


Interplay of RNA m6A Modification-Related Geneset in Pan-Cancer

 ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Geneset Collection, Protein-Protein Interaction (PPI), and Gene Annotation of m6A Modifiers
2.2. Data Collection and Processing
2.3. Cox Regression Analysis
2.4. Copy Number Variants (CNVs), Single Nucleotide Variations (SNVs), and Methylation Level Analysis
2.5. Consensus Cluster Analysis
2.6. Risk Model Construction
2.7. Immune Cell Infiltration, Stemness Features, and Tumor Heterogeneity Analysis
2.8. Immune Regulator and Immune Checkpoint Gene Analysis
2.9. Immune Score and miRNA Analysis
2.10. Drug Prediction
2.11. Statistical Analyses
2.12. Identifying m6A-Related Biomarkers by Three Machine Learning Algorithms
3. Results
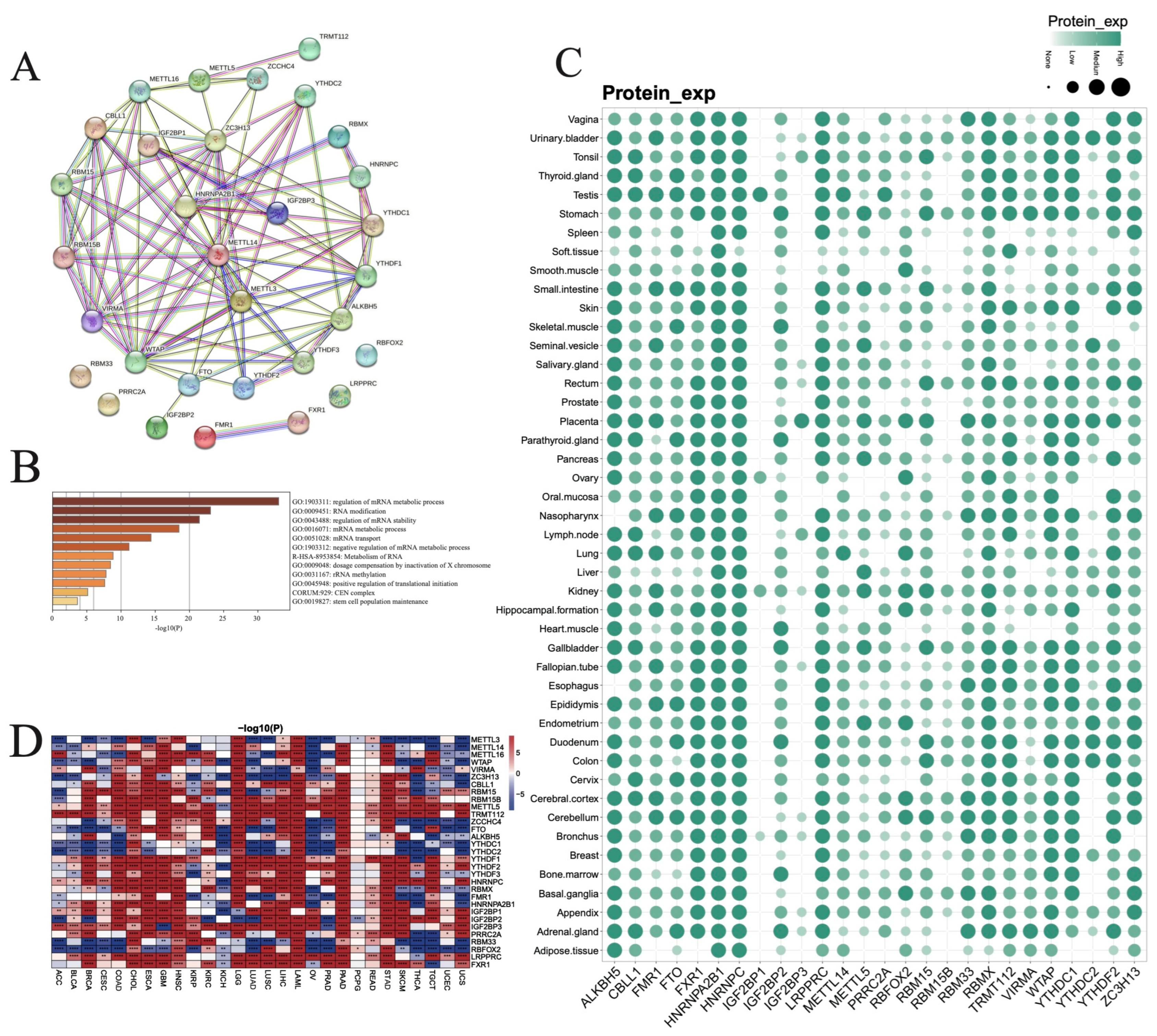
3.1. Expression Pattern and Functional Analysis of RNA m6A Modification Regulators in Pan-Cancer and Normal Tissues
3.2. Correlations among RNA m6A Modification Regulators in Pan-Cancer
3.3. Overall Survival (OS) Status Based on the Expression of RNA m6A Modification Regulators in Pan-Cancer
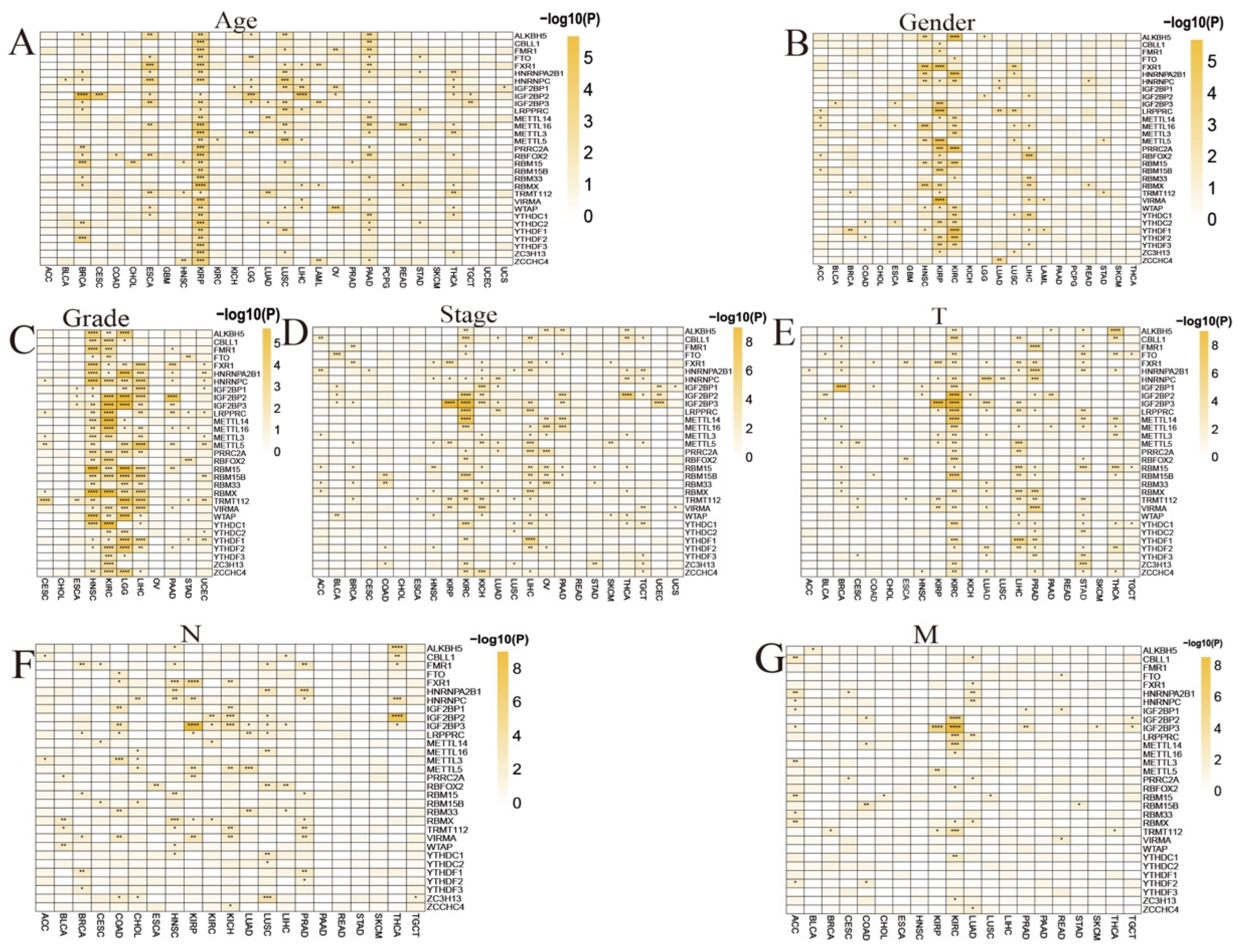
3.4. Clinical Significance of RNA m6A Modification Regulators in Pan-Cancer
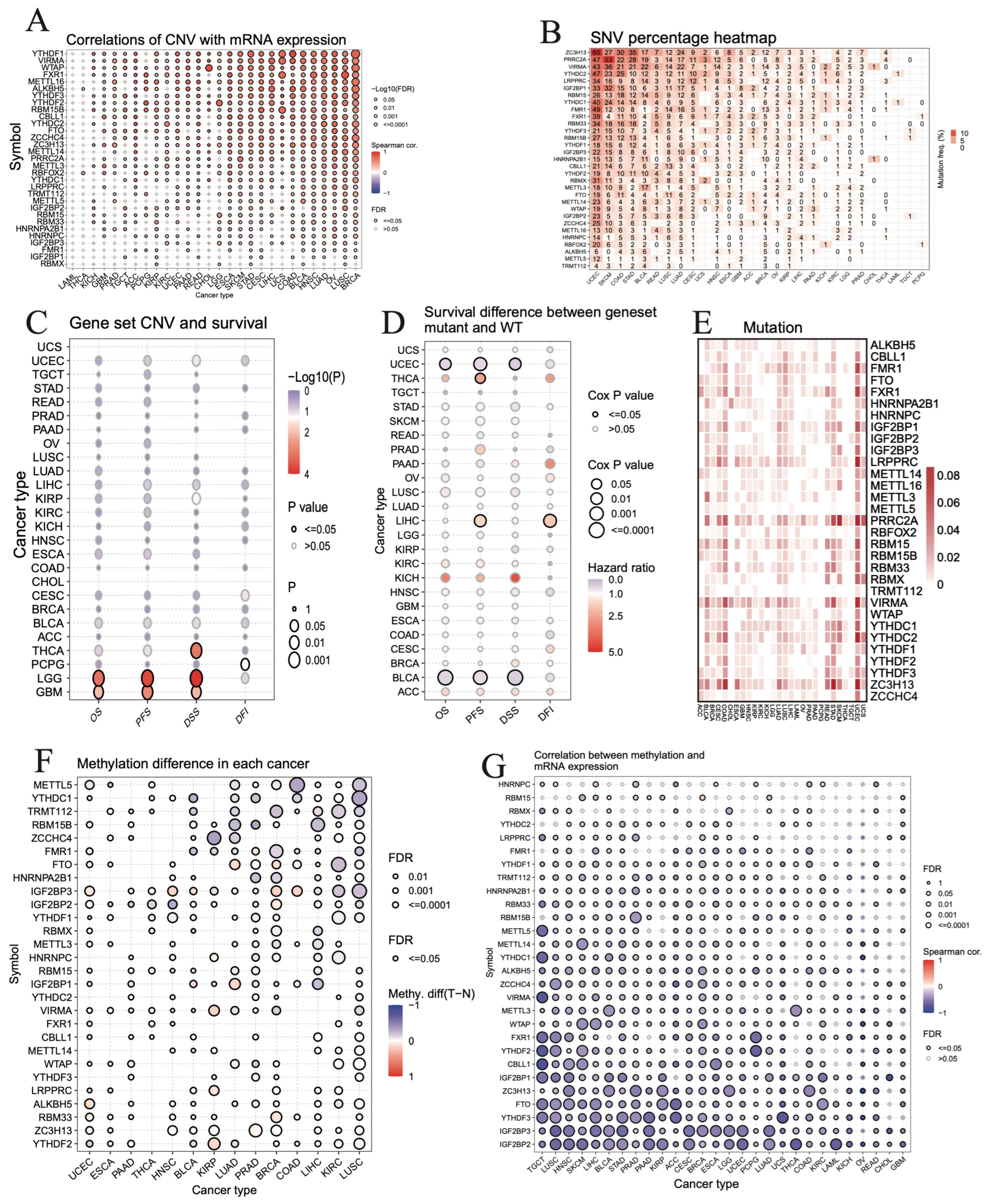
3.5. Correlation between Genetic or Epigenetic Alterations and Expression Levels of m6A Regulators in Pan-Cancer
3.6. Distinguishing Different Clusters of RNA m6A Regulators in TCGA Pan-Cancer Cohort
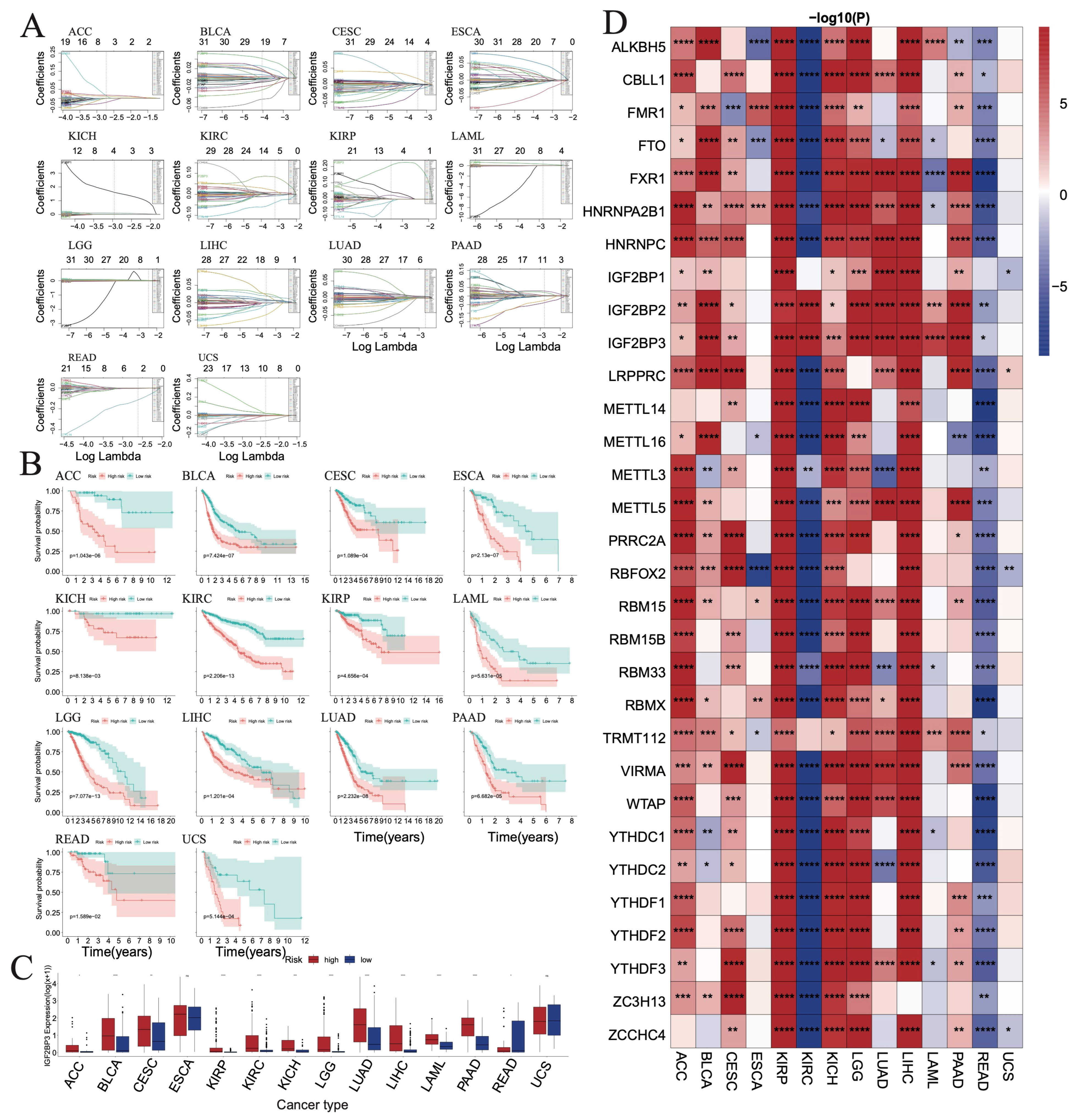
3.7. Evaluating the Prognostic Value of RNA m6A Regulators in TCGA Pan-Cancer Cohort
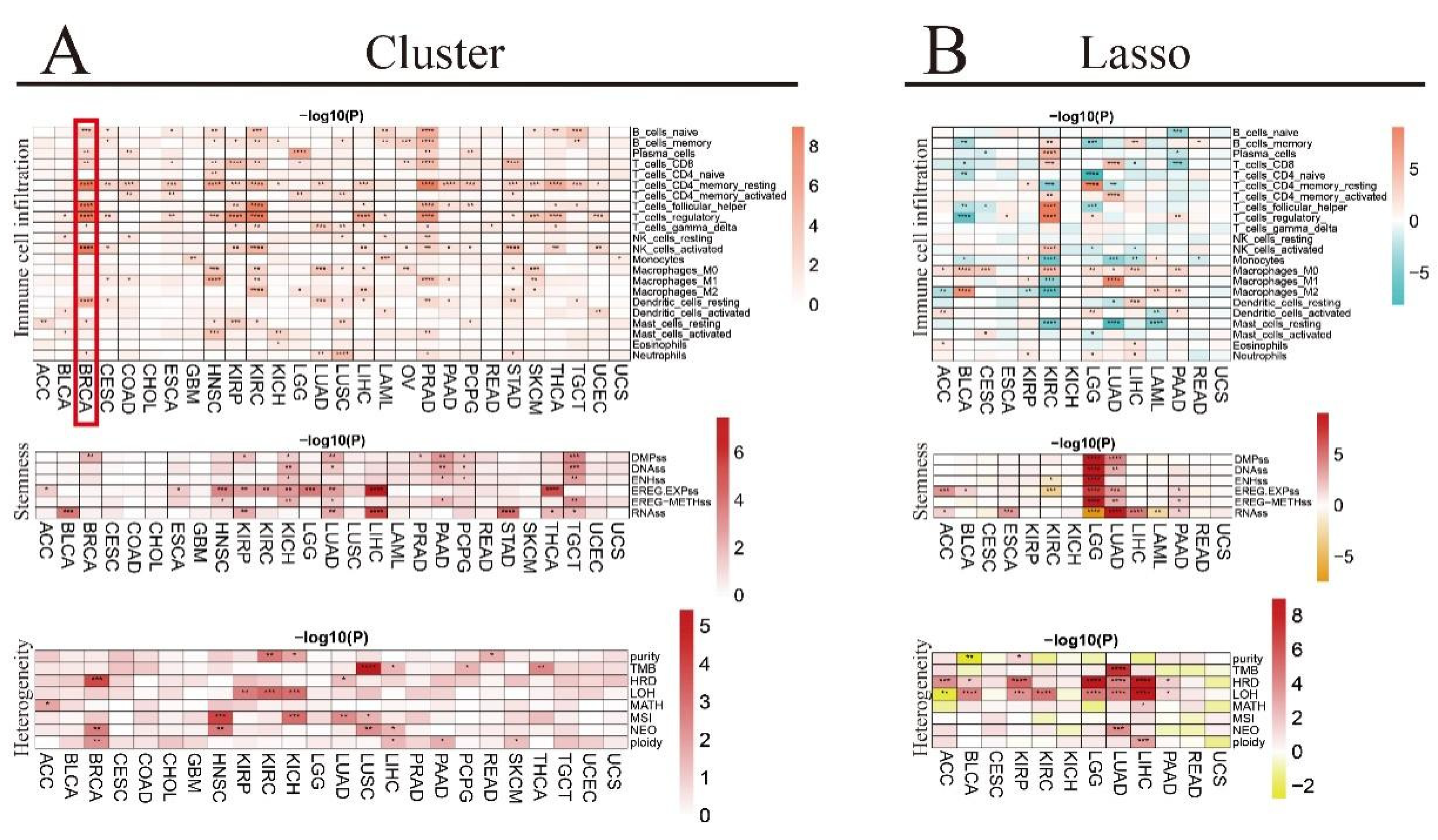
3.8. Correlation between Immune Infiltrating Score and RNA m6A Regulators in TCGA Pan-Cancer Cohort
3.9. Association of Tumor Microenvironment (TME) Infiltrating Cells with RNA m6A Regulators in Pan-Cancer by CIBERSORT
3.10. Association of Tumor Stemness Score with RNA m6A Regulators in TCGA Pan-Cancer Cohort
3.11. Association of Genomic Heterogeneity with RNA m6A Regulators in TCGA Pan-Cancer Cohort
3.12. Association of Immune Regulatory or Checkpoint Genes with RNA m6A Regulators in TCGA Pan-Cancer Cohort
3.13. Correlation between microRNAs (miRNAs) and the RNA m6A Regulators in TCGA Pan-Cancer
3.14. Drug Prediction Based on m6A Modification Genes in TCGA Pan-Cancer Cohort
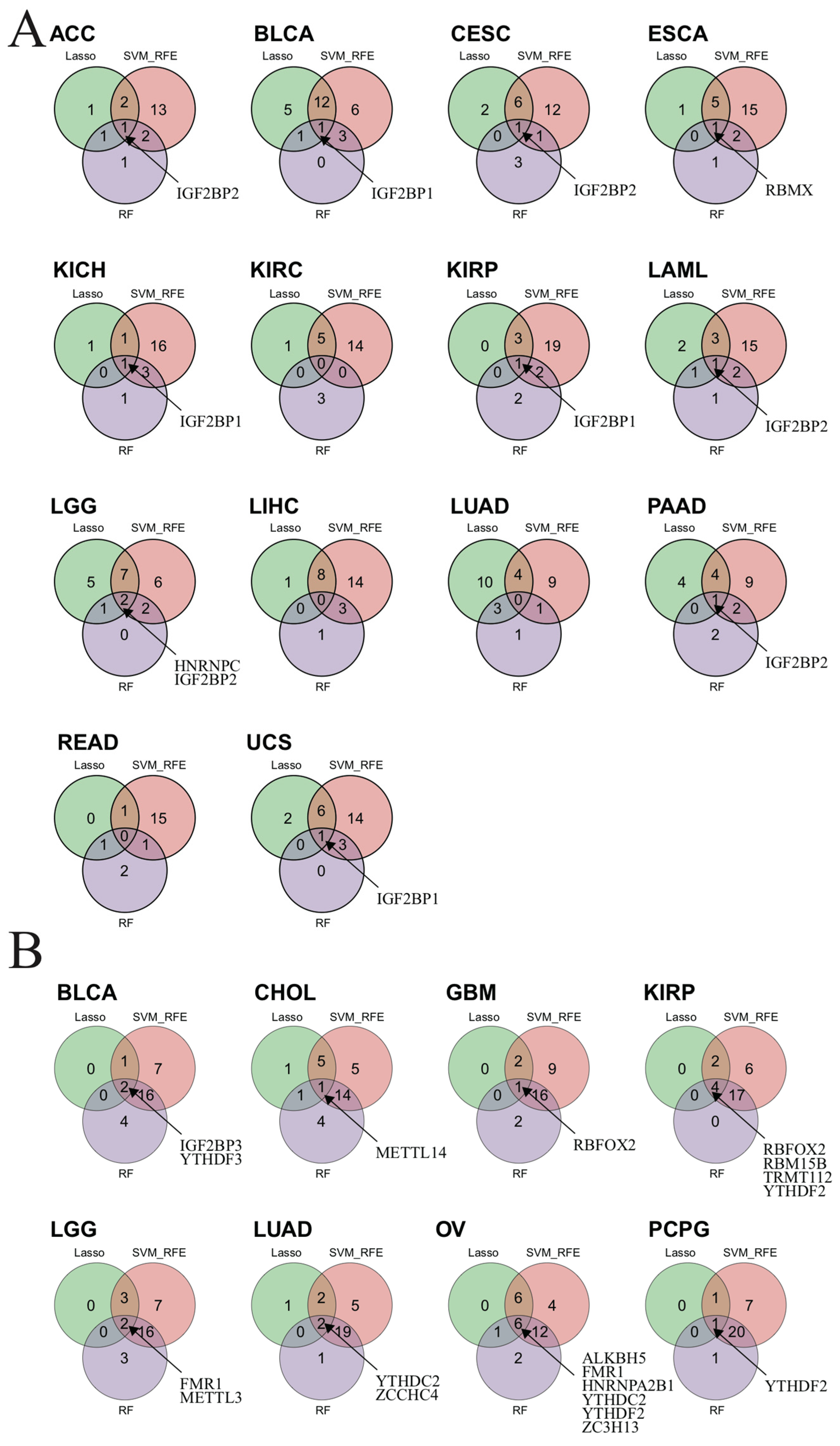
3.15. Identified m6A-Related Biomarkers by Three Machine Learning Algorithms
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Code Availability Statement
Conflicts of Interest
References
- Linder, B.; Grozhik, A.V.; Olarerin-George, A.O.; Meydan, C.; Mason, C.E.; Jaffrey, S.R. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat. Methods 2015, 12, 767–772. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, S.; Mumbach, M.R.; Jovanovic, M.; Wang, T.; Maciag, K.; Bushkin, G.G.; Mertins, P.; Ter-Ovanesyan, D.; Habib, N.; Cacchiarelli, D.; et al. Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5′ sites. Cell Rep. 2014, 8, 284–296. [Google Scholar] [CrossRef] [PubMed]
- Mattick, J.S.; Amaral, P.P.; Carninci, P.; Carpenter, S.; Chang, H.Y.; Chen, L.L.; Chen, R.; Dean, C.; Dinger, M.E.; Fitzgerald, K.A.; et al. Long non-coding RNAs: Definitions, functions, challenges and recommendations. Nat. Rev. Mol. Cell Biol. 2023, 24, 430–447. [Google Scholar] [CrossRef]
- Meyer, K.D.; Saletore, Y.; Zumbo, P.; Elemento, O.; Mason, C.E.; Jaffrey, S.R. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell 2012, 149, 1635–1646. [Google Scholar] [CrossRef]
