
Hypoxia Dysregulates the Transcription of Myoendothelial Junction Proteins Involved with Nitric Oxide Production in Brain Endothelial Cells


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture, Chemically Induced Hypoxia, and NO Detection
2.3. Statistics
3. Results
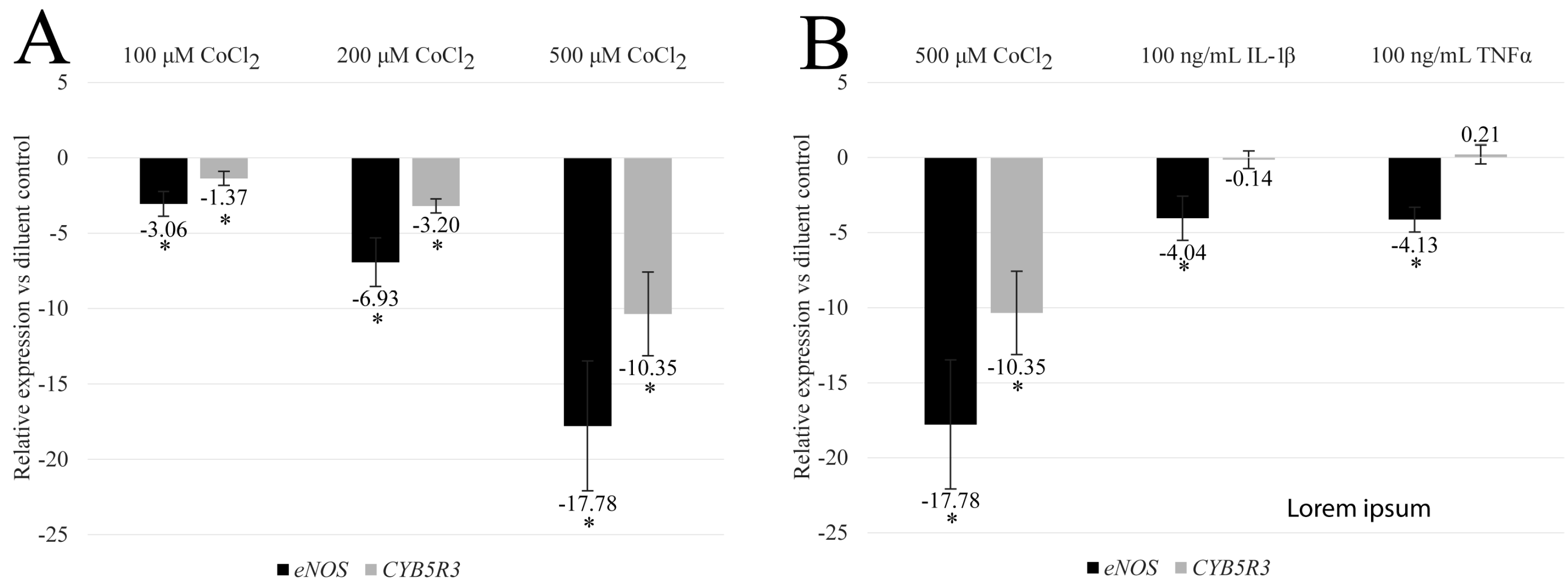
3.1. CoCl2 Reduces the Transcription of eNOS and CYB5R3 in HBMEC
3.2. CoCl2-Induced eNOS and CYB5R3 Transcriptional Programs Differ from Inflammatory Programs
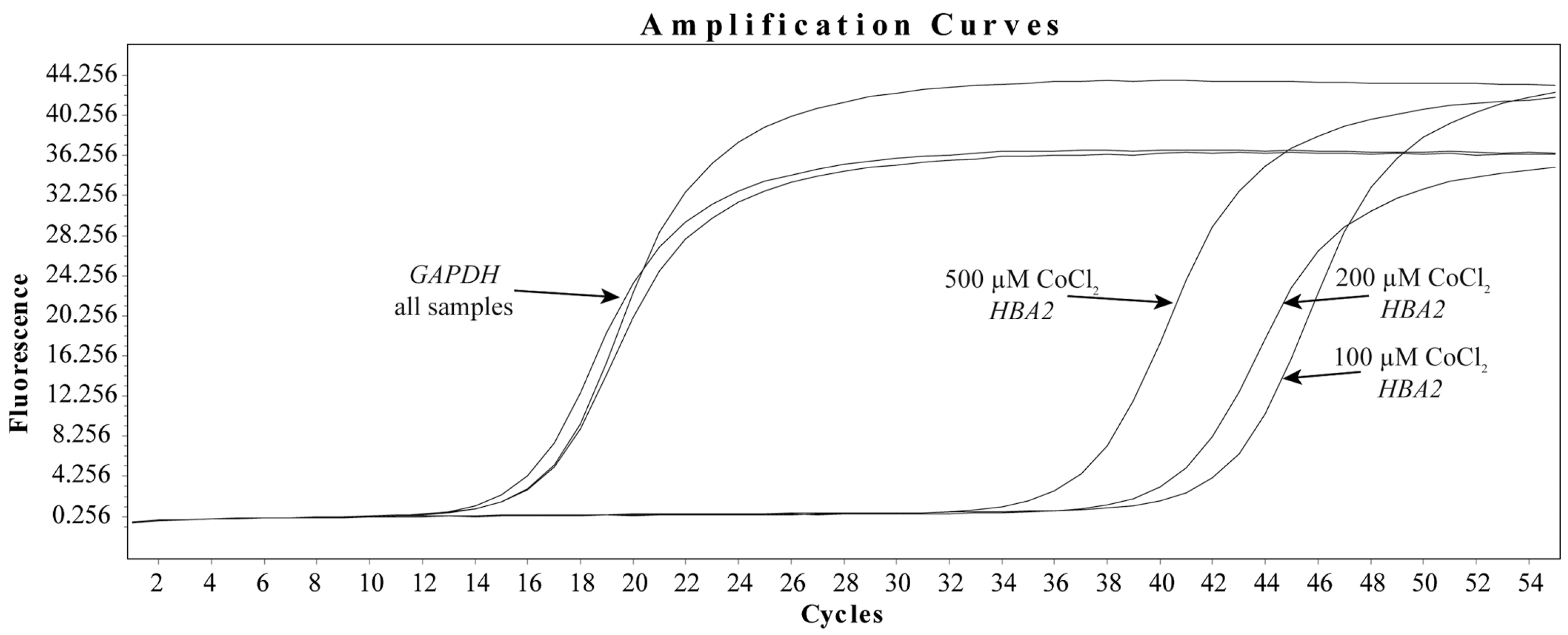
3.3. CoCl2 Induces the Transcription of HBA2 in HBMEC
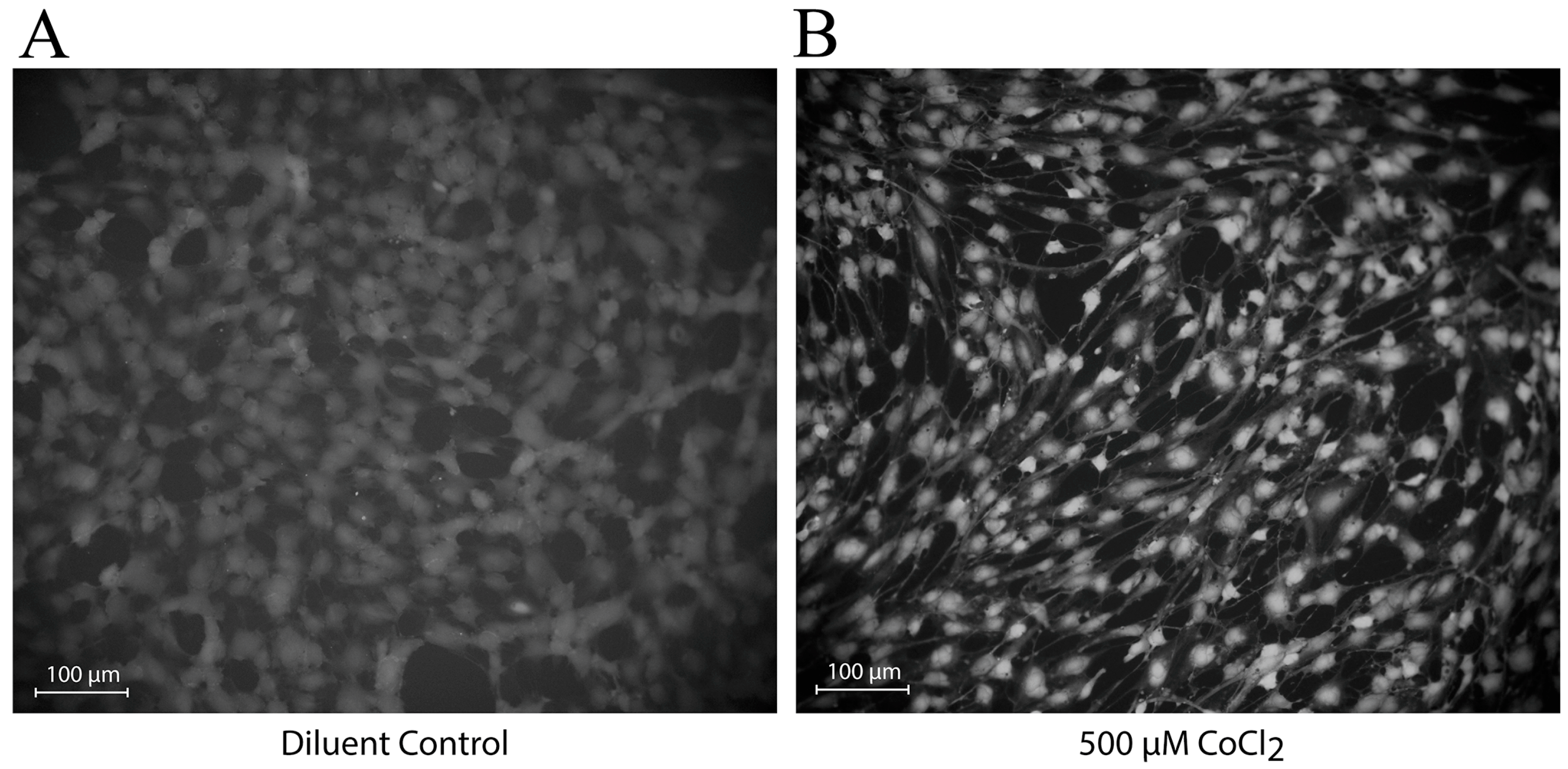
3.4. CoCl2 Induces Changes in the Localization of NO Production
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhao, Y.; Vanhoutte, P.M.; Leung, S.W.S. Vascular Nitric Oxide: Beyond ENOS. J. Pharmacol. Sci. 2015, 129, 83–94. [Google Scholar] [CrossRef] [PubMed]
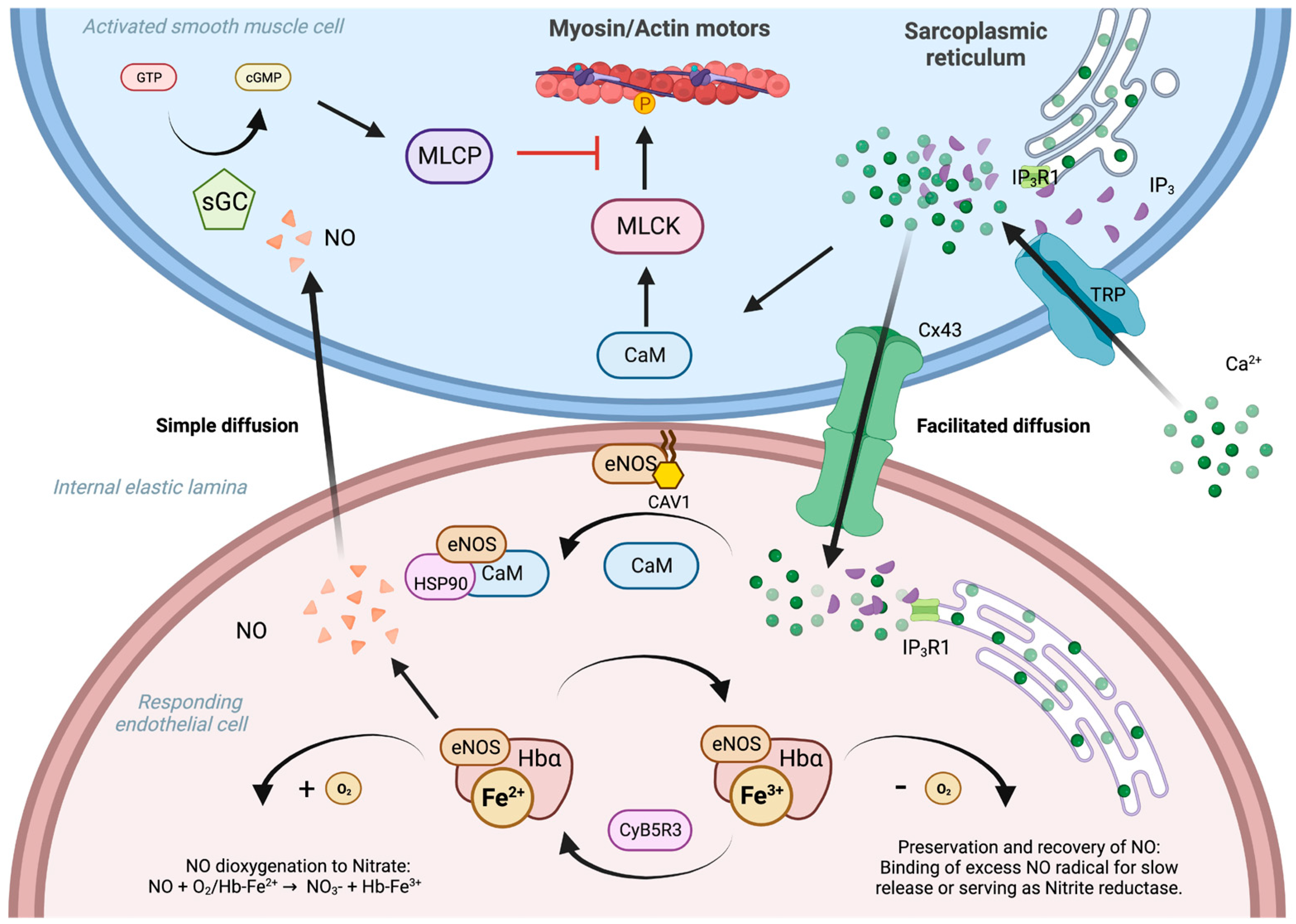
- Straub, A.C.; Zeigler, A.C.; Isakson, B.E. The Myoendothelial Junction: Connections That Deliver the Message. Physiology 2014, 29, 242–249. [Google Scholar] [CrossRef]
- Butcher, J.T.; Johnson, T.; Beers, J.; Columbus, L.; Isakson, B.E. Hemoglobin α in the Blood Vessel Wall. Free Radic. Biol. Med. 2014, 73, 136–142. [Google Scholar] [CrossRef]
