
Unveiling the Molecular Mechanisms of Glioblastoma through an Integrated Network-Based Approach
 , , , ,
, , , ,  ,
,
Abstract
1. Introduction
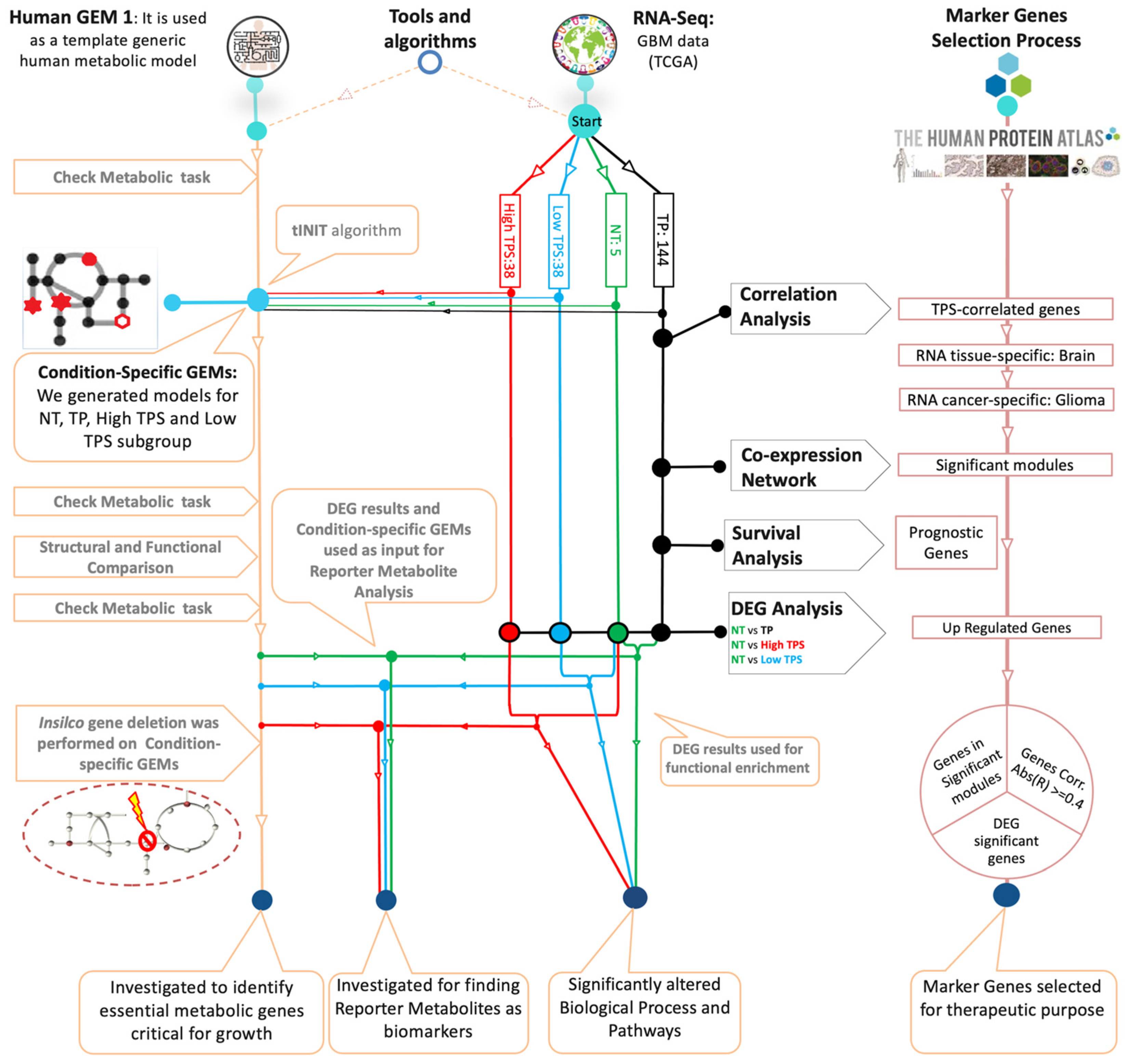
2. Materials and Methods
2.1. Data Collection and Processing
2.2. Transcriptome Analysis
2.3. Genome-Scale Metabolic Modelling
2.4. Reporter Metabolite Analysis Using GEMs
2.5. Gene Essentiality Analysis Using GEMs
2.6. Survival Analysis and TPS Correlation
2.7. Gene Co-Expression Network Analysis
3. Results
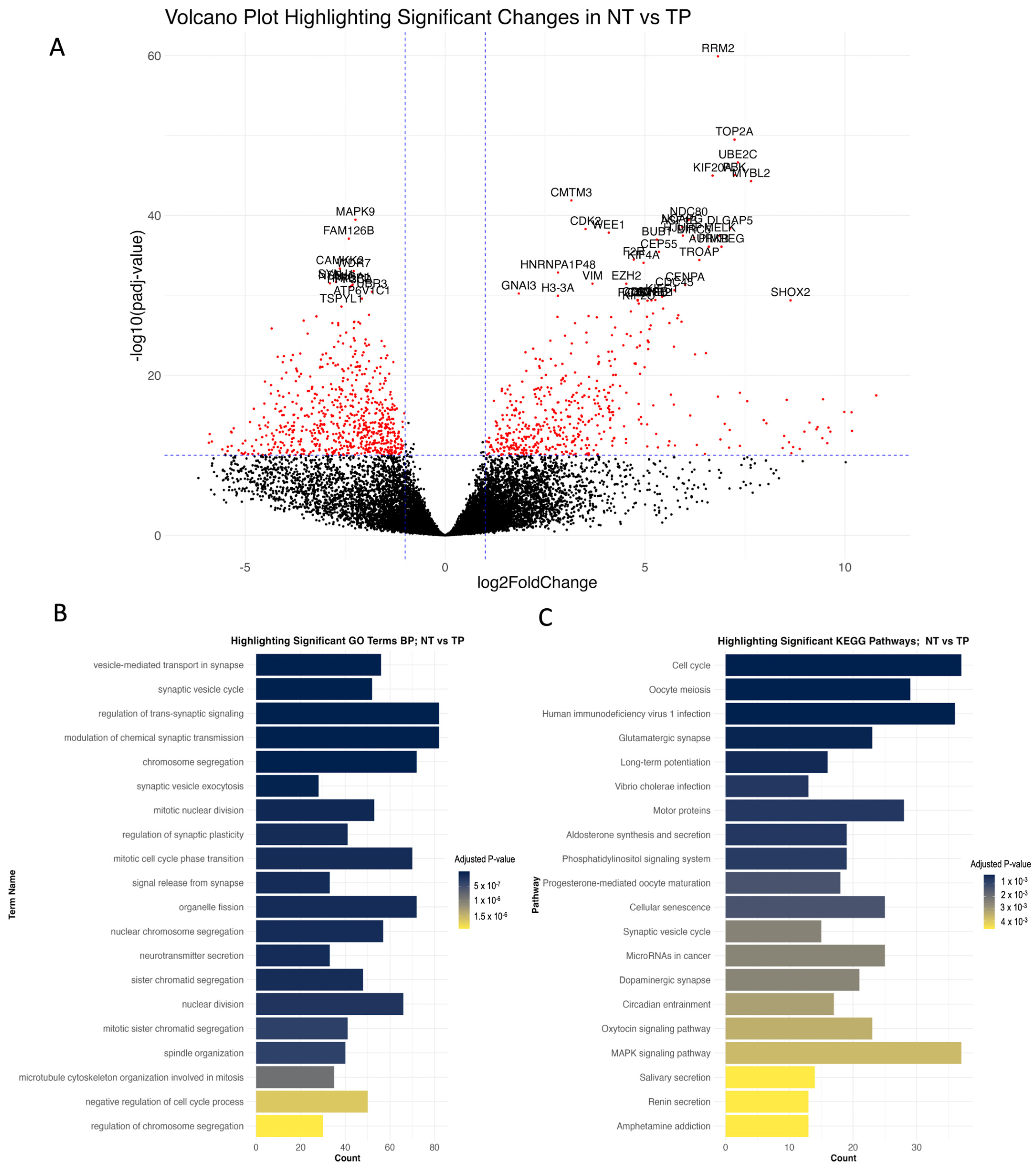
3.1. Gene-Set Enrichment Analysis
3.2. Structural Comparison of Generated Metabolic Models
3.3. Reporter Metabolite Analysis
3.4. Gene Essentiality Analysis
