
Pharmacological Approaches Using Diabetic Drugs Repurposed for Alzheimer’s Disease

Abstract
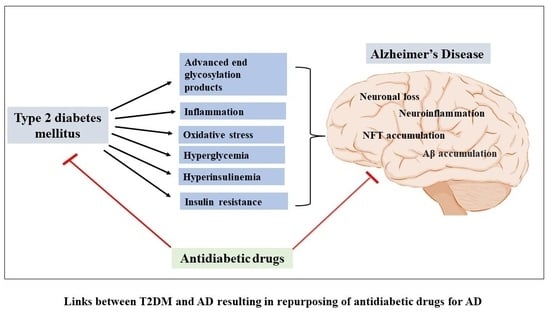
:
1. Introduction
2. Method
Literature Search
3. Discussion
3.1. Metformin
3.2. Insulin
3.3. Incretins
3.4. DPP4 Inhibitors
3.5. PPAR-γ Agonists
3.6. SGLT2 Inhibitors
4. Conclusions

{kind=link}
| Classes of Diabetic Drugs Being Repurposed for AD | Specific Drugs in the Class Being Explored for AD | Summary of Clinical Studies |
|---|---|---|
| metformin |
| |
| Insulin |
| |
| Incretins/GLP1 Receptor Agonists | Liraglutide, semaglutide, exenatide |
|
| DPP4 Inhibitors | Linagliptin, sitagliptin, saxagliptin, vildagliptin |
|
| p-par gamma agonists | Rosiglitazone, piaglitazone |
|
| SGLT2 Inhibitors | Empagliflozin, cangliflozin, dapagliflozin |
|
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Finder, V.H. Alzheimer’s disease: A general introduction and pathomechanism. J. Alzheimers Dis. 2010, 22 (Suppl. S3), 5–19. [Google Scholar] [CrossRef] [PubMed]
- Duyckaerts, C.; Delatour, B.; Potier, M.C. Classification and basic pathology of Alzheimer disease. Acta Neuropathol. 2009, 118, 5–36. [Google Scholar] [CrossRef] [PubMed]
- Fish, P.V.; Steadman, D.; Bayle, E.D.; Whiting, P. New approaches for the treatment of Alzheimer’s disease. Bioorganic. Med. Chem. Lett. 2019, 29, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Hunsberger, H.C.; Pinky, P.D.; Smith, W.; Suppiramaniam, V.; Reed, M.N. The role of APOE4 in Alzheimer’s disease: Strategies for future therapeutic interventions. Neuronal Signal. 2019, 3, NS20180203. [Google Scholar] [CrossRef]
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas. Diabetes Res. Clin. Pract. 2019, 157, 107843. [Google Scholar] [CrossRef] [PubMed]
- Ong, K.L.; Stafford, L.K.; McLaughlin, S.A.; Boyko, E.J.; Vollset, S.E.; Smith, A.E.; Dalton, B.E.; Duprey, J.; Cruz, J.A.; Hagins, H. Global, regional, and national burden of diabetes from 1990 to 2021, with projections of prevalence to 2050: A systematic analysis for the Global Burden of Disease Study 2021. Lancet 2023, 402, 203–234. [Google Scholar] [CrossRef]
- Kahn, S.E.; Cooper, M.E.; Del Prato, S. Pathophysiology and treatment of type 2 diabetes: Perspectives on the past, present, and future. Lancet 2014, 383, 1068–1083. [Google Scholar] [CrossRef]
- Weyer, C.; Bogardus, C.; Mott, D.M.; Pratley, R.E. The natural history of insulin secretory dysfunction and insulin resistance in the pathogenesis of type 2 diabetes mellitus. J. Clin. Investig. 1999, 104, 787–794. [Google Scholar] [CrossRef]
- Stumvoll, M.; Goldstein, B.J.; Van Haeften, T.W. Type 2 diabetes: Principles of pathogenesis and therapy. Lancet 2005, 365, 1333–1346. [Google Scholar] [CrossRef]
- Papatheodorou, K.; Banach, M.; Bekiari, E.; Rizzo, M.; Edmonds, M. Complications of diabetes 2017. J. Diabetes Res. 2018, 2018, 3086167. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Xu, W.; Ou, Y.-N.; Cao, X.-P.; Tan, M.-S.; Tan, L.; Yu, J.-T. Diabetes mellitus and risks of cognitive impairment and dementia: A systematic review and meta-analysis of 144 prospective studies. Ageing Res. Rev. 2019, 55, 100944. [Google Scholar] [CrossRef]
- Luchsinger, J.A.; Reitz, C.; Patel, B.; Tang, M.-X.; Manly, J.J.; Mayeux, R. Relation of diabetes to mild cognitive impairment. Arch. Neurol. 2007, 64, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Biessels, G.J.; Staekenborg, S.; Brunner, E.; Brayne, C.; Scheltens, P. Risk of dementia in diabetes mellitus: A systematic review. Lancet Neurol. 2006, 5, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Peters, S.A.; Woodward, M.; Mejia Arango, S.; Batty, G.D.; Beckett, N.; Beiser, A.; Borenstein, A.R.; Crane, P.K.; Haan, M. Type 2 diabetes as a risk factor for dementia in women compared with men: A pooled analysis of 2.3 million people comprising more than 100,000 cases of dementia. Diabetes Care 2016, 39, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Arvanitakis, Z.; Wilson, R.S.; Bienias, J.L.; Evans, D.A.; Bennett, D.A. Diabetes mellitus and risk of Alzheimer disease and decline in cognitive function. Arch. Neurol. 2004, 61, 661–666. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.-C.; Woung, L.-C.; Tsai, M.-T.; Liu, C.-C.; Su, Y.-H.; Li, C.-Y. Risk of Alzheimer’s disease in relation to diabetes: A population-based cohort study. Neuroepidemiology 2012, 38, 237–244. [Google Scholar] [CrossRef]
- Ott, A.; Stolk, R.; Hofman, A.; van Harskamp, F.; Grobbee, D.; Breteler, M. Association of diabetes mellitus and dementia: The Rotterdam Study. Diabetologia 1996, 39, 1392–1397. [Google Scholar] [CrossRef]
- Janson, J.; Laedtke, T.; Parisi, J.E.; O’Brien, P.; Petersen, R.C.; Butler, P.C. Increased risk of type 2 diabetes in Alzheimer disease. Diabetes 2004, 53, 474–481. [Google Scholar] [CrossRef]
- Lu, F.-P.; Lin, K.-P.; Kuo, H.-K. Diabetes and the risk of multi-system aging phenotypes: A systematic review and meta-analysis. PLoS ONE 2009, 4, e4144. [Google Scholar] [CrossRef]
- Blázquez, E.; Velázquez, E.; Hurtado-Carneiro, V.; Ruiz-Albusac, J.M. Insulin in the brain: Its pathophysiological implications for States related with central insulin resistance, type 2 diabetes and Alzheimer’s disease. Front. Endocrinol. 2014, 5, 161. [Google Scholar] [CrossRef]
- Michailidis, M.; Moraitou, D.; Tata, D.A.; Kalinderi, K.; Papamitsou, T.; Papaliagkas, V. Alzheimer’s disease as type 3 diabetes: Common pathophysiological mechanisms between Alzheimer’s disease and type 2 diabetes. Int. J. Mol. Sci. 2022, 23, 2687. [Google Scholar] [CrossRef] [PubMed]
- De la Monte, S.M.; Wands, J.R. Alzheimer’s disease is type 3 diabetes—Evidence reviewed. J. Diabetes Sci. Technol. 2008, 2, 1101–1113. [Google Scholar] [CrossRef] [PubMed]