- Dominissini, D.; Moshitch-Moshkovitz, S.; Schwartz, S.; Salmon-Divon, M.; Ungar, L.; Osenberg, S.; Cesarkas, K.; Jacob-Hirsch, J.; Amariglio, N.; Kupiec, M.; et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 2012, 485, 201–206. [Google Scholar] [CrossRef]
- Xu, W.; He, C.; Kaye, E.G.; Li, J.; Mu, M.; Nelson, G.M.; Dong, L.; Wang, J.; Wu, F.; Shi, Y.G.; et al. Dynamic control of chromatin-associated m6A methylation regulates nascent RNA synthesis. Mol. Cell 2022, 82, 1156–1168.e7. [Google Scholar] [CrossRef]
- Li, Z.; Weng, H.; Su, R.; Weng, X.; Zuo, Z.; Li, C.; Huang, H.; Nachtergaele, S.; Dong, L.; Hu, C.; et al. FTO Plays an Oncogenic Role in Acute Myeloid Leukemia as a N6-Methyladenosine RNA Demethylase. Cancer Cell 2017, 31, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Ping, X.L.; Sun, B.F.; Wang, L.; Xiao, W.; Yang, X.; Wang, W.J.; Adhikari, S.; Shi, Y.; Lv, Y.; Chen, Y.S.; et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 2014, 24, 177–189. [Google Scholar] [CrossRef]
- Mendel, M.; Chen, K.M.; Homolka, D.; Gos, P.; Pandey, R.R.; McCarthy, A.A.; Pillai, R.S. Methylation of structured RNA by the m6A writer METTL16 is essential for mouse embryonic development. Mol. Cell 2018, 71, 986–1000.e11. [Google Scholar] [CrossRef]
- Wen, J.; Lv, R.; Ma, H.; Shen, H.; He, C.; Wang, J.; Jiao, F.; Liu, H.; Yang, P.; Tan, L.; et al. ZC3H13 regulates nuclear RNA m6A methylation and mouse embryonic stem cell self-renewal. Mol. Cell 2018, 69, 1028–1038.e6. [Google Scholar] [CrossRef]
- Ruzicka, K.; Zhang, M.; Campilho, A.; Bodi, Z.; Kashif, M.; Saleh, M.; Eeckhout, D.; El-Showk, S.; Li, H.; Zhong, S.; et al. Identification of factors required for m6A mRNA methylation in Arabidopsis reveals a role for the conserved E3 ubiquitin ligase HAKAI. New Phytol. 2017, 215, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Patil, D.P.; Chen, C.K.; Pickering, B.F.; Chow, A.; Jackson, C.; Guttman, M.; Jaffrey, S.R. m6A RNA methylation promotes XIST-mediated transcriptional repression. Nature 2016, 537, 369–373. [Google Scholar] [CrossRef] [PubMed]
- van Tran, N.; Ernst, F.G.M.; Hawley, B.R.; Zorbas, C.; Ulryck, N.; Hackert, P.; Bohnsack, K.E.; Bohnsack, M.T.; Jaffrey, S.R.; Graille, M.; et al. The human 18S rRNA m6A methyltransferase METTL5 is stabilized by TRMT112. Nucleic Acids Res. 2019, 47, 7719–7733. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Fu, Y.; Zhao, X.; Dai, Q.; Zheng, G.; Yang, Y.; Yi, C.; Lindahl, T.; Pan, T.; Yang, Y.G.; et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 2011, 7, 885–887. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.; Dahl, J.A.; Niu, Y.; Fedorcsak, P.; Huang, C.M.; Li, C.J.; Vagbo, C.B.; Shi, Y.; Wang, W.L.; Song, S.H.; et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol. Cell 2013, 49, 18–29. [Google Scholar] [CrossRef]
- Xiao, W.; Adhikari, S.; Dahal, U.; Chen, Y.S.; Hao, Y.J.; Sun, B.F.; Sun, H.Y.; Li, A.; Ping, X.L.; Lai, W.Y.; et al. Nuclear m6A reader YTHDC1 regulates mRNA splicing. Mol. Cell 2016, 61, 507–519. [Google Scholar] [CrossRef]
- Hsu, P.J.; Zhu, Y.; Ma, H.; Guo, Y.; Shi, X.; Liu, Y.; Qi, M.; Lu, Z.; Shi, H.; Wang, J.; et al. Ythdc2 is an N6-methyladenosine binding protein that regulates mammalian spermatogenesis. Cell Res. 2017, 27, 1115–1127. [Google Scholar] [CrossRef]
- Wang, X.; Zhao, B.S.; Roundtree, I.A.; Lu, Z.; Han, D.; Ma, H.; Weng, X.; Chen, K.; Shi, H.; He, C. N6-methyladenosine Modulates Messenger RNA Translation Efficiency. Cell 2015, 161, 1388–1399. [Google Scholar] [CrossRef]