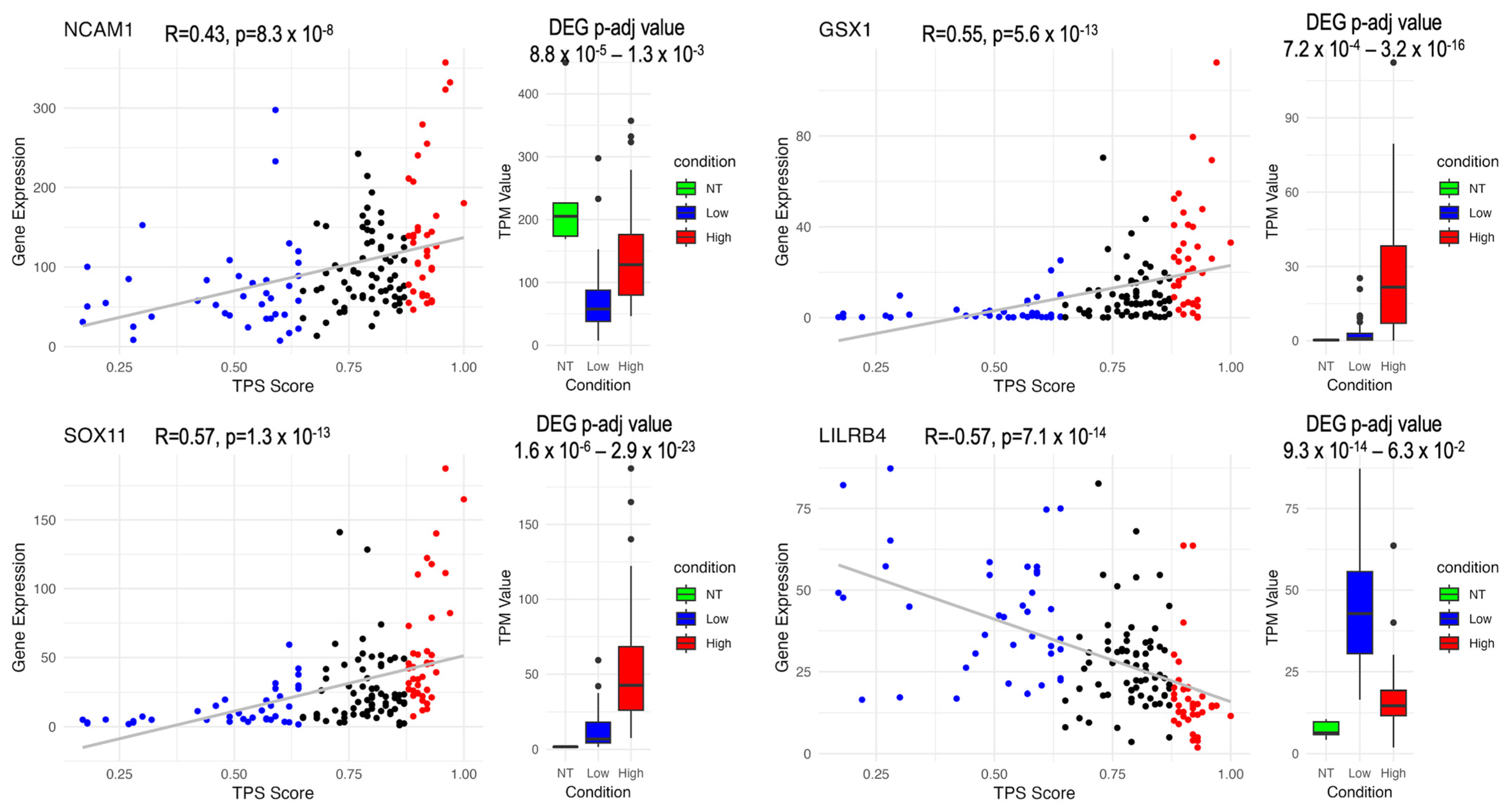
3.5. Survival Analysis and Correlation Analysis
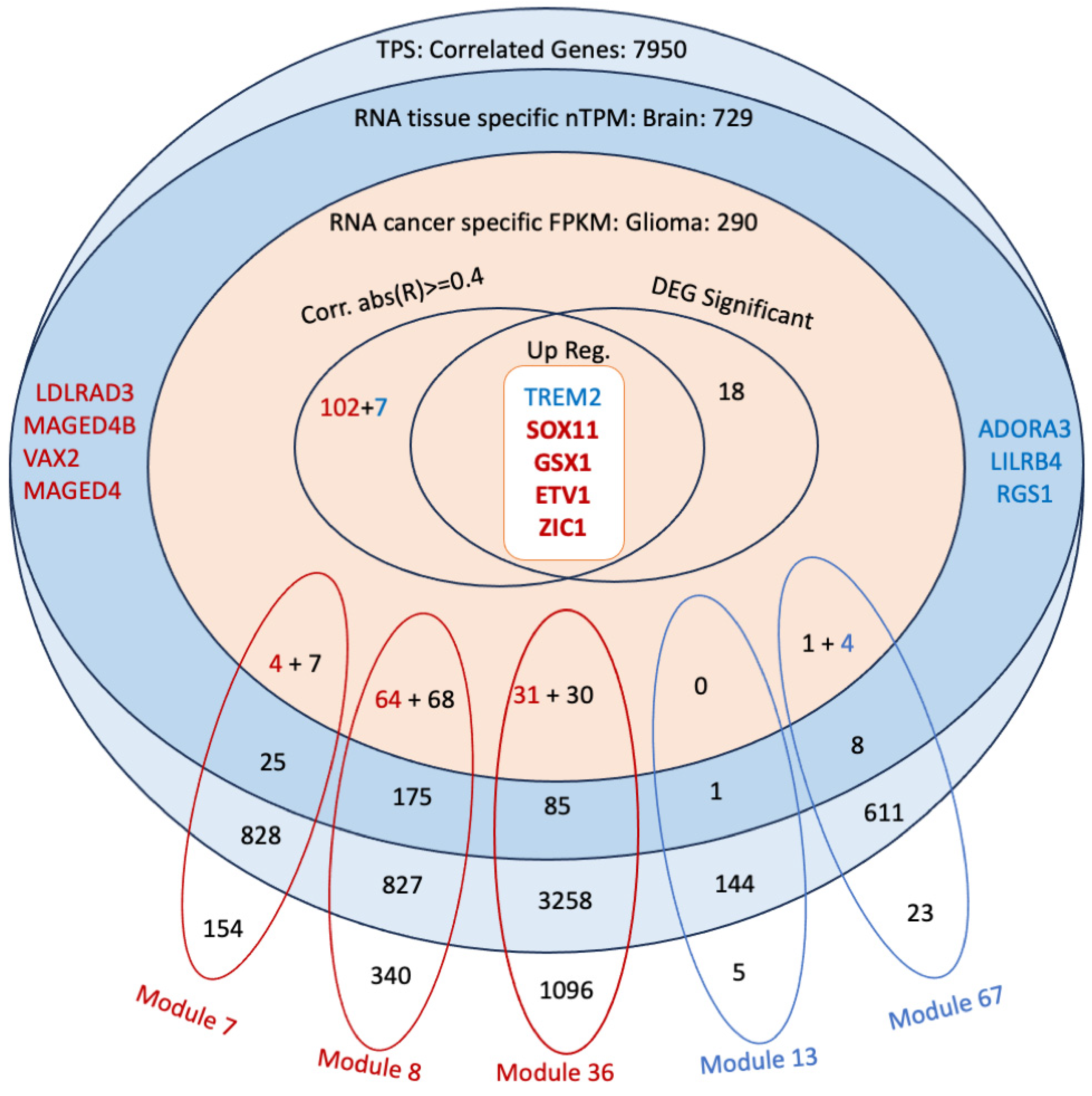
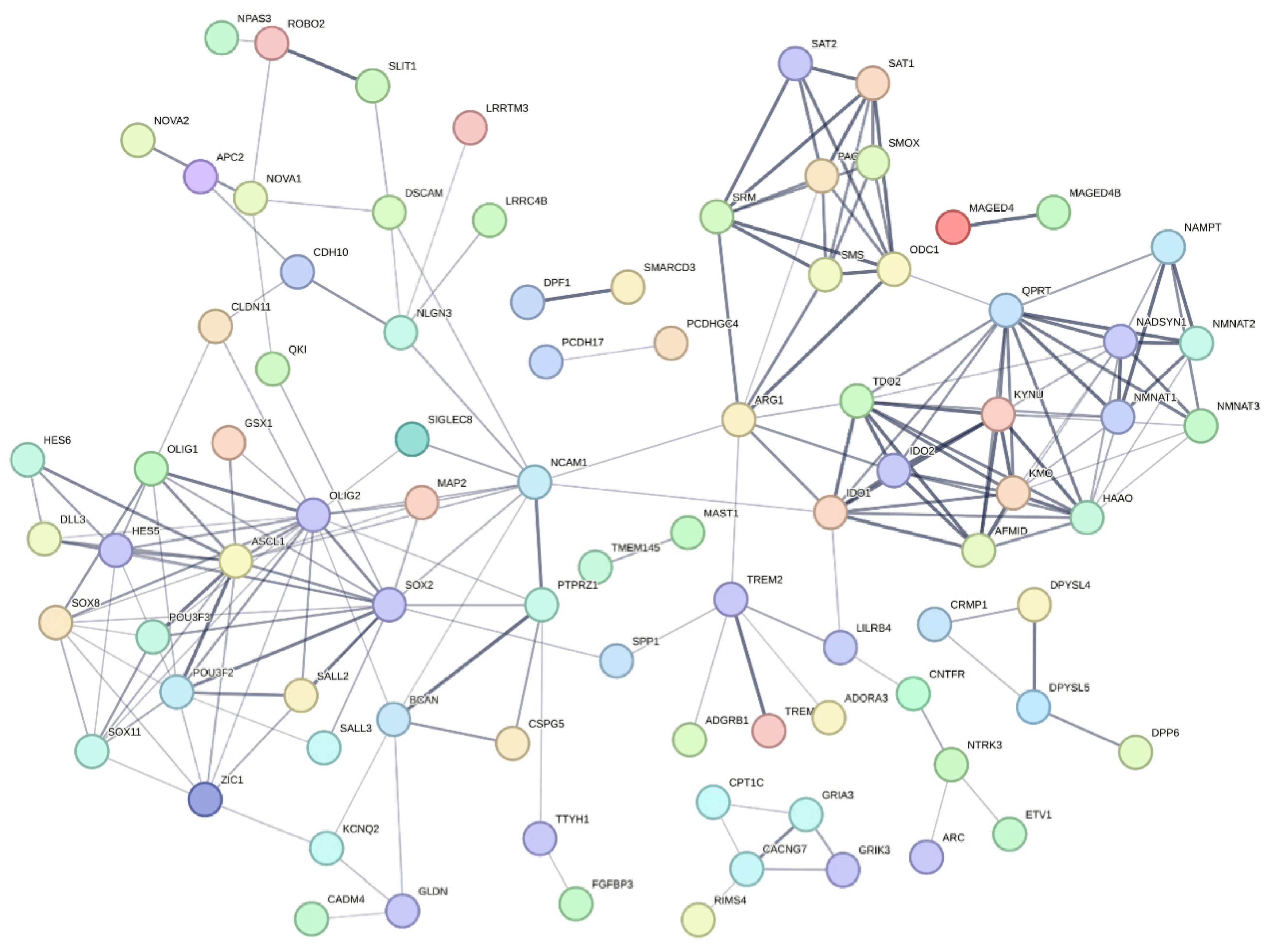
3.6. Co-Expression Analysis Reveals Significant Modules Relevant to the Correlated Genes
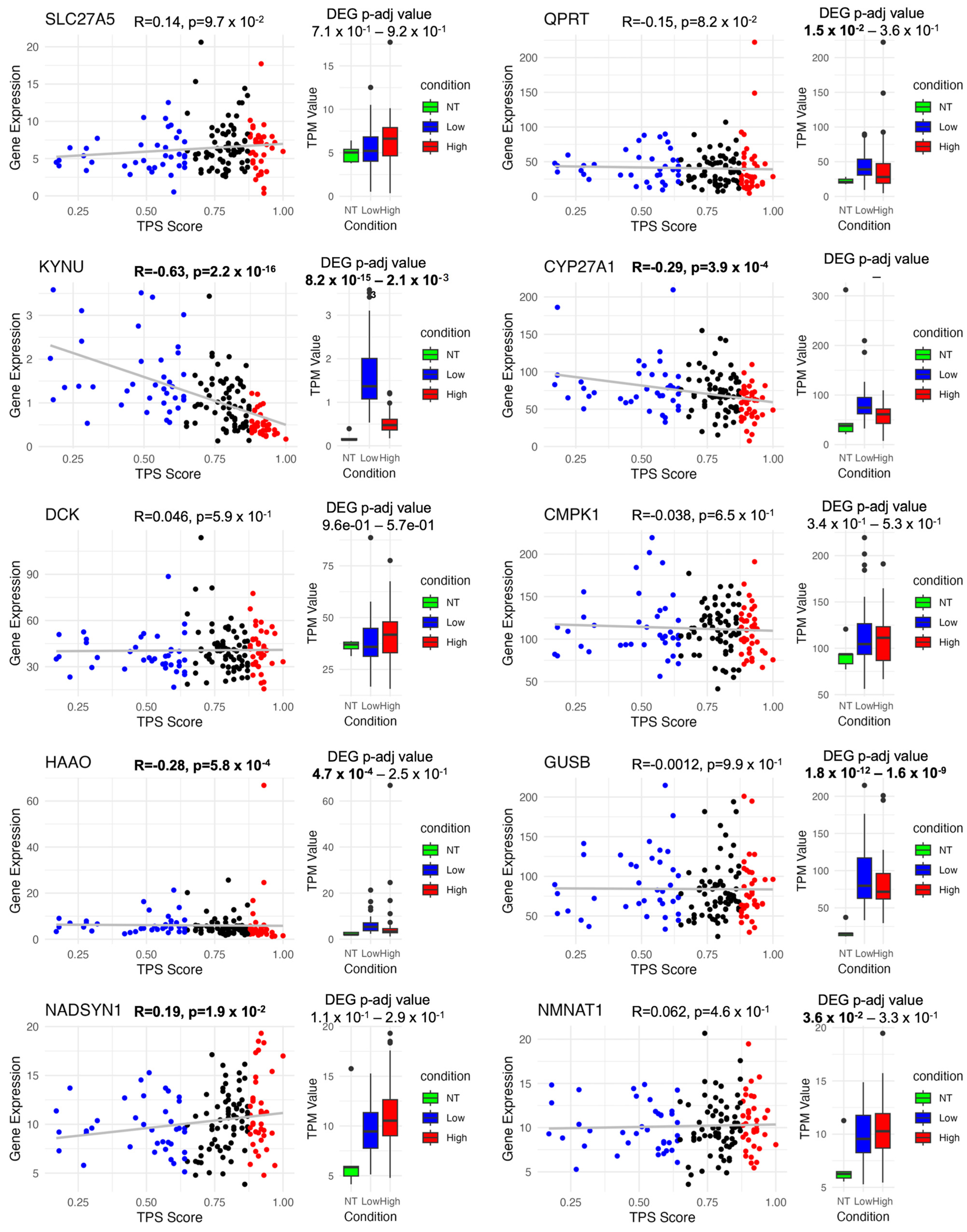
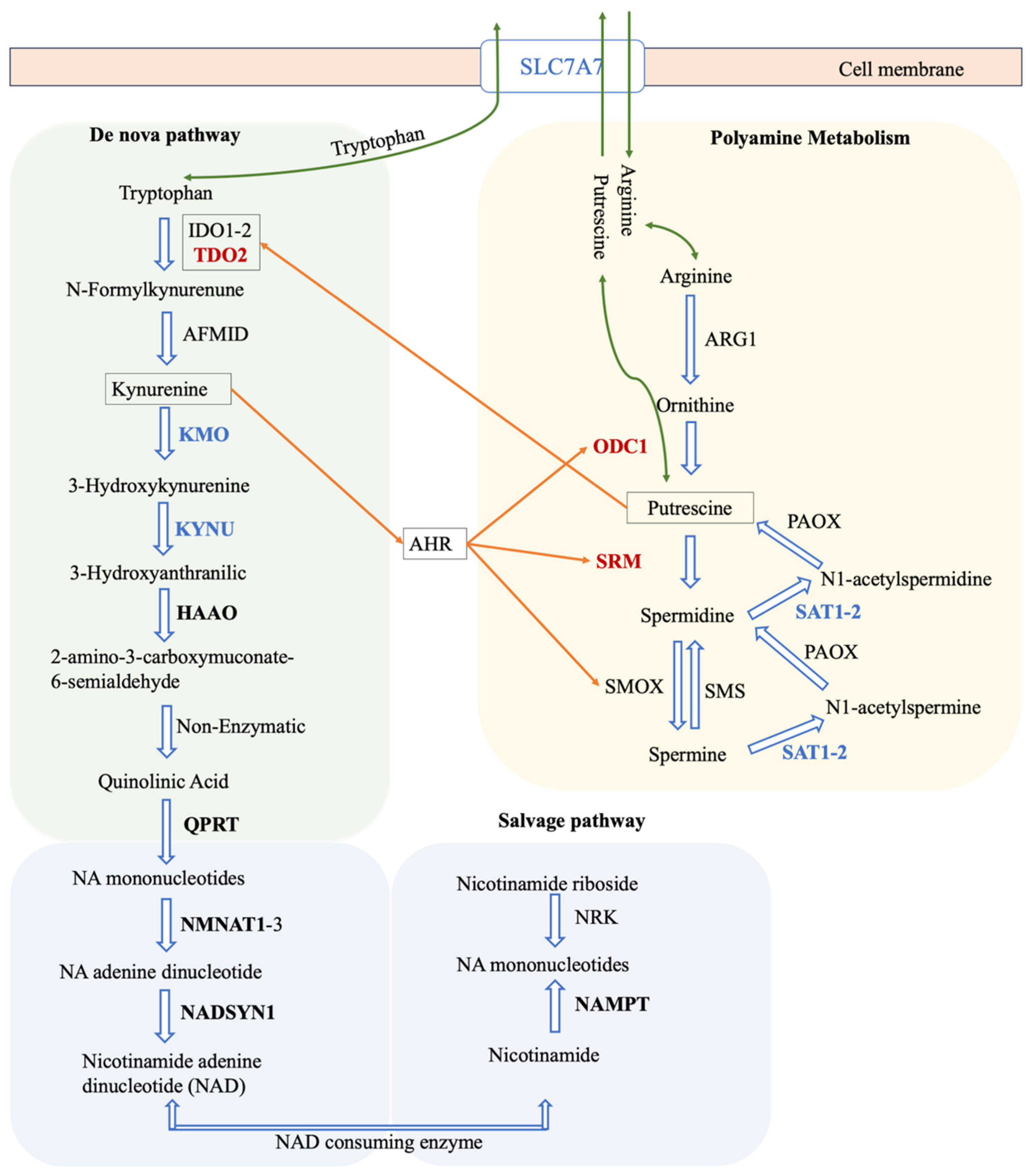
3.7. Discovery of Potential Genes for the Development of GBM Therapy
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fernandes, C.; Costa, A.; Osorio, L.; Lago, R.C.; Linhares, P.; Carvalho, B.; Caeiro, C. Current Standards of Care in Glioblastoma Therapy. In Glioblastoma; De Vleeschouwer, S., Ed.; Codon Publication: Brisbane, Australia, 2017. [Google Scholar]