- Vitek, M.P.; Bhattacharya, K.; Glendening, J.M.; Stopa, E.; Vlassara, H.; Bucala, R.; Manogue, K.; Cerami, A. Advanced glycation end products contribute to amyloidosis in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1994, 91, 4766–4770. [Google Scholar] [CrossRef] [PubMed]
- An, F.-M.; Chen, S.; Xu, Z.; Yin, L.; Wang, Y.; Liu, A.-R.; Yao, W.-B.; Gao, X.-D. Glucagon-like peptide-1 regulates mitochondrial biogenesis and tau phosphorylation against advanced glycation end product-induced neuronal insult: Studies in vivo and in vitro. Neuroscience 2015, 300, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.-G.; Zhang, W.; Sima, A.A. Alzheimer-like changes in rat models of spontaneous diabetes. Diabetes 2007, 56, 1817–1824. [Google Scholar] [CrossRef] [PubMed]
- Son, S.M.; Song, H.; Byun, J.; Park, K.S.; Jang, H.C.; Park, Y.J.; Mook-Jung, I. Accumulation of autophagosomes contributes to enhanced amyloidogenic APP processing under insulin-resistant conditions. Autophagy 2012, 8, 1842–1844. [Google Scholar] [CrossRef] [PubMed]
- Mehla, J.; Chauhan, B.C.; Chauhan, N.B. Experimental induction of type 2 diabetes in aging-accelerated mice triggered Alzheimer-like pathology and memory deficits. J. Alzheimer’s Dis. 2014, 39, 145–162. [Google Scholar] [CrossRef]
- Clodfelder-Miller, B.J.; Zmijewska, A.A.; Johnson, G.V.; Jope, R.S. Tau is hyperphosphorylated at multiple sites in mouse brain in vivo after streptozotocin-induced insulin deficiency. Diabetes 2006, 55, 3320–3325. [Google Scholar] [CrossRef]
- Planel, E.; Tatebayashi, Y.; Miyasaka, T.; Liu, L.; Wang, L.; Herman, M.; Yu, W.H.; Luchsinger, J.A.; Wadzinski, B.; Duff, K.E. Insulin dysfunction induces in vivo tau hyperphosphorylation through distinct mechanisms. J. Neurosci. 2007, 27, 13635–13648. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, L.; Lu, S.; Wang, D.; Liu, X.-D.; Xie, L.; Wang, G. Impaired amyloid β-degrading enzymes in brain of streptozotocin-induced diabetic rats. J. Endocrinol. Investig. 2011, 34, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Farris, W.; Mansourian, S.; Chang, Y.; Lindsley, L.; Eckman, E.A.; Frosch, M.P.; Eckman, C.B.; Tanzi, R.E.; Selkoe, D.J.; Guénette, S. Insulin-degrading enzyme regulates the levels of insulin, amyloid β-protein, and the β-amyloid precursor protein intracellular domain in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 4162–4167. [Google Scholar] [CrossRef] [PubMed]
- Claxton, A.; Baker, L.D.; Hanson, A.; Trittschuh, E.H.; Cholerton, B.; Morgan, A.; Callaghan, M.; Arbuckle, M.; Behl, C.; Craft, S. Long-acting intranasal insulin detemir improves cognition for adults with mild cognitive impairment or early-stage Alzheimer’s disease dementia. J. Alzheimer’s Dis. 2015, 44, 897–906. [Google Scholar] [CrossRef] [PubMed]
- Batista, A.F.; Forny-Germano, L.; Clarke, J.R.; Lyra e Silva, N.M.; Brito-Moreira, J.; Boehnke, S.E.; Winterborn, A.; Coe, B.C.; Lablans, A.; Vital, J.F. The diabetes drug liraglutide reverses cognitive impairment in mice and attenuates insulin receptor and synaptic pathology in a non-human primate model of Alzheimer’s disease. J. Pathol. 2018, 245, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Bomfim, T.R.; Forny-Germano, L.; Sathler, L.B.; Brito-Moreira, J.; Houzel, J.-C.; Decker, H.; Silverman, M.A.; Kazi, H.; Melo, H.M.; McClean, P.L. An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer’s disease–associated Aβ oligomers. J. Clin. Investig. 2012, 122, 1339–1353. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.L.; Chen, F.Q.; Xu, W.J.; Yue, W.Z.; Yuan, G.; Yang, Y. Early intervention with glucagon-like peptide 1 analog liraglutide prevents tau hyperphosphorylation in diabetic db/db mice. J. Neurochem. 2015, 135, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Association, A.D. 9. Pharmacologic approaches to glycemic treatment: Standards of Medical Care in Diabetes—2021. Diabetes Care 2021, 44, S111–S124. [Google Scholar] [CrossRef] [PubMed]
- Hundal, R.S.; Krssak, M.; Dufour, S.; Laurent, D.; Lebon, V.; Chandramouli, V.; Inzucchi, S.E.; Schumann, W.C.; Petersen, K.F.; Landau, B.R. Mechanism by which metformin reduces glucose production in type 2 diabetes. Diabetes 2000, 49, 2063–2069. [Google Scholar] [CrossRef]
- Shu, Y.; Sheardown, S.A.; Brown, C.; Owen, R.P.; Zhang, S.; Castro, R.A.; Ianculescu, A.G.; Yue, L.; Lo, J.C.; Burchard, E.G. Effect of genetic variation in the organic cation transporter 1 (OCT1) on metformin action. J. Clin. Investig. 2007, 117, 1422–1431. [Google Scholar] [CrossRef]
- El-Mir, M.-Y.; Nogueira, V.; Fontaine, E.; Avéret, N.; Rigoulet, M.; Leverve, X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J. Biol. Chem. 2000, 275, 223–228. [Google Scholar] [CrossRef]
- Owen, M.R.; Doran, E.; Halestrap, A.P. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J. 2000, 348, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Foretz, M.; Hébrard, S.; Leclerc, J.; Zarrinpashneh, E.; Soty, M.; Mithieux, G.; Sakamoto, K.; Andreelli, F.; Viollet, B. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J. Clin. Investig. 2010, 120, 2355–2369. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G. AMP-activated/SNF1 protein kinases: Conserved guardians of cellular energy. Nat. Rev. Mol. Cell Biol. 2007, 8, 774–785. [Google Scholar] [CrossRef] [PubMed]
- Stephenne, X.; Foretz, M.; Taleux, N.; Van Der Zon, G.; Sokal, E.; Hue, L.; Viollet, B.; Guigas, B. Metformin activates AMP-activated protein kinase in primary human hepatocytes by decreasing cellular energy status. Diabetologia 2011, 54, 3101–3110. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, W.H.; Braga, C.F.; Lós, D.B.; Araújo, S.M.R.; França, M.R.; Duarte-Silva, E.; Rodrigues, G.B.; Rocha, S.W.S.; Peixoto, C.A. Metformin prevents p-tau and amyloid plaque deposition and memory impairment in diabetic mice. Exp. Brain Res. 2021, 239, 2821–2839. [Google Scholar] [CrossRef]
