
Randomized Phase II Cancer Clinical Trials to Validate Predictive Biomarkers


Abstract
:1. Introduction
2. Material and Methods
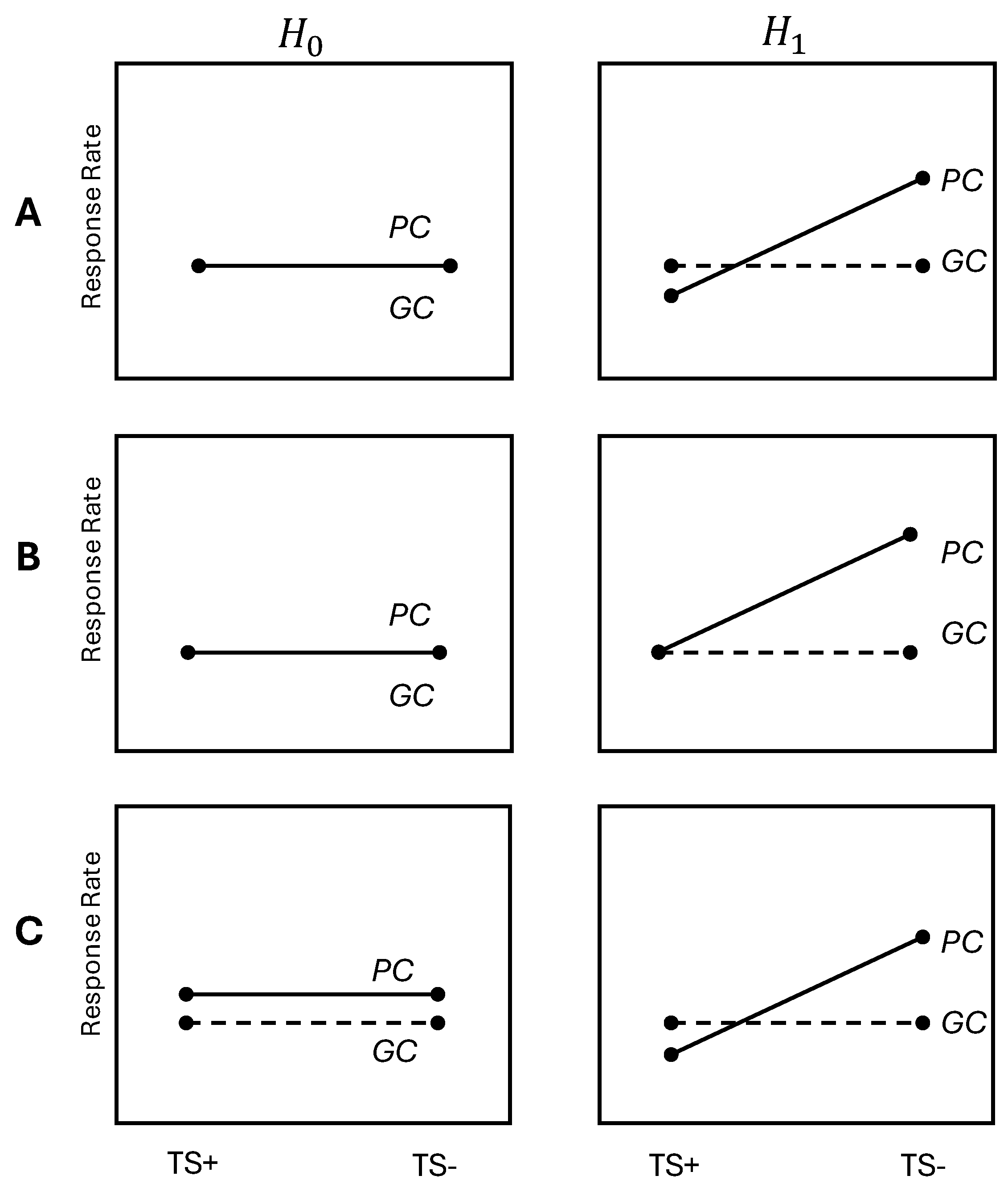
2.1. Interaction Based on Logit-Transformed RRs
- Type I error rate and power: ;
- Expected RRs: ;
- Allocation proportion for arm : ;
- Prevalence of biomarker status : .