- Zhang, Y.; Xia, Q.; Lin, J. Identification of the potential oncogenes in glioblastoma based on bioinformatic analysis and elucidation of the underlying mechanisms. Oncol. Rep. 2018, 40, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Kotecha, R.; Odia, Y.; Khosla, A.A.; Ahluwalia, M.S. Key Clinical Principles in the Management of Glioblastoma. JCO Oncol. Pract. 2023, 19, 180–189. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Patil, N.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2013-2017. Neuro Oncol. 2020, 22, iv1–iv96. [Google Scholar] [CrossRef] [PubMed]
- Kaynar, A.; Altay, O.; Li, X.; Zhang, C.; Turkez, H.; Uhlen, M.; Shoaie, S.; Mardinoglu, A. Systems Biology Approaches to Decipher the Underlying Molecular Mechanisms of Glioblastoma Multiforme. Int. J. Mol. Sci. 2021, 22, 13213. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Li, M.; Dey, R.; Chen, Y. Nanomaterials for cancer therapy: Current progress and perspectives. J. Hematol. Oncol. 2021, 14, 85. [Google Scholar] [CrossRef] [PubMed]
- Sousa, Â.; Barud, H.D.S.; Xu, Z.P.; Soares, C.P. Nanotechnology for Precision Cancer Therapy: Advances in gene therapy, immunotherapy, and 3D bioprinting. Front. Nanotechnol. 2021, 3, 776898. [Google Scholar] [CrossRef]
- Tian, H.; Zhang, T.; Qin, S.; Huang, Z.; Zhou, L.; Shi, J.; Nice, E.C.; Xie, N.; Huang, C.; Shen, Z. Enhancing the therapeutic efficacy of nanoparticles for cancer treatment using versatile targeted strategies. J. Hematol. Oncol. 