- Xu, X.; Sun, Y.; Cen, X.; Shan, B.; Zhao, Q.; Xie, T.; Wang, Z.; Hou, T.; Xue, Y.; Zhang, M. Metformin activates chaperone-mediated autophagy and improves disease pathologies in an Alzheimer disease mouse model. Protein Cell 2021, 12, 769–787. [Google Scholar] [CrossRef]
- Ou, Z.; Kong, X.; Sun, X.; He, X.; Zhang, L.; Gong, Z.; Huang, J.; Xu, B.; Long, D.; Li, J. Metformin treatment prevents amyloid plaque deposition and memory impairment in APP/PS1 mice. Brain Behav. Immun. 2018, 69, 351–363. [Google Scholar] [CrossRef]
- Chen, Y.; Zhao, S.; Fan, Z.; Li, Z.; Zhu, Y.; Shen, T.; Li, K.; Yan, Y.; Tian, J.; Liu, Z. Metformin attenuates plaque-associated tau pathology and reduces amyloid-β burden in APP/PS1 mice. Alzheimer’s Res. Ther. 2021, 13, 40. [Google Scholar] [CrossRef]
- Ng, T.P.; Feng, L.; Yap, K.B.; Lee, T.S.; Tan, C.H.; Winblad, B. Long-term metformin usage and cognitive function among older adults with diabetes. J. Alzheimer’s Dis. 2014, 41, 61–68. [Google Scholar] [CrossRef]
- Sluggett, J.K.; Koponen, M.; Bell, J.S.; Taipale, H.; Tanskanen, A.; Tiihonen, J.; Uusitupa, M.; Tolppanen, A.-M.; Hartikainen, S. Metformin and risk of Alzheimer’s disease among community-dwelling people with diabetes: A national case-control study. J. Clin. Endocrinol. Metab. 2020, 105, e963–e972. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Xu, M.; Walker, V.; Yuan, J.; Korologou-Linden, R.; Robinson, J.; Huang, P.; Burgess, S.; Au Yeung, S.L.; Luo, S. Evaluating the efficacy and mechanism of metformin targets on reducing Alzheimer’s disease risk in the general population: A Mendelian randomisation study. Diabetologia 2022, 65, 1664–1675. [Google Scholar] [CrossRef] [PubMed]
- Luchsinger, J.A.; Perez, T.; Chang, H.; Mehta, P.; Steffener, J.; Pradabhan, G.; Ichise, M.; Manly, J.; Devanand, D.P.; Bagiella, E. Metformin in amnestic mild cognitive impairment: Results of a pilot randomized placebo controlled clinical trial. J. Alzheimer’s Dis. 2016, 51, 501–514. [Google Scholar] [CrossRef] [PubMed]
- Luo, A.; Ning, P.; Lu, H.; Huang, H.; Shen, Q.; Zhang, D.; Xu, F.; Yang, L.; Xu, Y. Association between metformin and Alzheimer’s disease: A systematic review and meta-analysis of clinical observational studies. J. Alzheimer’s Dis. 2022, 88, 1311–1323. [Google Scholar] [CrossRef]
- De Meyts, P. The insulin receptor and its signal transduction network. In Endotext [Internet]; MDText.com, Inc.: South Dartmouth, MA, USA, 2016. [Google Scholar]
- Pessin, J.E.; Saltiel, A.R. Signaling pathways in insulin action: Molecular targets of insulin resistance. J. Clin. Investig. 2000, 106, 165–169. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef]
- Tzatsos, A. Raptor binds the SAIN (Shc and IRS-1 NPXY binding) domain of insulin receptor substrate-1 (IRS-1) and regulates the phosphorylation of IRS-1 at Ser-636/639 by mTOR. J. Biol. Chem. 2009, 284, 22525–22534. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, F. Tissue-specific insulin signaling in the regulation of metabolism and aging. IUBMB Life 2014, 66, 485–495. [Google Scholar] [CrossRef]
- Havrankova, J.; Roth, J.; Brownstein, M. Insulin receptors are widely distributed in the central nervous system of the rat. Nature 1978, 272, 827–829. [Google Scholar] [CrossRef]
- Unger, J.; McNeill, T.; Moxley, R., III; White, M.; Moss, A.; Livingston, J. Distribution of insulin receptor-like immunoreactivity in the rat forebrain. Neuroscience 1989, 31, 143–157. [Google Scholar] [CrossRef]
- Havrankova, J.; Schmechel, D.; Roth, J.; Brownstein, M. Identification of insulin in rat brain. Proc. Natl. Acad. Sci. USA 1978, 75, 5737–5741. [Google Scholar] [CrossRef] [PubMed]
- Havrankova, J.; Roth, J. Concentrations of insulin and of insulin receptors in the brain are independent of peripheral insulin levels: Studies of obese and streptozotocin-treated rodents. J. Clin. Investig. 1979, 64, 636–642. [Google Scholar] [CrossRef] [PubMed]
- Reitz, C.; Tosto, G.; Mayeux, R.; Luchsinger, J.A.; NIA-LOAD/NCRAD Family Study Group; Alzheimer’s Disease Neuroimaging Initiative. Genetic variants in the Fat and Obesity Associated (FTO) gene and risk of Alzheimer’s disease. PLoS ONE 2012, 7, e50354. [Google Scholar] [CrossRef]
- Keller, L.; Xu, W.; Wang, H.-X.; Winblad, B.; Fratiglioni, L.; Graff, C. The obesity related gene, FTO, interacts with APOE, and is associated with Alzheimer’s disease risk: A prospective cohort study. J. Alzheimer’s Dis. 2011, 23, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Nakano, H. Signaling crosstalk between NF-κB and JNK. Trends Immunol. 2004, 25, 402–405. [Google Scholar] [CrossRef] [PubMed]
- Sears, B.; Perry, M. The role of fatty acids in insulin resistance. Lipids Health Dis. 2015, 14, 121. [Google Scholar] [CrossRef]
- Craft, S.; Watson, G.S. Insulin and neurodegenerative disease: Shared and specific mechanisms. Lancet Neurol. 2004, 3, 169–178. [Google Scholar] [CrossRef]
- Benedict, C.; Brooks, S.J.; Kullberg, J.; Burgos, J.; Kempton, M.J.; Nordenskjöld, R.; Nylander, R.; Kilander, L.; Craft, S.; Larsson, E.-M. Impaired insulin sensitivity as indexed by the HOMA score is associated with deficits in verbal fluency and temporal lobe gray matter volume in the elderly. Diabetes Care 2012, 35, 488–494. [Google Scholar] [CrossRef]
- Steen, E.; Terry, B.M.; J Rivera, E.; Cannon, J.L.; Neely, T.R.; Tavares, R.; Xu, X.J.; Wands, J.R.; de La Monte, S.M. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease–is this type 3 diabetes? J. Alzheimer’s Dis. 2005, 7, 63–80. [Google Scholar] [CrossRef]