2.2. Interaction Based on Raw RRs
3. Numerical Analysis
3.1. Real Study Example
3.2. Simulations
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| TS | Thymidylate Synthase |
| PC | Pemetrexed/Cisplatin |
| GC | Gemcitabin/Cisplatin |
| NSCLC | Non-small-cell Lung Cancer |
| RR | Response Rate |
References
- Yang, Z.-Y.; Zhao, K.; Luo, D.; Yin, Z.; Zhou, C.; Chen, J.; Zhang, C. Carcinoembryonic antigen levels are increased with pulmonary output in pulmonary hypertension due to congenital heart disease. J. Int. Med. Res. 2020, 48, 0300060520964378. [Google Scholar]
- Tolmeijer, S.H.; Koornstra, R.H.; de Groot, J.W.B.; Geerlings, M.J.; van Rens, D.H.; Boers-Sonderen, M.J.; Schalken, J.A.; Gerritsen, W.R.; Ligtenberg, M.J.; Mehra, N. Plasma BRAF mutation detection for the diagnostic and monitoring trajectory of patients with LDH-high stage IV melanoma. Cancers 2021, 13, 3913. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Liu, S.; Maghsoudloo, M.; Shasaltaneh, M.D.; Kaboli, P.J.; Zhang, C.; Deng, Y.; Heidari, H.; Entezari, M.; Fu, S.; et al. PLA1A expression as a diagnostic marker of BRAF-mutant metastasis in melanoma cancer. Sci. Rep. 2021, 11, 6056. [Google Scholar] [CrossRef] [PubMed]
- Malapelle, U.; Rossi, G.; Pisapia, P.; Barberis, M.; Buttitta, F.; Castiglione, F.; Cecere, F.L.; Grimaldi, A.M.; Iaccarino, A.; Marchetti, A.; et al. BRAF as a positive predictive biomarker: Focus on lung cancer and melanoma patients. Crit. Rev. Oncol. 2020, 156, 103118. [Google Scholar] [CrossRef] [PubMed]
- Freidlin, B.; McShane, L.M.; Korn, E.L. Randomized clinical trials with biomarkers: Design issues. J. Natl. Cancer Inst. 2010, 102, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Freidlin, B.; McShane, L.M.; Polley, M.Y.C.; Korn, E.L. Randomized phase II trial designs with biomarkers. J. Clin. Oncol. 2012, 30, 3304. [Google Scholar] [CrossRef] [PubMed]
- Foppa, I.; Spiegelman, D. Power and sample size calculations for case-control studies of gene-environment interactions with a polytomous exposure variable. Am. J. Epidemiol. 1997, 146, 596–604. [Google Scholar] [CrossRef] [PubMed]
- Demidenko, E. Sample size and optimal design for logistic regression with binary interaction. Stat. Med. 2008, 27, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Ozasa, H.; Oguri, T.; Uemura, T.; Miyazaki, M.; Maeno, K.; Sato, S.; Ueda, R. Significance of thymidylate synthase for resistance to pemetrexed in lung cancer. Cancer Sci. 2010, 101, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.M.; Ahn, J.S.; Jung, S.H.; Sun, J.; Ha, S.Y.; Han, J.; Park, K.; Ahn, M.J. Pemetrexed plus cisplatin versus gemcitabine plus cisplatin according to thymidylate synthase expression in nonsquamous non–small-cell lung cancer: A biomarker-stratified randomized phase II trial. J. Clin. Oncol. 