- Sharma, A.; Bernatchez, P.N.; De Haan, J.B. Targeting Endothelial Dysfunction in Vascular Complications Associated with Diabetes. Int. J. Vasc. Med. 2012, 2012, 750126. [Google Scholar] [CrossRef]
- Keller, T.C.S.; Lechauve, C.; Keller, A.S.; Broseghini-Filho, G.B.; Butcher, J.T.; Askew Page, H.R.; Islam, A.; Tan, Z.Y.; DeLalio, L.J.; Brooks, S.; et al. Endothelial Alpha Globin Is a Nitrite Reductase. Nat. Commun. 2022, 13, 6405. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, P.; Rengarajan, T.; Thangavel, J.; Nishigaki, Y.; Sakthisekaran, D.; Sethi, G.; Nishigaki, I. The Vascular Endothelium and Human Diseases. Int. J. Biol. Sci. 2013, 9, 1057–1069. [Google Scholar] [CrossRef] [PubMed]
- Intengan, H.D.; Schiffrin, E.L. Structure and Mechanical Properties of Resistance Arteries in Hypertension. Hypertension 2000, 36, 312–318. [Google Scholar] [CrossRef] [PubMed]
- Findlay, J.M.; Nisar, J.; Darsaut, T. Cerebral Vasospasm: A Review. Can. J. Neurol. Sci. 2015, 43, 15–32. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Sánchez, J.; Chánez-Cárdenas, M.E. The Use of Cobalt Chloride as a Chemical Hypoxia Model. J. Appl. Toxicol. 2019, 39, 556–570. [Google Scholar] [CrossRef] [PubMed]