2022, 15, 132. [Google Scholar] [CrossRef]
- De Simone, M.; Iaconetta, G.; Palermo, G.; Fiorindi, A.; Schaller, K.; De Maria, L. Clustering Functional Magnetic Resonance Imaging Time Series in Glioblastoma Characterization: A Review of the Evolution, Applications, and Potentials. Brain Sci. 2024, 14, 296. [Google Scholar] [CrossRef] [PubMed]
- De Simone, M.; Conti, V.; Palermo, G.; De Maria, L.; Iaconetta, G. Advancements in Glioma Care: Focus on Emerging Neurosurgical Techniques. Biomedicines 2024, 12, 8. [Google Scholar] [CrossRef]
- De Vleeschouwer, S.; Bergers, G. Glioblastoma: To Target the Tumor Cell or the Microenvironment? In Glioblastoma; De Vleeschouwer, S., Ed.; Codon Publication: Brisbane, Australia, 2017. [Google Scholar]
- Hambardzumyan, D.; Bergers, G. Glioblastoma: Defining Tumor Niches. Trends Cancer 2015, 1, 252–265. [Google Scholar] [CrossRef]
- Ho, I.A.W.; Shim, W.S.N. Contribution of the Microenvironmental Niche to Glioblastoma Heterogeneity. Biomed. Res. Int. 2017, 2017, 9634172. [Google Scholar] [CrossRef] [PubMed]
- Matias, D.; Balca-Silva, J.; da Graca, G.C.; Wanjiru, C.M.; Macharia, L.W.; Nascimento, C.P.; Roque, N.R.; Coelho-Aguiar, J.M.; Pereira, C.M.; Dos Santos, M.F.; et al. Microglia/Astrocytes-Glioblastoma Crosstalk: Crucial Molecular Mechanisms and Microenvironmental Factors. Front. Cell Neurosci. 2018, 12, 235. [Google Scholar] [CrossRef]