- Wang, X.; Lu, Z.; Gomez, A.; Hon, G.C.; Yue, Y.; Han, D.; Fu, Y.; Parisien, M.; Dai, Q.; Jia, G.; et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 2014, 505, 117–120. [Google Scholar] [CrossRef]
- Li, A.; Chen, Y.S.; Ping, X.L.; Yang, X.; Xiao, W.; Yang, Y.; Sun, H.Y.; Zhu, Q.; Baidya, P.; Wang, X.; et al. Cytoplasmic m6A reader YTHDF3 promotes mRNA translation. Cell Res. 2017, 27, 444–447. [Google Scholar] [CrossRef]
- Liu, N.; Dai, Q.; Zheng, G.; He, C.; Parisien, M.; Pan, T. N6-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature 2015, 518, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Zhou, K.I.; Parisien, M.; Dai, Q.; Diatchenko, L.; Pan, T. N6-methyladenosine alters RNA structure to regulate binding of a low-complexity protein. Nucleic Acids Res. 2017, 45, 6051–6063. [Google Scholar] [CrossRef]
- Edupuganti, R.R.; Geiger, S.; Lindeboom, R.G.H.; Shi, H.; Hsu, P.J.; Lu, Z.; Wang, S.Y.; Baltissen, M.P.A.; Jansen, P.; Rossa, M.; et al. N6-methyladenosine (m6A) recruits and repels proteins to regulate mRNA homeostasis. Nat. Struct. Mol. Biol. 2017, 24, 870–878. [Google Scholar] [CrossRef]
- Alarcon, C.R.; Goodarzi, H.; Lee, H.; Liu, X.; Tavazoie, S.; Tavazoie, S.F. HNRNPA2B1 is a mediator of m6A-dependent nuclear RNA processing events. Cell 2015, 162, 1299–1308. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Weng, H.; Sun, W.; Qin, X.; Shi, H.; Wu, H.; Zhao, B.S.; Mesquita, A.; Liu, C.; Yuan, C.L.; et al. Recognition of RNA N6-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat. Cell Biol. 2018, 20, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Li, A.; Sun, B.; Sun, J.G.; Zhang, J.; Zhang, T.; Chen, Y.; Xiao, Y.; Gao, Y.; Zhang, Q.; et al. A novel m6A reader Prrc2a controls oligodendroglial specification and myelination. Cell Res. 2019, 29, 23–41. [Google Scholar] [CrossRef]
- Yu, F.; Zhu, A.C.; Liu, S.; Gao, B.; Wang, Y.; Khudaverdyan, N.; Yu, C.; Wu, Q.; Jiang, Y.; Song, J.; et al. RBM33 is a unique m6A RNA-binding protein that regulates ALKBH5 demethylase activity and substrate selectivity. Mol. Cell 2023, 83, 2003–2019.e6. [Google Scholar] [CrossRef]
- Dou, X.; Xiao, Y.; Shen, C.; Wang, K.; Wu, T.; Liu, C.; Li, Y.; Yu, X.; Liu, J.; Dai, Q.; et al. RBFOX2 recognizes N6-methyladenosine to suppress transcription and block myeloid leukaemia differentiation. Nat. Cell Biol. 2023, 25, 1359–1368. [Google Scholar] [CrossRef]
- Arguello, A.E.; DeLiberto, A.N.; Kleiner, R.E. RNA chemical proteomics reveals the N6-Methyladenosine (m6A)-regulated protein-RNA interactome. J. Am. Chem. Soc. 2017, 139, 17249–17252. [Google Scholar] [CrossRef]
- Deng, S.; Zhang, J.; Su, J.; Zuo, Z.; Zeng, L.; Liu, K.; Zheng, Y.; Huang, X.; Bai, R.; Zhuang, L.; et al. RNA m6A regulates transcription via DNA demethylation and chromatin accessibility. Nat. Genet. 2022, 54, 1427–1437. [Google Scholar] [CrossRef]
- Sommer, S.; Lavi, U.; Darnell, J.E., Jr. The absolute frequency of labeled N-6-methyladenosine in HeLa cell messenger RNA decreases with label time. J. Mol. Biol. 1978, 124, 487–499. [Google Scholar] [CrossRef] [PubMed]
- Ries, R.J.; Zaccara, S.; Klein, P.; Olarerin-George, A.; Namkoong, S.; Pickering, B.F.; Patil, D.P.; Kwak, H.; Lee, J.H.; Jaffrey, S.R. m6A enhances the phase separation potential of mRNA. Nature 2019, 571, 424–428. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, F.L.; Holaska, J.M.; Zhang, Z.; Sharma, A.; Manilal, S.; Holt, I.; Stamm, S.; Wilson, K.L.; Morris, G.E. Emerin interacts in vitro with the splicing-associated factor, YT521-B. Eur. J. Biochem. 2003, 270, 2459–2466. [Google Scholar] [CrossRef] [PubMed]