- Fassbender, K.; Hodapp, B.; Rossol, S.; Bertsch, T.; Schmeck, J.; Schütt, S.; Fritzinger, M.; Horn, P.; Vajkoczy, P.; Kreisel, S.; et al. Inflammatory Cytokines in Subarachnoid Haemorrhage: Association with Abnormal Blood Flow Velocities in Basal Cerebral Arteries. J. Neurol. Neurosurg. Psychiatry 2001, 70, 534–537. [Google Scholar] [CrossRef] [PubMed]
- Hanafy, K.A.; Grobelny, B.; Fernandez, L.; Kurtz, P.; Connolly, E.S.; Mayer, S.A.; Schindler, C.; Badjatia, N. Brain Interstitial Fluid TNF-α after Subarachnoid Hemorrhage. J. Neurol. Sci. 2010, 291, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Janaszak-Jasiecka, A.; Siekierzycka, A.; Bartoszewska, S.; Serocki, M.; Dobrucki, L.W.; Collawn, J.F.; Kalinowski, L.; Bartoszewski, R. ENOS Expression and NO Release during Hypoxia Is Inhibited by MiR-200b in Human Endothelial Cells. Angiogenesis 2018, 21, 711–724. [Google Scholar] [CrossRef] [PubMed]
- Min, J.; Jin, Y.M.; Moon, J.S.; Sung, M.S.; Jo, S.A.; Jo, I. Hypoxia-Induced Endothelial NO Synthase Gene Transcriptional Activation Is Mediated through the Tax-Responsive Element in Endothelial Cells. Hypertension 2006, 47, 1189–1196. [Google Scholar] [CrossRef] [PubMed]