- Rivera, E.J.; Goldin, A.; Fulmer, N.; Tavares, R.; Wands, J.R.; de la Monte, S.M. Insulin and insulin-like growth factor expression and function deteriorate with progression of Alzheimer’s disease: Link to brain reductions in acetylcholine. J. Alzheimer’s Dis. 2005, 8, 247–268. [Google Scholar] [CrossRef]
- Starks, E.J.; Patrick O’Grady, J.; Hoscheidt, S.M.; Racine, A.M.; Carlsson, C.M.; Zetterberg, H.; Blennow, K.; Okonkwo, O.C.; Puglielli, L.; Asthana, S. Insulin resistance is associated with higher cerebrospinal fluid tau levels in asymptomatic APOE ε4 carriers. J. Alzheimer’s Dis. 2015, 46, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Freiherr, J.; Hallschmid, M.; Frey, W.H.; Brünner, Y.F.; Chapman, C.D.; Hölscher, C.; Craft, S.; De Felice, F.G.; Benedict, C. Intranasal insulin as a treatment for Alzheimer’s disease: A review of basic research and clinical evidence. CNS Drugs 2013, 27, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhang, J.; Zhang, B.; Gong, C.-X. Targeting insulin signaling for the treatment of Alzheimer’s disease. Curr. Top. Med. Chem. 2016, 16, 485–492. [Google Scholar] [CrossRef] [PubMed]
- Ho, L.; Qin, W.; Pompl, P.N.; Xiang, Z.; Wang, J.; Zhao, Z.; Peng, Y.; Cambareri, G.; Rocher, A.; Mobbs, C.V. Diet-induced insulin resistance promotes amyloidosis in a transgenic mouse model of Alzheimer’s disease. FASEB J. 2004, 18, 902–904. [Google Scholar] [CrossRef] [PubMed]
- Willette, A.A.; Johnson, S.C.; Birdsill, A.C.; Sager, M.A.; Christian, B.; Baker, L.D.; Craft, S.; Oh, J.; Statz, E.; Hermann, B.P. Insulin resistance predicts brain amyloid deposition in late middle-aged adults. Alzheimer’s Dement. 2015, 11, 504–510.e1. [Google Scholar] [CrossRef] [PubMed]
- Lochhead, J.J.; Wolak, D.J.; Pizzo, M.E.; Thorne, R.G. Rapid transport within cerebral perivascular spaces underlies widespread tracer distribution in the brain after intranasal administration. J. Cereb. Blood Flow Metab. 2015, 35, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Reger, M.A.; Watson, G.S.; Green, P.S.; Wilkinson, C.W.; Baker, L.D.; Cholerton, B.; Fishel, M.A.; Plymate, S.; Breitner, J.; DeGroodt, W. Intranasal insulin improves cognition and modulates β-amyloid in early AD. Neurology 2008, 70, 440–448. [Google Scholar] [CrossRef] [PubMed]
- Reger, M.A.; Watson, G.; Green, P.S.; Baker, L.D.; Cholerton, B.; Fishel, M.A.; Plymate, S.R.; Cherrier, M.M.; Schellenberg, G.D.; Frey Ii, W.H. Intranasal insulin administration dose-dependently modulates verbal memory and plasma amyloid-β in memory-impaired older adults. J. Alzheimer’s Dis. 2008, 13, 323–331. [Google Scholar] [CrossRef]
- Craft, S.; Baker, L.D.; Montine, T.J.; Minoshima, S.; Watson, G.S.; Claxton, A.; Arbuckle, M.; Callaghan, M.; Tsai, E.; Plymate, S.R. Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: A pilot clinical trial. Arch. Neurol. 2012, 69, 29–38. [Google Scholar] [CrossRef]
- Craft, S.; Claxton, A.; Baker, L.D.; Hanson, A.J.; Cholerton, B.; Trittschuh, E.H.; Dahl, D.; Caulder, E.; Neth, B.; Montine, T.J. Effects of regular and long-acting insulin on cognition and Alzheimer’s disease biomarkers: A pilot clinical trial. J. Alzheimer’s Dis. 2017, 57, 1325–1334. [Google Scholar] [CrossRef]
- Kellar, D.; Lockhart, S.; Aisen, P.; Raman, R.; Rissman, R.; Brewer, J.; Craft, S. Intranasal insulin reduces white matter hyperintensity progression in association with improvements in cognition and CSF biomarker profiles in mild cognitive impairment and Alzheimer’s disease. J. Prev. Alzheimer’s Dis. 2021, 8, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Craft, S.; Raman, R.; Chow, T.W.; Rafii, M.S.; Sun, C.-K.; Rissman, R.A.; Donohue, M.C.; Brewer, J.B.; Jenkins, C.; Harless, K. Safety, efficacy, and feasibility of intranasal insulin for the treatment of mild cognitive impairment and Alzheimer disease dementia: A randomized clinical trial. JAMA Neurol. 2020, 77, 1099–1109. [Google Scholar] [CrossRef] [PubMed]
- Bayliss, W.M.; Starling, E.H. The mechanism of pancreatic secretion. J. Physiol. 1902, 28, 325. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.; Mutt, V.; Pederson, R. Further purification of a polypeptide demonstrating enterogastrone activity. J. Physiol. 1970, 209, 57. [Google Scholar] [CrossRef] [PubMed]
- Gutniak, M.; Ørkov, C.; Holst, J.J.; Ahrén, B.; Efendić, S. Antidiabetogenic effect of glucagon-like peptide-1 (7–36) amide in normal subjects and patients with diabetes mellitus. N. Engl. J. Med. 1992, 326, 1316–1322. [Google Scholar] [CrossRef] [PubMed]
- Doyle, M.E.; Egan, J.M. Mechanisms of action of glucagon-like peptide 1 in the pancreas. Pharmacol. Ther. 2007, 113, 546–593. [Google Scholar] [CrossRef] [PubMed]
- Calsolaro, V.; Edison, P. Novel GLP-1 (glucagon-like peptide-1) analogues and insulin in the treatment for Alzheimer’s disease and other neurodegenerative diseases. CNS Drugs 2015, 29, 1023–1039. [Google Scholar] [CrossRef]
- Salcedo, I.; Tweedie, D.; Li, Y.; Greig, N.H. Neuroprotective and neurotrophic actions of glucagon-like peptide-1: An emerging opportunity to treat neurodegenerative and cerebrovascular disorders. Br. J. Pharmacol. 2012, 166, 1586–1599. [Google Scholar] [CrossRef]
- Ji, C.; Xue, G.-F.; Li, G.; Li, D.; Hölscher, C. Neuroprotective effects of glucose-dependent insulinotropic polypeptide in Alzheimer’s disease. Rev. Neurosci. 2016, 27, 61–70. [Google Scholar] [CrossRef]
- Qin, Z.; Sun, Z.; Huang, J.; Hu, Y.; Wu, Z.; Mei, B. Mutated recombinant human glucagon-like peptide-1 protects SH-SY5Y cells from apoptosis induced by amyloid-β peptide (1–42). Neurosci. Lett. 2008, 444, 217–221. [Google Scholar] [CrossRef]