- Roundtree, I.A.; Luo, G.Z.; Zhang, Z.; Wang, X.; Zhou, T.; Cui, Y.; Sha, J.; Huang, X.; Guerrero, L.; Xie, P.; et al. YTHDC1 mediates nuclear export of N6-methyladenosine methylated mRNAs. Elife 2017, 6, e31311. [Google Scholar] [CrossRef]
- Vu, L.P.; Pickering, B.F.; Cheng, Y.; Zaccara, S.; Nguyen, D.; Minuesa, G.; Chou, T.; Chow, A.; Saletore, Y.; MacKay, M.; et al. The N6-methyladenosine (m6A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat. Med. 2017, 23, 1369–1376. [Google Scholar] [CrossRef]
- Du, M.; Zhang, Y.; Mao, Y.; Mou, J.; Zhao, J.; Xue, Q.; Wang, D.; Huang, J.; Gao, S.; Gao, Y. MiR-33a suppresses proliferation of NSCLC cells via targeting METTL3 mRNA. Biochem. Biophys. Res. Commun. 2017, 482, 582–589. [Google Scholar] [CrossRef]
- Chen, M.; Wei, L.; Law, C.T.; Tsang, F.H.; Shen, J.; Cheng, C.L.; Tsang, L.H.; Ho, D.W.; Chiu, D.K.; Lee, J.M.; et al. RNA N6-methyladenosine methyltransferase-like 3 promotes liver cancer progression through YTHDF2-dependent posttranscriptional silencing of SOCS2. Hepatology 2018, 67, 2254–2270. [Google Scholar] [CrossRef]
- Kennedy, L.; Hargrove, L.; Demieville, J.; Francis, N.; Seils, R.; Villamaria, S.; Francis, H. Recent Advances in Understanding Cholangiocarcinoma. F1000Research 2017, 6, 1818. [Google Scholar] [CrossRef]
- Xue, C.; Chu, Q.; Zheng, Q.; Jiang, S.; Bao, Z.; Su, Y.; Lu, J.; Li, L. Role of main RNA modifications in cancer: N6-methyladenosine, 5-methylcytosine, and pseudouridine. Signal Transduct. Target. Ther. 2022, 7, 142. [Google Scholar] [CrossRef]
- Benci, J.L.; Xu, B.; Qiu, Y.; Wu, T.J.; Dada, H.; Twyman-Saint Victor, C.; Cucolo, L.; Lee, D.S.M.; Pauken, K.E.; Huang, A.C.; et al. Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell 2016, 167, 1540–1554.e12. [Google Scholar] [CrossRef]
- Zheng, Q.; Hou, J.; Zhou, Y.; Li, Z.; Cao, X. The RNA helicase DDX46 inhibits innate immunity by entrapping m6A-demethylated antiviral transcripts in the nucleus. Nat. Immunol. 2017, 18, 1094–1103. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Bajpai, A.K.; Davuluri, S.; Tiwary, K.; Narayanan, S.; Oguru, S.; Basavaraju, K.; Dayalan, D.; Thirumurugan, K.; Acharya, K.K. Systematic comparison of the protein-protein interaction databases from a user’s perspective. J. Biomed. Inform. 2020, 103, 103380. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef] [PubMed]
- Leek, J.T.; Johnson, W.E.; Parker, H.S.; Jaffe, A.E.; Storey, J.D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 2012, 28, 882–883. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, B.; Zhang, Z.; Ge, J.; Xu, L.; Mao, J.; Zhou, X.; Mao, L.; Xu, Q.; Sang, M. Predicting Prognosis and Immunotherapy Response in Multiple Cancers Based on the Association of PANoptosis-Related Genes with Tumor Heterogeneity. Genes 2023, 14, 1994. [Google Scholar] [CrossRef]
- Tomczak, K.; Czerwinska, P.; Wiznerowicz, M. The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Contemp. Oncol. 2015, 19, A68–A77. [Google Scholar] [CrossRef]
- Schaschl, H.; Wallner, B. Population-specific, recent positive directional selection suggests adaptation of human male reproductive genes to different environmental conditions. BMC Evol. Biol. 2020, 20, 27. [Google Scholar] [CrossRef]
- Wilkerson, M.D.; Hayes, D.N. ConsensusClusterPlus: A class discovery tool with confidence assessments and item tracking. Bioinformatics 2010, 26, 1572–1573. [Google Scholar] [CrossRef]