- Straub, A.C.; Lohman, A.W.; Billaud, M.; Johnstone, S.R.; Dwyer, S.T.; Lee, M.Y.; Bortz, P.S.; Best, A.K.; Columbus, L.; Gaston, B.; et al. Endothelial Cell Expression of Haemoglobin α Regulates Nitric Oxide Signalling. Nature 2012, 491, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Rahaman, M.M.; Nguyen, A.T.; Miller, M.P.; Hahn, S.A.; Sparacino-Watkins, C.; Jobbagy, S.; Carew, N.T.; Cantu-Medellin, N.; Wood, K.C.; Baty, C.J.; et al. Cytochrome B5 Reductase 3 Modulates Soluble Guanylate Cyclase Redox State and CGMP Signaling. Circ. Res. 2017, 121, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Denton, C.C.; Shah, P.; Suriany, S.; Liu, H.; Thuptimdang, W.; Sunwoo, J.; Chalacheva, P.; Veluswamy, S.; Kato, R.; Wood, J.C.; et al. Loss of Alpha-Globin Genes in Human Subjects Is Associated with Improved Nitric Oxide-Mediated Vascular Perfusion. Am. J. Hematol. 2021, 96, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Keller, T.C.S.; Butcher, J.T.; Broseghini-Filho, G.B.; Marziano, C.; DeLalio, L.J.; Rogers, S.; Ning, B.; Martin, J.N.; Chechova, S.; Cabot, M.; et al. Modulating Vascular Hemodynamics with an Alpha Globin Mimetic Peptide (HbαX). Hypertension 2016, 68, 1494–1503. [Google Scholar] [CrossRef] [PubMed]