- Perry, T.; Lahiri, D.K.; Sambamurti, K.; Chen, D.; Mattson, M.P.; Egan, J.M.; Greig, N.H. Glucagon-like peptide-1 decreases endogenous amyloid-β peptide (Aβ) levels and protects hippocampal neurons from death induced by Aβ and iron. J. Neurosci. Res. 2003, 72, 603–612. [Google Scholar] [CrossRef] [PubMed]
- McClean, P.L.; Parthsarathy, V.; Faivre, E.; Hölscher, C. The diabetes drug liraglutide prevents degenerative processes in a mouse model of Alzheimer’s disease. J. Neurosci. 2011, 31, 6587–6594. [Google Scholar] [CrossRef] [PubMed]
- Seshadri, S.; Beiser, A.; Selhub, J.; Jacques, P.F.; Rosenberg, I.H.; D’Agostino, R.B.; Wilson, P.W.; Wolf, P.A. Plasma homocysteine as a risk factor for dementia and Alzheimer’s disease. N. Engl. J. Med. 2002, 346, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Liu, Y.-H.; Zhang, C.-E.; Wang, Q.; Wei, Z.; Mousseau, D.D.; Wang, J.-Z.; Tian, Q.; Liu, G.-P. Folate/vitamin-B12 prevents chronic hyperhomocysteinemia-induced tau hyperphosphorylation and memory deficits in aged rats. J. Alzheimer’s Dis. 2011, 27, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xie, J.-Z.; Xu, X.-Y.; Hu, J.; Xu, T.; Jin, S.; Yang, S.-J.; Wang, J.-Z. Liraglutide ameliorates hyperhomocysteinemia-induced Alzheimer-like pathology and memory deficits in rats via multi-molecular targeting. Neurosci. Bull. 2019, 35, 724–734. [Google Scholar] [CrossRef]
- Qi, L.; Ke, L.; Liu, X.; Liao, L.; Ke, S.; Liu, X.; Wang, Y.; Lin, X.; Zhou, Y.; Wu, L. Subcutaneous administration of liraglutide ameliorates learning and memory impairment by modulating tau hyperphosphorylation via the glycogen synthase kinase-3β pathway in an amyloid β protein induced alzheimer disease mouse model. Eur. J. Pharmacol. 2016, 783, 23–32. [Google Scholar] [CrossRef]
- Wiciński, M.; Socha, M.; Malinowski, B.; Wódkiewicz, E.; Walczak, M.; Górski, K.; Słupski, M.; Pawlak-Osińska, K. Liraglutide and its Neuroprotective properties—Focus on possible biochemical mechanisms in Alzheimer’s disease and cerebral ischemic events. Int. J. Mol. Sci. 2019, 20, 1050. [Google Scholar] [CrossRef]
- Duarte, A.I.; Candeias, E.; Alves, I.N.; Mena, D.; Silva, D.F.; Machado, N.J.; Campos, E.J.; Santos, M.S.; Oliveira, C.R.; Moreira, P.I. Liraglutide protects against brain amyloid-β1–42 accumulation in female mice with early Alzheimer’s disease-like pathology by partially rescuing oxidative/nitrosative stress and inflammation. Int. J. Mol. Sci. 2020, 21, 1746. [Google Scholar] [CrossRef]
- Carranza-Naval, M.J.; Del Marco, A.; Hierro-Bujalance, C.; Alves-Martinez, P.; Infante-Garcia, C.; Vargas-Soria, M.; Herrera, M.; Barba-Cordoba, B.; Atienza-Navarro, I.; Lubian-Lopez, S. Liraglutide reduces vascular damage, neuronal loss, and cognitive impairment in a mixed murine model of Alzheimer’s disease and type 2 diabetes. Front. Aging Neurosci. 2021, 13, 741923. [Google Scholar] [CrossRef]
- Chen, S.; Sun, J.; Zhao, G.; Guo, A.; Chen, Y.; Fu, R.; Deng, Y. Liraglutide improves water maze learning and memory performance while reduces hyperphosphorylation of tau and neurofilaments in APP/PS1/Tau triple transgenic mice. Neurochem. Res. 2017, 42, 2326–2335. [Google Scholar] [CrossRef]
- McClean, P.L.; Jalewa, J.; Hölscher, C. Prophylactic liraglutide treatment prevents amyloid plaque deposition, chronic inflammation and memory impairment in APP/PS1 mice. Behav. Brain Res. 2015, 293, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Hansen, H.H.; Fabricius, K.; Barkholt, P.; Niehoff, M.L.; Morley, J.E.; Jelsing, J.; Pyke, C.; Knudsen, L.B.; Farr, S.A.; Vrang, N. The GLP-1 receptor agonist liraglutide improves memory function and increases hippocampal CA1 neuronal numbers in a senescence-accelerated mouse model of Alzheimer’s disease. J. Alzheimer’s Dis. 2015, 46, 877–888. [Google Scholar] [CrossRef] [PubMed]
- Edison, P.; Femminella, G.D.; Ritchie, C.W.; Holmes, C.; Walker, Z.; Ridha, B.H.; Raza, S.; Livingston, N.R.; Nowell, J.; Busza, G. Evaluation of liraglutide in the treatment of Alzheimer’s disease. Alzheimer’s Dement. 2021, 17, e057848. [Google Scholar] [CrossRef]
- Chang, Y.-F.; Zhang, D.; Hu, W.-M.; Liu, D.-X.; Li, L. Semaglutide-mediated protection against Aβ correlated with enhancement of autophagy and inhibition of apotosis. J. Clin. Neurosci. 2020, 81, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Atri, A.; Feldman, H.H.; Hansen, C.T.; Honore, J.B.; Johannsen, P.; Knop, F.K.; Poulsen, P.; Raket, L.L.; Sano, M.; Soininen, H. evoke and evoke+: Design of two large-scale, double-blind, placebo-controlled, phase 3 studies evaluating the neuroprotective effects of semaglutide in early Alzheimer’s disease. Alzheimer’s Dement. 2022, 18, e062415. [Google Scholar] [CrossRef]
- Song, X.; Sun, Y.; Wang, Z.; Su, Y.; Wang, Y.; Wang, X. Exendin-4 alleviates β-Amyloid peptide toxicity via DAF-16 in a Caenorhabditis elegans model of Alzheimer’s disease. Front. Aging Neurosci. 2022, 14, 955113. [Google Scholar] [CrossRef] [PubMed]
- Rachmany, L.; Tweedie, D.; Li, Y.; Rubovitch, V.; Holloway, H.W.; Miller, J.; Hoffer, B.J.; Greig, N.H.; Pick, C.G. Exendin-4 induced glucagon-like peptide-1 receptor activation reverses behavioral impairments of mild traumatic brain injury in mice. Age 2013, 35, 1621–1636. [Google Scholar] [CrossRef] [PubMed]
- Zago, A.M.; Carvalho, F.B.; Rahmeier, F.L.; Santin, M.; Guimarães, G.R.; Gutierres, J.M.; Fernandes, M.d.C. Exendin-4 Prevents Memory Loss and Neuronal Death in Rats with Sporadic Alzheimer-Like Disease. Mol. Neurobiol. 2023. Online ahead of print. [Google Scholar] [CrossRef]