- Friedman, J.; Hastie, T.; Tibshirani, R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J. Stat. Softw. 2010, 33, 1–22. [Google Scholar] [CrossRef]
- Hothorn, T.; Lausen, B. On the exact distribution of maximally selected rank statistics. Comput. Stat. Data Anal. 2003, 43, 121–137. [Google Scholar] [CrossRef]
- Zeng, D.; Ye, Z.; Shen, R.; Yu, G.; Wu, J.; Xiong, Y.; Zhou, R.; Qiu, W.; Huang, N.; Sun, L.; et al. IOBR: Multi-Omics Immuno-Oncology Biological Research to Decode Tumor Microenvironment and Signatures. Front. Immunol. 2021, 12, 687975. [Google Scholar] [CrossRef] [PubMed]
- Malta, T.M.; Sokolov, A.; Gentles, A.J.; Burzykowski, T.; Poisson, L.; Weinstein, J.N.; Kaminska, B.; Huelsken, J.; Omberg, L.; Gevaert, O.; et al. Machine Learning Identifies Stemness Features Associated with Oncogenic Dedifferentiation. Cell 2018, 173, 338–354.e15. [Google Scholar] [CrossRef]
- Mayakonda, A.; Lin, D.C.; Assenov, Y.; Plass, C.; Koeffler, H.P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018, 28, 1747–1756. [Google Scholar] [CrossRef]
- Bonneville, R.; Krook, M.A.; Kautto, E.A.; Miya, J.; Wing, M.R.; Chen, H.Z.; Reeser, J.W.; Yu, L.; Roychowdhury, S. Landscape of Microsatellite Instability across 39 Cancer Types. JCO Precis. Oncol. 2017, 2017, PO.17.00073. [Google Scholar] [CrossRef] [PubMed]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef]
- Hu, J.; Yu, A.; Othmane, B.; Qiu, D.; Li, H.; Li, C.; Liu, P.; Ren, W.; Chen, M.; Gong, G.; et al. Siglec15 shapes a non-inflamed tumor microenvironment and predicts the molecular subtype in bladder cancer. Theranostics 2021, 11, 3089–3108. [Google Scholar] [CrossRef]
- Yoshihara, K.; Shahmoradgoli, M.; Martinez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Trevino, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.A.; Thompson, I.R.; et al. Genomics of Drug Sensitivity in Cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2013, 41, D955–D961. [Google Scholar] [CrossRef]
- Maeser, D.; Gruener, R.F.; Huang, R.S. oncoPredict: An R package for predicting in vivo or cancer patient drug response and biomarkers from cell line screening data. Brief Bioinform. 2021, 22, bbab260. [Google Scholar] [CrossRef]
- Huang, M.L.; Hung, Y.H.; Lee, W.M.; Li, R.K.; Jiang, B.R. SVM-RFE based feature selection and Taguchi parameters optimization for multiclass SVM classifier. Sci. World J. 2014, 2014, 795624. [Google Scholar] [CrossRef] [PubMed]
- Rigatti, S.J. Random forest. J. Insur. Med. 2017, 47, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Xu, J.; Hu, X.; Chen, Q.; Tu, L.; Huang, J.; Cui, J. Diagnostic method of diabetes based on support vector machine and tongue images. BioMed Res. Int. 2017, 2017, 7961494. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Danli, M.; Yu, H.; Zhuo, Z.; Ye, Z. N6-methyladenosine (m6A) as a regulator of carcinogenesis and drug resistance by targeting epithelial-mesenchymal transition and cancer stem cells. Heliyon 2023, 9, e14001. [Google Scholar] [CrossRef]
- He, L.; Li, H.; Wu, A.; Peng, Y.; Shu, G.; Yin, G. Functions of N6-methyladenosine and its role in cancer. Mol. Cancer 2019, 18, 176. [Google Scholar] [CrossRef]
- Cheng, M.; Sheng, L.; Gao, Q.; Xiong, Q.; Zhang, H.; Wu, M.; Liang, Y.; Zhu, F.; Zhang, Y.; Zhang, X.; et al. The m6A methyltransferase METTL3 promotes bladder cancer progression via AFF4/NF-kappaB/MYC signaling network. Oncogene 2019, 38, 3667–3680. [Google Scholar] [CrossRef]
