesis, and tumor growth | Knockdown | Xenografts with Notch1 downregulation reached 6 x the starting volume in 18.3 days, while control xenografts took 13.4 days. |
| Eisemann et al. [83] 2019 | PDPN | PDPN | mediates the maturation and integrity of the developing vasculature in the murine brain in interaction with C-type lectin-like receptor 2 on platelets | Knockout | Similar rates of proliferation, apoptosis, angiogenesis, and invasion were observed in control and podoplanin-deleted tumors. |
| Szymura et al. [84] 2020 | DDX39B | NF-κB | regulation of the extracellular ECM and promotes angiogenesis | Knockdown | CRISPR-mediated DDX39B depletion increased p65 phosphorylation, while MAVS knockdown reduced this phosphorylation; loss of DDX39B rendered U87 cells highly resistant to TMZ. |
| Lu et al. [85] 2019 | BIG1, BIG2 | VEGF | angiogenesis | Knockdown | BIG1 and BIG2 knockdown significantly decreased VEGF mRNA and protein levels in GBM U251 cells and HUVECs. |
| Lee et al. [86] 2023 | ANGPT2 | VEGFR2 | normal-to-tumor vascular transition | Knockout | Treatment with the agonistic anti-Tie2 antibody, 4E2, resulted in vascular normalization throughout GBM tissues. |
| Inflammation | |||||
| Nakazawa et al. [55] | CIS | NKCs | Enhances NKCs effects | Knockout | The NK mock group showed longer survival compared to the NB group (mOS: 41.0 days vs. 56.5 days). The NK dCIS group exhibited prolonged OS compared to the NK mock group (mOS: 79.5 days). |
| Wei et al. [87] | OPN | M2 macrophages | M2 macrophages reduction and T-lymphocite effector activity elevation | Knockout | OPN deficiency in innate immune or glioma cells reduced M2 macrophages and elevated T cell effector activity infiltrating the glioma. |
| Chen et al. [88] | AIM2 | IL-1β, IL-18 | Pyroptosis (infammatory programmed cell death) | Knockdown | AIM2 immunoreactivity concentrated in the tumor core in the absence of PCNA immunodetection, showing a predominant 52 kDa immunoreactive band on western blot. |
| Reference Year | Targeted Gene | Targeted Molecules/Focus | Targeted Function | CRISPR-Cas9 Gene Editing | Therapy Efficiency or Outcome |
|---|---|---|---|---|---|
| Wu et al. [89] 2020 | ALDH1A3 | ALDHs | TMZ resistance | Knockdown | The observed difference was particularly significant at dosages ≤ 300 μM. |
| Han et al. [90] 2023 | MGMT | MGMT | TMZ resistance | Knockdown | T98G and LN18 cells displayed a dose-dependent decrease in viability, with IC50 values of 475.6 µM and 424.7 µM, respectively. |
| Tong et al. [91] 2023 | MUC1 | EGFRvIII | TMZ resistance | Knockdown | EGFRvIII was localized in the nucleus after TMZ treatment, consistent with its reported role in assisting DNA damage repair during chemotherapy and radiation. |
| Liu et al. [92] 2023 | GSS | Angiopep-2 | Radiotherapy resistance | Knockout | GSS perturbation in glioma cells demonstrated significant antitumor activity when combined with radiotherapy. |
| Rocha et al. [93] 2020 | MSH2, PTCH2, CLCA2, FZD6, CTNNB1, NRF2 | Transmembrane proteins | TMZ resistance | Knockout | Silencing the top three genes (MSH2, PTCH2, and CLCA2) confirmed cell protection from TMZ-induced death. |
| Yin et al. [94] | HPRT1 | AMPK | TMZ resistance | Knockout | Combining HPRT1 depletion with TMZ treatment achieved the longest survival extension. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Begagić, E.; Bečulić, H.; Đuzić, N.; Džidić-Krivić, A.; Pugonja, R.; Muharemović, A.; Jaganjac, B.; Salković, N.; Sefo, H.; Pojskić, M. CRISPR/Cas9-Mediated Gene Therapy for Glioblastoma: A Scoping Review. Biomedicines 2024, 12, 238. https://doi.org/10.3390/biomedicines12010238
Begagić E, Bečulić H, Đuzić N, Džidić-Krivić A, Pugonja R, Muharemović A, Jaganjac B, Salković N, Sefo H, Pojskić M. CRISPR/Cas9-Mediated Gene Therapy for Glioblastoma: A Scoping Review. Biomedicines. 2024; 12(1):238. https://doi.org/10.3390/biomedicines12010238
Chicago/Turabian StyleBegagić, Emir, Hakija Bečulić, Nermin Đuzić, Amina Džidić-Krivić, Ragib Pugonja, Asja Muharemović, Belma Jaganjac, Naida Salković, Haso Sefo, and Mirza Pojskić. 2024. "CRISPR/Cas9-Mediated Gene Therapy for Glioblastoma: A Scoping Review" Biomedicines 12, no. 1: 238. https://doi.org/10.3390/biomedicines12010238
APA StyleBegagić, E., Bečulić, H., Đuzić, N., Džidić-Krivić, A., Pugonja, R., Muharemović, A., Jaganjac, B., Salković, N., Sefo, H., & Pojskić, M. (2024). CRISPR/Cas9-Mediated Gene Therapy for Glioblastoma: A Scoping Review. Biomedicines, 12(1), 238. https://doi.org/10.3390/biomedicines12010238