- Mullins, R.J.; Mustapic, M.; Chia, C.W.; Carlson, O.; Gulyani, S.; Tran, J.; Li, Y.; Mattson, M.P.; Resnick, S.; Egan, J.M. A pilot study of exenatide actions in Alzheimer’s disease. Curr. Alzheimer Res. 2019, 16, 741–752. [Google Scholar] [CrossRef]
- Gerstein, H.C.; Colhoun, H.M.; Dagenais, G.R.; Diaz, R.; Lakshmanan, M.; Pais, P.; Probstfield, J.; Riesmeyer, J.S.; Riddle, M.C.; Rydén, L. Dulaglutide and cardiovascular outcomes in type 2 diabetes (REWIND): A double-blind, randomised placebo-controlled trial. Lancet 2019, 394, 121–130. [Google Scholar] [CrossRef]
- Cukierman-Yaffe, T.; Gerstein, H.C.; Colhoun, H.M.; Diaz, R.; García-Pérez, L.-E.; Lakshmanan, M.; Bethel, A.; Xavier, D.; Probstfield, J.; Riddle, M.C. Effect of dulaglutide on cognitive impairment in type 2 diabetes: An exploratory analysis of the REWIND trial. Lancet Neurol. 2020, 19, 582–590. [Google Scholar] [CrossRef] [PubMed]
- Mulvihill, E.E.; Drucker, D.J. Pharmacology, physiology, and mechanisms of action of dipeptidyl peptidase-4 inhibitors. Endocr. Rev. 2014, 35, 992–1019. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, N.; Ali, J.; Parvez, S.; Zameer, S.; Najmi, A.K.; Akhtar, M. Linagliptin, a DPP-4 inhibitor, ameliorates Aβ (1−42) peptides induced neurodegeneration and brain insulin resistance (BIR) via insulin receptor substrate-1 (IRS-1) in rat model of Alzheimer’s disease. Neuropharmacology 2021, 195, 108662. [Google Scholar] [CrossRef] [PubMed]
- Kosaraju, J.; Holsinger, R.D.; Guo, L.; Tam, K.Y. Linagliptin, a dipeptidyl peptidase-4 inhibitor, mitigates cognitive deficits and pathology in the 3xTg-AD mouse model of Alzheimer’s disease. Mol. Neurobiol. 2017, 54, 6074–6084. [Google Scholar] [CrossRef] [PubMed]
- Hooper, C.; Killick, R.; Lovestone, S. The GSK3 hypothesis of Alzheimer’s disease. J. Neurochem. 2008, 104, 1433–1439. [Google Scholar] [CrossRef] [PubMed]
- Kornelius, E.; Lin, C.L.; Chang, H.H.; Li, H.H.; Huang, W.N.; Yang, Y.S.; Lu, Y.L.; Peng, C.H.; Huang, C.N. DPP-4 inhibitor linagliptin attenuates Aβ-induced cytotoxicity through activation of AMPK in neuronal cells. CNS Neurosci. Ther. 2015, 21, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-J.; Guo, X.; Li, C.-J.; Li, D.-Q.; Zhang, J.; Yang, Y.; Kong, Y.; Guo, H.; Liu, D.-M.; Chen, L.-M. Dipeptidyl peptidase-4 inhibitor, vildagliptin, inhibits pancreatic beta cell apoptosis in association with its effects suppressing endoplasmic reticulum stress in db/db mice. Metabolism 2015, 64, 226–235. [Google Scholar] [CrossRef]
- Li, Y.; Guo, Y.; Tang, J.; Jiang, J.; Chen, Z. New insights into the roles of CHOP-induced apoptosis in ER stress. Acta Biochim. Biophys. Sin. 2014, 46, 629–640. [Google Scholar] [CrossRef]
- d’Amico, M.; Di Filippo, C.; Marfella, R.; Abbatecola, A.M.; Ferraraccio, F.; Rossi, F.; Paolisso, G. Long-term inhibition of dipeptidyl peptidase-4 in Alzheimer’s prone mice. Exp. Gerontol. 2010, 45, 202–207. [Google Scholar] [CrossRef]
- Kosaraju, J.; Gali, C.C.; Khatwal, R.B.; Dubala, A.; Chinni, S.; Holsinger, R.D.; Madhunapantula, V.S.R.; Nataraj, S.K.M.; Basavan, D. Saxagliptin: A dipeptidyl peptidase-4 inhibitor ameliorates streptozotocin induced Alzheimer’s disease. Neuropharmacology 2013, 72, 291–300. [Google Scholar] [CrossRef]
- Kosaraju, J.; Murthy, V.; Khatwal, R.B.; Dubala, A.; Chinni, S.; Muthureddy Nataraj, S.K.; Basavan, D. Vildagliptin: An anti-diabetes agent ameliorates cognitive deficits and pathology observed in streptozotocin-induced Alzheimer’s disease. J. Pharm. Pharmacol. 2013, 65, 1773–1784. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, M.R.; Barbieri, M.; Boccardi, V.; Angellotti, E.; Marfella, R.; Paolisso, G. Dipeptidyl peptidase-4 inhibitors have protective effect on cognitive impairment in aged diabetic patients with mild cognitive impairment. J. Gerontol. Ser. A Biomed. Sci. Med. Sci. 2014, 69, 1122–1131. [Google Scholar] [CrossRef] [PubMed]
- Isik, A.T.; Soysal, P.; Yay, A.; Usarel, C. The effects of sitagliptin, a DPP-4 inhibitor, on cognitive functions in elderly diabetic patients with or without Alzheimer’s disease. Diabetes Res. Clin. Pract. 2017, 123, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Tontonoz, P.; Spiegelman, B.M. Fat and beyond: The diverse biology of PPARγ. Annu. Rev. Biochem. 2008, 77, 289–312. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, Y.; Shimohama, S.; Koike, H.; Kakimura, J.-I.; Matsuoka, Y.; Nomura, Y.; Gebicke-Haerter, P.J.; Taniguchi, T. Increased expression of cyclooxygenases and peroxisome proliferator-activated receptor-γ in Alzheimer’s disease brains. Biochem. Biophys. Res. Commun. 1999, 254, 582–586. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.L.; Pee, H.N.; Yang, S.; Ho, P.C. Influence of drug transporters and stereoselectivity on the brain penetration of pioglitazone as a potential medicine against Alzheimer’s disease. Sci. Rep. 2015, 5, 9000. [Google Scholar] [CrossRef]
- Moosecker, S.; Gomes, P.; Dioli, C.; Yu, S.; Sotiropoulos, I.; Almeida, O.F. Activated PPARγ abrogates misprocessing of amyloid precursor protein, tau missorting and synaptotoxicity. Front. Cell. Neurosci. 2019, 13, 239. [Google Scholar] [CrossRef]
- Seok, H.; Lee, M.; Shin, E.; Yun, M.R.; Lee, Y.-H.; Moon, J.H.; Kim, E.; Lee, P.H.; Lee, B.-W.; Kang, E.S. Low-dose pioglitazone can ameliorate learning and memory impairment in a mouse model of dementia by increasing LRP1 expression in the hippocampus. Sci. Rep. 2019, 9, 4414. [Google Scholar] [CrossRef]
- Dhavan, R.; Tsai, L.-H. A decade of CDK5. Nat. Rev. Mol. Cell Biol. 2001, 2, 749–759. [Google Scholar] [CrossRef]
- Cheung, Z.H.; Ip, N.Y. Cdk5: A multifaceted kinase in neurodegenerative diseases. Trends Cell Biol. 2012, 22, 169–175. [Google Scholar] [CrossRef]