- Liu, J.; Eckert, M.A.; Harada, B.T.; Liu, S.M.; Lu, Z.; Yu, K.; Tienda, S.M.; Chryplewicz, A.; Zhu, A.C.; Yang, Y.; et al. m6A mRNA methylation regulates AKT activity to promote the proliferation and tumorigenicity of endometrial cancer. Nat. Cell Biol. 2018, 20, 1074–1083. [Google Scholar] [CrossRef]
- Li, W.; Gao, Y.; Jin, X.; Wang, H.; Lan, T.; Wei, M.; Yan, W.; Wang, G.; Li, Z.; Zhao, Z.; et al. Comprehensive analysis of N6-methylandenosine regulators and m6A-related RNAs as prognosis factors in colorectal cancer. Mol. Ther. Nucleic Acids 2022, 27, 598–610. [Google Scholar] [CrossRef]
- Zhao, H.; Xu, Y.; Xie, Y.; Zhang, L.; Gao, M.; Li, S.; Wang, F. m6A Regulators Is Differently Expressed and Correlated with Immune Response of Esophageal Cancer. Front. Cell Dev. Biol. 2021, 9, 650023. [Google Scholar] [CrossRef]
- Luo, Y.; Sun, X.; Xiong, J. Characterization of m6A Regulator-Mediated Methylation Modification Patterns and Tumor Microenvironment Infiltration in Ovarian Cancer. Front. Cell Dev. Biol. 2021, 9, 794801. [Google Scholar] [CrossRef]
- Zhu, D.; Zhou, J.; Zhao, J.; Jiang, G.; Zhang, X.; Zhang, Y.; Dong, M. ZC3H13 suppresses colorectal cancer proliferation and invasion via inactivating Ras-ERK signaling. J. Cell. Physiol. 2019, 234, 8899–8907. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Wang, J.Z.; Wei, J.F.; Lu, C. Role of m6A methyltransferase component VIRMA in multiple human cancers (Review). Cancer Cell Int. 2021, 21, 172. [Google Scholar] [CrossRef]
- Banerji, J.; Sands, J.; Strominger, J.L.; Spies, T. A gene pair from the human major histocompatibility complex encodes large proline-rich proteins with multiple repeated motifs and a single ubiquitin-like domain. Proc. Natl. Acad. Sci. USA 1990, 87, 2374–2378. [Google Scholar] [CrossRef]
- Pan, Q.; Ning, Y.; Chen, L.Z.; Zhang, S.; Liu, Z.Z.; Yang, X.X.; Wei, W.; Wei, H.; Li, Q.G.; Yue, H.N.; et al. Association of MHC class-III gene polymorphisms with ER-positive breast cancer in Chinese Han population. Genet. Mol. Res. 2012, 11, 4299–4306. [Google Scholar] [CrossRef]
- Walsh, K.M.; Gorlov, I.P.; Hansen, H.M.; Wu, X.; Spitz, M.R.; Zhang, H.; Lu, E.Y.; Wenzlaff, A.S.; Sison, J.D.; Wei, C.; et al. Fine-mapping of the 5p15.33, 6p22.1-p21.31, and 15q25.1 regions identifies functional and histology-specific lung cancer susceptibility loci in African-Americans. Cancer Epidemiol. Biomark. Prev. 2013, 22, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Nieters, A.; Conde, L.; Slager, S.L.; Brooks-Wilson, A.; Morton, L.; Skibola, D.R.; Novak, A.J.; Riby, J.; Ansell, S.M.; Halperin, E.; et al. PRRC2A and BCL2L11 gene variants influence risk of non-Hodgkin lymphoma: Results from the InterLymph consortium. Blood 2012, 120, 4645–4648. [Google Scholar] [CrossRef]
- Liu, K.; Gao, Y.; Gan, K.; Wu, Y.; Xu, B.; Zhang, L.; Chen, M. Prognostic Roles of N6-Methyladenosine METTL3 in Different Cancers: A System Review and Meta-Analysis. Cancer Control 2021, 28, 1073274821997455. [Google Scholar] [CrossRef]
- Guan, Q.; Lin, H.; Miao, L.; Guo, H.; Chen, Y.; Zhuo, Z.; He, J. Functions, mechanisms, and therapeutic implications of METTL14 in human cancer. J. Hematol. Oncol. 2022, 15, 13. [Google Scholar] [CrossRef] [PubMed]
- Gong, P.J.; Shao, Y.C.; Yang, Y.; Song, W.J.; He, X.; Zeng, Y.F.; Huang, S.R.; Wei, L.; Zhang, J.W. Analysis of N6-Methyladenosine Methyltransferase Reveals METTL14 and ZC3H13 as Tumor Suppressor Genes in Breast Cancer. Front. Oncol. 2020, 10, 578963. [Google Scholar] [CrossRef]
- Su, Z.; Xu, L.; Dai, X.; Zhu, M.; Chen, X.; Li, Y.; Li, J.; Ge, R.; Cheng, B.; Wang, Y. Prognostic and clinicopathological value of m6A regulators in human cancers: A meta-analysis. Aging 2022, 14, 8818–8838. [Google Scholar] [CrossRef]