2015, 33, 2450–2456. [Google Scholar] [CrossRef] [PubMed]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.M.; Han, J.; Ahn, J.S.; Park, K.; Ahn, M.J. Significance of thymidylate synthase and thyroid transcription factor 1 expression in patients with nonsquamous non-small cell lung cancer treated with pemetrexed-based chemotherapy. J. Thorac. Oncol. 2011, 6, 1392–1399. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Scenario | ||||||||
|---|---|---|---|---|---|---|---|---|
| A1 | 0.2 | 0.2 | 0.2 | 0.2 | 0.2 | 0.2 | 0.1 | 0.4 |
| A2 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.2 | 0.5 |
| A3 | 0.4 | 0.4 | 0.4 | 0.4 | 0.4 | 0.4 | 0.3 | 0.6 |
| A4 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.4 | 0.7 |
| B1 | 0.2 | 0.2 | 0.2 | 0.2 | 0.2 | 0.2 | 0.2 | 0.55 |
| B2 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.65 |
| B3 | 0.4 | 0.4 | 0.4 | 0.4 | 0.4 | 0.4 | 0.4 | 0.75 |
| B4 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.85 |
| C1 | 0.2 | 0.2 | 0.3 | 0.3 | 0.2 | 0.2 | 0.1 | 0.5 |
| C2 | 0.3 | 0.3 | 0.4 | 0.4 | 0.3 | 0.3 | 0.2 | 0.6 |
| C3 | 0.4 | 0.4 | 0.5 | 0.5 | 0.4 | 0.4 | 0.3 | 0.7 |
| C4 | 0.5 | 0.5 | 0.6 | 0.6 | 0.5 | 0.5 | 0.4 | 0.8 |
| Scenario | ||||||||
|---|---|---|---|---|---|---|---|---|
| A1 | 1.792 | 228 | 0.1005 | 0.9180 | 0.30 | 188 | 0.1048 | 0.9033 |
| A2 | 1.386 | 272 | 0.1013 | 0.9028 | 0.30 | 244 | 0.1038 | 0.8932 |
| A3 | 1.253 | 288 | 0.1043 | 0.9086 | 0.30 | 272 | 0.0984 | 0.8983 |
| A4 | 1.253 | 284 | 0.1077 | 0.9074 | 0.30 | 276 | 0.1030 | 0.8958 |
| B1 | 1.587 | 236 | 0.1023 | 0.9004 | 0.35 | 156 | 0.1091 | 0.8949 |
| B2 | 1.466 | 228 | 0.1004 | 0.9095 | 0.35 | 184 | 0.1025 | 0.8986 |
| B3 | 1.504 | 208 | 0.0974 | 0.9094 | 0.35 | 196 | 0.1092 | 0.9001 |
| B4 | 1.735 | 172 | 0.0979 | 0.9142 | 0.35 | 188 | 0.1003 | 0.8974 |
| C1 | 1.792 | 152 | 0.1011 | 0.8962 | 0.30 | 108 | 0.1085 | 0.8912 |
| C2 | 2.197 | 164 | 0.0997 | 0.9057 | 0.40 | 136 | 0.1088 | 0.902 |
| C3 | 1.792 | 164 | 0.096 | 0.9056 | 0.40 | 148 | 0.1083 | 0.8972 |
| C4 | 1.792 | 152 | 0.108 | 0.9143 | 0.40 | 148 | 0.1019 | 0.8969 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, B.; Sun, J.-M.; Ahn, M.-J.; Jung, S.-H. Randomized Phase II Cancer Clinical Trials to Validate Predictive Biomarkers. Biomedicines 2024, 12, 2185. https://doi.org/10.3390/biomedicines12102185
Zhang B, Sun J-M, Ahn M-J, Jung S-H. Randomized Phase II Cancer Clinical Trials to Validate Predictive Biomarkers. Biomedicines. 2024; 12(10):2185. https://doi.org/10.3390/biomedicines12102185
Chicago/Turabian StyleZhang, Baoshan, Jong-Mu Sun, Myung-Ju Ahn, and Sin-Ho Jung. 2024. "Randomized Phase II Cancer Clinical Trials to Validate Predictive Biomarkers" Biomedicines 12, no. 10: 2185. https://doi.org/10.3390/biomedicines12102185