- Quan, Q.; Qian, Y.; Li, X.; Li, M. Pioglitazone reduces β amyloid levels via inhibition of PPARγ phosphorylation in a neuronal model of Alzheimer’s disease. Front. Aging Neurosci. 2019, 11, 178. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, S.; Sun, W.; Li, J. Anti-diabetes drug pioglitazone ameliorates synaptic defects in AD transgenic mice by inhibiting cyclin-dependent kinase5 activity. PLoS ONE 2015, 10, e0123864. [Google Scholar] [CrossRef] [PubMed]
- Toba, J.; Nikkuni, M.; Ishizeki, M.; Yoshii, A.; Watamura, N.; Inoue, T.; Ohshima, T. PPARγ agonist pioglitazone improves cerebellar dysfunction at pre-Aβ deposition stage in APPswe/PS1dE9 Alzheimer’s disease model mice. Biochem. Biophys. Res. Commun. 2016, 473, 1039–1044. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Chen, Z.; Cao, M.; Li, R.; Wang, Z.; Zhang, M. Pioglitazone ameliorates Aβ42 deposition in rats with diet-induced insulin resistance associated with AKT/GSK3β activation. Mol. Med. Rep. 2017, 15, 2588–2594. [Google Scholar] [CrossRef] [PubMed]
- Kunze, L.H.; Ruch, F.; Biechele, G.; Eckenweber, F.; Wind-Mark, K.; Dinkel, L.; Feyen, P.; Bartenstein, P.; Ziegler, S.; Paeger, L. Long-Term Pioglitazone Treatment Has No Significant Impact on Microglial Activation and Tau Pathology in P301S Mice. Int. J. Mol. Sci. 2023, 24, 10106. [Google Scholar] [CrossRef] [PubMed]
- Hanyu, H.; Sato, T.; Kiuchi, A.; Sakurai, H.; Iwamoto, T. Pioglitazone improved cognition in a pilot study on patients with Alzheimer’s disease and mild cognitive impairment with diabetes mellitus. J. Am. Geriatr. Soc. 2009, 57, 177–179. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Hanyu, H.; Hirao, K.; Kanetaka, H.; Sakurai, H.; Iwamoto, T. Efficacy of PPAR-γ agonist pioglitazone in mild Alzheimer disease. Neurobiol. Aging 2011, 32, 1626–1633. [Google Scholar] [CrossRef]
- Escribano, L.; Simón, A.-M.; Pérez-Mediavilla, A.; Salazar-Colocho, P.; Del Río, J.; Frechilla, D. Rosiglitazone reverses memory decline and hippocampal glucocorticoid receptor down-regulation in an Alzheimer’s disease mouse model. Biochem. Biophys. Res. Commun. 2009, 379, 406–410. [Google Scholar] [CrossRef]
- Escribano, L.; Simón, A.-M.; Gimeno, E.; Cuadrado-Tejedor, M.; Lopez de Maturana, R.; García-Osta, A.; Ricobaraza, A.; Pérez-Mediavilla, A.; Del Río, J.; Frechilla, D. Rosiglitazone rescues memory impairment in Alzheimer’s transgenic mice: Mechanisms involving a reduced amyloid and tau pathology. Neuropsychopharmacology 2010, 35, 1593–1604. [Google Scholar] [CrossRef]
- Jahrling, J.B.; Hernandez, C.M.; Denner, L.; Dineley, K.T. PPARγ recruitment to active ERK during memory consolidation is required for Alzheimer’s disease-related cognitive enhancement. J. Neurosci. 2014, 34, 4054–4063. [Google Scholar] [CrossRef]
- Watson, G.S.; Cholerton, B.A.; Reger, M.A.; Baker, L.D.; Plymate, S.R.; Asthana, S.; Fishel, M.A.; Kulstad, J.J.; Green, P.S.; Cook, D.G. Preserved cognition in patients with early Alzheimer disease and amnestic mild cognitive impairment during treatment with rosiglitazone: A preliminary study. Am. J. Geriatr. Psychiatry 2005, 13, 950–958. [Google Scholar] [CrossRef] [PubMed]
- Risner, M.; Saunders, A.; Altman, J.; Ormandy, G.; Craft, S.; Foley, I.; Zvartau-Hind, M.; Hosford, D.; Roses, A. Efficacy of rosiglitazone in a genetically defined population with mild-to-moderate Alzheimer’s disease. Pharm. J. 2006, 6, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Abbatecola, A.M.; Lattanzio, F.; Molinari, A.M.; Cioffi, M.; Mansi, L.; Rambaldi, P.; DiCioccio, L.; Cacciapuoti, F.; Canonico, R.; Paolisso, G. Rosiglitazone and cognitive stability in older individuals with type 2 diabetes and mild cognitive impairment. Diabetes Care 2010, 33, 1706–1711. [Google Scholar] [CrossRef] [PubMed]
- Gold, M.; Alderton, C.; Zvartau-Hind, M.; Egginton, S.; Saunders, A.M.; Irizarry, M.; Craft, S.; Landreth, G.; Linnamägi, Ü.; Sawchak, S. Rosiglitazone monotherapy in mild-to-moderate Alzheimer’s disease: Results from a randomized, double-blind, placebo-controlled phase III study. Dement. Geriatr. Cogn. Disord. 2010, 30, 131–146. [Google Scholar] [CrossRef] [PubMed]
- Maeshiba, Y.; Kiyota, Y.; Yamashita, K.; Yoshimura, Y.; Motohashi, M.; Tanayama, S. Disposition of the new antidiabetic agent pioglitazone in rats, dogs, and monkeys. Arzneimittelforschung 1997, 47, 29–34. [Google Scholar]
- Cheung, B.M. Behind the rosiglitazone controversy. Expert Rev. Clin. Pharmacol. 2010, 3, 723–725. [Google Scholar] [CrossRef]
- Cassis, P.; Locatelli, M.; Cerullo, D.; Corna, D.; Buelli, S.; Zanchi, C.; Villa, S.; Morigi, M.; Remuzzi, G.; Benigni, A. SGLT2 inhibitor dapagliflozin limits podocyte damage in proteinuric nondiabetic nephropathy. JCI Insight 2018, 3, e98720. [Google Scholar] [CrossRef]
- American Diabetes Association Professional Practice Committee. 9. Pharmacologic approaches to glycemic treatment: Standards of Medical Care in Diabetes—2022. Diabetes Care 2022, 45, S125–S143. [Google Scholar] [CrossRef]
- Zelniker, T.A.; Wiviott, S.D.; Raz, I.; Im, K.; Goodrich, E.L.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Furtado, R.H. SGLT2 inhibitors for primary and secondary prevention of cardiovascular and renal outcomes in type 2 diabetes: A systematic review and meta-analysis of cardiovascular outcome trials. Lancet 2019, 393, 31–39. [Google Scholar] [CrossRef]
- Rossing, P.; Caramori, M.L.; Chan, J.C.; Heerspink, H.J.; Hurst, C.; Khunti, K.; Liew, A.; Michos, E.D.; Navaneethan, S.D.; Olowu, W.A. Executive summary of the KDIGO 2022 Clinical Practice Guideline for Diabetes Management in Chronic Kidney Disease: An update based on rapidly emerging new evidence. Kidney Int. 2022, 102, 990–999. [Google Scholar] [CrossRef]