- Zheng, F.; Du, F.; Qian, H.; Zhao, J.; Wang, X.; Yue, J.; Hu, N.; Si, Y.; Xu, B.; Yuan, P. Expression and clinical prognostic value of m6A RNA methylation modification in breast cancer. Biomark. Res. 2021, 9, 28. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Liu, J.; Li, S.; Xu, P. Analysis of m6A regulators related immune characteristics in ankylosing spondylitis by integrated bioinformatics and computational strategies. Sci. Rep. 2024, 14, 2724. [Google Scholar] [CrossRef]
- Quan, Y.; Zhang, X.; Ping, H. Construction of a risk prediction model using m6A RNA methylation regulators in prostate cancer: Comprehensive bioinformatic analysis and histological validation. Cancer Cell Int. 2022, 22, 33. [Google Scholar] [CrossRef]
- Ding, Y.; Bajpai, A.K.; Wu, F.; Lu, W.; Xu, L.; Mao, J.; Li, Q.; Pan, Q.; Lu, L.; Wang, X. 5-methylcytosine RNA modification regulators-based patterns and features of immune microenvironment in acute myeloid leukemia. Aging 2024, 16, 2340–2361. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Gao, W.; Liu, Z.; Yu, S.; Jian, H.; Hou, Z.; Zeng, P. Comprehensive analysis of m6A regulators associated with immune infiltration in Hepatitis B virus-related hepatocellular carcinoma. BMC Gastroenterol. 2023, 23, 259. [Google Scholar] [CrossRef]
- Lee, S.; Seo, M.H.; Shin, Y. The lasso for high dimensional regression with a possible change point. J. R. Stat. Soc. Ser. B Stat. Methodol. 2016, 78, 193–210. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Jiang, X.; Li, X.; Yan, D.; Liu, J.; Yang, J.; Yan, S. The methylation modification of m6A regulators contributes to the prognosis of head and neck squamous cell carcinoma. Ann. Transl. Med. 2021, 9, 1346. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Wu, G.; Yang, L.; Wu, Q.; Yuan, L. The predictive significance of a 5-m6A RNA methylation regulator signature in colorectal cancer. Heliyon 2023, 9, e20172. [Google Scholar] [CrossRef]
- Ji, L.; Chen, S.; Gu, L.; Zhang, X. Exploration of Potential Roles of m6A Regulators in Colorectal Cancer Prognosis. Front. Oncol. 2020, 10, 768. [Google Scholar] [CrossRef]
- Li, W.; Liu, J.; Ma, Z.; Zhai, X.; Cheng, B.; Zhao, H. m6A RNA Methylation Regulators Elicit Malignant Progression and Predict Clinical Outcome in Hepatocellular Carcinoma. Dis. Markers 2021, 2021, 8859590. [Google Scholar] [CrossRef]
- Klingbeil, K.D.; Tang, J.P.; Graham, D.S.; Lofftus, S.Y.; Jaiswal, A.K.; Lin, T.L.; Frias, C.; Chen, L.Y.; Nakasaki, M.; Dry, S.M.; et al. IGF2BP3 as a Prognostic Biomarker in Well-Differentiated/Dedifferentiated Liposarcoma. Cancers 2023, 15, 4489. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Zheng, X.; Sun, Y.; Yu, J.; Sheng, M.; Yan, S.; Zhu, Q.; Lan, Q. Identification of IGF2BP3 as an Adverse Prognostic Biomarker of Gliomas. Front. Genet. 2021, 12, 743738. [Google Scholar] [CrossRef]
- Huang, W.; Zhu, L.; Huang, H.; Li, Y.; Wang, G.; Zhang, C. IGF2BP3 overexpression predicts poor prognosis and correlates with immune infiltration in bladder cancer. BMC Cancer 2023, 23, 116. [Google Scholar] [CrossRef]
- Bense, R.D.; Sotiriou, C.; Piccart-Gebhart, M.J.; Haanen, J.; van Vugt, M.; de Vries, E.G.E.; Schroder, C.P.; Fehrmann, R.S.N. Relevance of Tumor-Infiltrating Immune Cell Composition and Functionality for Disease Outcome in Breast Cancer. J. Natl. Cancer Inst. 2017, 109, djw192. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Zhang, J.; Zeng, D.; Sun, H.; Rong, X.; Shi, M.; Bin, J.; Liao, Y.; Liao, W. Immune cell infiltration as a biomarker for the diagnosis and prognosis of stage I-III colon cancer. Cancer Immunol. Immunother. 2019, 68, 433–442. [Google Scholar] [CrossRef]