- Straub, A.C.; Butcher, J.T.; Billaud, M.; Mutchler, S.M.; Artamonov, M.V.; Nguyen, A.T.; Johnson, T.; Best, A.K.; Miller, M.P.; Palmer, L.A.; et al. Hemoglobin α/ENOS Coupling at Myoendothelial Junctions Is Required for Nitric Oxide Scavenging During Vasoconstriction. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2594–2600. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thomas, G.; Banton, K.L.; Garrett, R.; Palacio, C.H.; Acuna, D.; Madayag, R.; Bar-Or, D. Hypoxia Dysregulates the Transcription of Myoendothelial Junction Proteins Involved with Nitric Oxide Production in Brain Endothelial Cells. Biomedicines 2024, 12, 75. https://doi.org/10.3390/biomedicines12010075
Thomas G, Banton KL, Garrett R, Palacio CH, Acuna D, Madayag R, Bar-Or D. Hypoxia Dysregulates the Transcription of Myoendothelial Junction Proteins Involved with Nitric Oxide Production in Brain Endothelial Cells. Biomedicines. 2024; 12(1):75. https://doi.org/10.3390/biomedicines12010075
Chicago/Turabian StyleThomas, Gregory, Kaysie L. Banton, Raymond Garrett, Carlos H. Palacio, David Acuna, Robert Madayag, and David Bar-Or. 2024. "Hypoxia Dysregulates the Transcription of Myoendothelial Junction Proteins Involved with Nitric Oxide Production in Brain Endothelial Cells" Biomedicines 12, no. 1: 75. https://doi.org/10.3390/biomedicines12010075