- Monzo, L.; Ferrari, I.; Cicogna, F.; Tota, C.; Calò, L. Sodium–glucose co-transporter-2 inhibitors eligibility in patients with heart failure with reduced ejection fraction. Int. J. Cardiol. 2021, 341, 56–59. [Google Scholar] [CrossRef] [PubMed]
- Pabel, S.; Hamdani, N.; Singh, J.; Sossalla, S. Potential mechanisms of SGLT2 inhibitors for the treatment of heart failure with preserved ejection fraction. Front. Physiol. 2021, 12, 752370. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.S.; Hirayama, B.A.; Timbol, G.; Liu, J.; Basarah, E.; Kepe, V.; Satyamurthy, N.; Huang, S.-C.; Wright, E.M.; Barrio, J.R. Functional expression of SGLTs in rat brain. Am. J. Physiol. Cell Physiol. 2010, 299, C1277–C1284. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.S.; Hirayama, B.A.; Timbol, G.; Liu, J.; Diez-Sampedro, A.; Kepe, V.; Satyamurthy, N.; Huang, S.-C.; Wright, E.M.; Barrio, J.R. Regional distribution of SGLT activity in rat brain in vivo. Am. J. Physiol. Cell Physiol. 2013, 304, C240–C247. [Google Scholar] [CrossRef] [PubMed]
- Sa-Nguanmoo, P.; Tanajak, P.; Kerdphoo, S.; Jaiwongkam, T.; Pratchayasakul, W.; Chattipakorn, N.; Chattipakorn, S.C. SGLT2-inhibitor and DPP-4 inhibitor improve brain function via attenuating mitochondrial dysfunction, insulin resistance, inflammation, and apoptosis in HFD-induced obese rats. Toxicol. Appl. Pharmacol. 2017, 333, 43–50. [Google Scholar] [CrossRef]
- Pawlos, A.; Broncel, M.; Woźniak, E.; Gorzelak-Pabiś, P. Neuroprotective effect of SGLT2 inhibitors. Molecules 2021, 26, 7213. [Google Scholar] [CrossRef] [PubMed]
- Bathina, S.; Das, U.N. Brain-derived neurotrophic factor and its clinical implications. Arch. Med. Sci. 2015, 11, 1164–1178. [Google Scholar] [CrossRef] [PubMed]
- Hierro-Bujalance, C.; Infante-Garcia, C.; Del Marco, A.; Herrera, M.; Carranza-Naval, M.J.; Suarez, J.; Alves-Martinez, P.; Lubian-Lopez, S.; Garcia-Alloza, M. Empagliflozin reduces vascular damage and cognitive impairment in a mixed murine model of Alzheimer’s disease and type 2 diabetes. Alzheimer’s Res. Ther. 2020, 12, 40. [Google Scholar] [CrossRef]
- Lin, B.; Koibuchi, N.; Hasegawa, Y.; Sueta, D.; Toyama, K.; Uekawa, K.; Ma, M.; Nakagawa, T.; Kusaka, H.; Kim-Mitsuyama, S. Glycemic control with empagliflozin, a novel selective SGLT2 inhibitor, ameliorates cardiovascular injury and cognitive dysfunction in obese and type 2 diabetic mice. Cardiovasc. Diabetol. 2014, 13, 148. [Google Scholar] [CrossRef]
- Majdi, A.; Sadigh-Eteghad, S.; Rahigh Aghsan, S.; Farajdokht, F.; Vatandoust, S.M.; Namvaran, A.; Mahmoudi, J. Amyloid-β, tau, and the cholinergic system in Alzheimer’s disease: Seeking direction in a tangle of clues. Rev. Neurosci. 2020, 31, 391–413. [Google Scholar] [CrossRef]
- Lian, W.; Fang, J.; Xu, L.; Zhou, W.; Kang, D.; Xiong, W.; Jia, H.; Liu, A.-L.; Du, G.-H. DL0410 ameliorates memory and cognitive impairments induced by scopolamine via increasing cholinergic neurotransmission in mice. Molecules 2017, 22, 410. [Google Scholar] [CrossRef] [PubMed]
- Arafa, N.M.; Ali, E.H.; Hassan, M.K. Canagliflozin prevents scopolamine-induced memory impairment in rats: Comparison with galantamine hydrobromide action. Chem. Biol. Interact. 2017, 277, 195–203. [Google Scholar] [CrossRef] [PubMed]
- MD Rizvi, S.; Shakil, S.; Biswas, D.; Shakil, S.; Shaikh, S.; Bagga, P.; A Kamal, M. Invokana (Canagliflozin) as a dual inhibitor of acetylcholinesterase and sodium glucose co-transporter 2: Advancement in Alzheimer’s disease-diabetes type 2 linkage via an enzoinformatics study. CNS Neurol. Disord. Drug Targets Former. Curr. Drug Targets-CNS Neurol. Disord. 2014, 13, 447–451. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-Y.; Iskander, C.; Wang, C.; Xiong, L.Y.; Shah, B.R.; Edwards, J.D.; Kapral, M.K.; Herrmann, N.; Lanctôt, K.L.; Masellis, M. Association of Sodium–Glucose Cotransporter 2 Inhibitors with Time to Dementia: A Population-Based Cohort Study. Diabetes Care 2023, 46, 297–304. [Google Scholar] [CrossRef]
- Wium-Andersen, I.K.; Osler, M.; Jørgensen, M.B.; Rungby, J.; Wium-Andersen, M.K. Antidiabetic medication and risk of dementia in patients with type 2 diabetes: A nested case–control study. Eur. J. Endocrinol. 2019, 181, 499–507. [Google Scholar] [CrossRef]
- Low, S.; Goh, K.S.; Ng, T.P.; Moh, A.; Ang, S.F.; Wang, J.; Ang, K.; Tang, W.E.; Lim, Z.; Subramaniam, T. Association between use of sodium-glucose co-transporter-2 (SGLT2) inhibitors and cognitive function in a longitudinal study of patients with type 2 diabetes. J. Alzheimer’s Dis. 2022, 87, 635–642. [Google Scholar] [CrossRef]
- Sabbagh, F.; Muhamad, I.I.; Niazmand, R.; Dikshit, P.K.; Kim, B.S. Recent progress in polymeric non-invasive insulin delivery. Int. J. Biol. Macromol. 2022, 203, 222–243. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adem, M.A.; Decourt, B.; Sabbagh, M.N. Pharmacological Approaches Using Diabetic Drugs Repurposed for Alzheimer’s Disease. Biomedicines 2024, 12, 99. https://doi.org/10.3390/biomedicines12010099
Adem MA, Decourt B, Sabbagh MN. Pharmacological Approaches Using Diabetic Drugs Repurposed for Alzheimer’s Disease. Biomedicines. 2024; 12(1):99. https://doi.org/10.3390/biomedicines12010099
Chicago/Turabian StyleAdem, Muna A., Boris Decourt, and Marwan N. Sabbagh. 2024. "Pharmacological Approaches Using Diabetic Drugs Repurposed for Alzheimer’s Disease" Biomedicines 12, no. 1: 99. https://doi.org/10.3390/biomedicines12010099
APA StyleAdem, M. A., Decourt, B., & Sabbagh, M. N. (2024). Pharmacological Approaches Using Diabetic Drugs Repurposed for Alzheimer’s Disease. Biomedicines, 12(1), 99. https://doi.org/10.3390/biomedicines12010099