- Zuo, S.; Wei, M.; Wang, S.; Dong, J.; Wei, J. Pan-Cancer Analysis of Immune Cell Infiltration Identifies a Prognostic Immune-Cell Characteristic Score (ICCS) in Lung Adenocarcinoma. Front. Immunol. 2020, 11, 1218. [Google Scholar] [CrossRef]
- Guo, Y.; Heng, Y.; Chen, H.; Huang, Q.; Wu, C.; Tao, L.; Zhou, L. Prognostic Values of METTL3 and Its Roles in Tumor Immune Microenvironment in Pan-Cancer. J. Clin. Med. 2022, 12, 155. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.W.; Han, S.W.; Bae, J.M.; Jang, H.; Han, H.; Kim, H.; Bang, D.; Jeong, S.Y.; Park, K.J.; Kang, G.H.; et al. Tumor Mutation Burden and Prognosis in Patients with Colorectal Cancer Treated with Adjuvant Fluoropyrimidine and Oxaliplatin. Clin. Cancer Res. 2019, 25, 6141–6147. [Google Scholar] [CrossRef]
- Qu, F.; Tsegay, P.S.; Liu, Y. N6-Methyladenosine, DNA Repair, and Genome Stability. Front. Mol. Biosci. 2021, 8, 645823. [Google Scholar] [CrossRef]
- Huang, W.; Chen, T.Q.; Fang, K.; Zeng, Z.C.; Ye, H.; Chen, Y.Q. N6-methyladenosine methyltransferases: Functions, regulation, and clinical potential. J. Hematol. Oncol. 2021, 14, 117. [Google Scholar] [CrossRef]
- Han, X.; Guo, J.; Fan, Z. Interactions between m6A modification and miRNAs in malignant tumors. Cell Death Dis. 2021, 12, 598. [Google Scholar] [CrossRef] [PubMed]
- Goa, K.L.; Faulds, D. Vinorelbine. A review of its pharmacological properties and clinical use in cancer chemotherapy. Drugs Aging 1994, 5, 200–234. [Google Scholar] [CrossRef] [PubMed]
- Salerni, B.L.; Bates, D.J.; Albershardt, T.C.; Lowrey, C.H.; Eastman, A. Vinblastine induces acute, cell cycle phase-independent apoptosis in some leukemias and lymphomas and can induce acute apoptosis in others when Mcl-1 is suppressed. Mol. Cancer Ther. 2010, 9, 791–802. [Google Scholar] [CrossRef] [PubMed]
- Dhyani, P.; Quispe, C.; Sharma, E.; Bahukhandi, A.; Sati, P.; Attri, D.C.; Szopa, A.; Sharifi-Rad, J.; Docea, A.O.; Mardare, I.; et al. Anticancer potential of alkaloids: A key emphasis to colchicine, vinblastine, vincristine, vindesine, vinorelbine and vincamine. Cancer Cell Int. 2022, 22, 206. [Google Scholar] [CrossRef]
- Omura, S.; Asami, Y.; Crump, A. Staurosporine: New lease of life for parent compound of today’s novel and highly successful anti-cancer drugs. J. Antibiot. 2018, 71, 688–701. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, B.; Hao, Y.; Liu, H.; Wu, J.; Lu, L.; Wang, X.; Bajpai, A.K.; Yang, X. Interplay of RNA m6A Modification-Related Geneset in Pan-Cancer. Biomedicines 2024, 12, 2211. https://doi.org/10.3390/biomedicines12102211
Zhang B, Hao Y, Liu H, Wu J, Lu L, Wang X, Bajpai AK, Yang X. Interplay of RNA m6A Modification-Related Geneset in Pan-Cancer. Biomedicines. 2024; 12(10):2211. https://doi.org/10.3390/biomedicines12102211
Chicago/Turabian StyleZhang, Boyu, Yajuan Hao, Haiyan Liu, Jiarun Wu, Lu Lu, Xinfeng Wang, Akhilesh K. Bajpai, and Xi Yang. 2024. "Interplay of RNA m6A Modification-Related Geneset in Pan-Cancer" Biomedicines 12, no. 10: 2211. https://doi.org/10.3390/biomedicines12102211
APA StyleZhang, B., Hao, Y., Liu, H., Wu, J., Lu, L., Wang, X., Bajpai, A. K., & Yang, X. (2024). Interplay of RNA m6A Modification-Related Geneset in Pan-Cancer. Biomedicines, 12(10), 2211. https://doi.org/10.3390/biomedicines12102211