- Eder, K.; Kalman, B. Molecular heterogeneity of glioblastoma and its clinical relevance. Pathol. Oncol. Res. 2014, 20, 777–787. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Wu, Y.; Fletcher, M.; Gu, Z.; Wang, Q.; Costa, B.; Bertoni, A.; Man, K.H.; Schlotter, M.; Felsberg, J.; Mangei, J.; et al. Glioblastoma epigenome profiling identifies SOX10 as a master regulator of molecular tumour subtype. Nat. Commun. 2020, 11, 6434. [Google Scholar] [CrossRef] [PubMed]
- Komori, T. Grading of adult diffuse gliomas according to the 2021 WHO Classification of Tumors of the Central Nervous System. Lab Investig. 2022, 102, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Ohgaki, H.; Kleihues, P. The definition of primary and secondary glioblastoma. Clin. Cancer Res. 2013, 19, 764–772. [Google Scholar] [CrossRef] [PubMed]
- Szopa, W.; Burley, T.A.; Kramer-Marek, G.; Kaspera, W. Diagnostic and Therapeutic Biomarkers in Glioblastoma: Current Status and Future Perspectives. Biomed. Res. Int. 2017, 2017, 8013575. [Google Scholar] [CrossRef] [PubMed]
- Turkez, H.; Tozlu, O.O.; Arslan, M.E.; Mardinoglu, A. Safety and Efficacy Assessments to Take Antioxidants in Glioblastoma Therapy: From In Vitro Experiences to Animal and Clinical Studies. Neurochem. Int. 2021, 150, 105168. [Google Scholar] [CrossRef]
- Behnan, J.; Finocchiaro, G.; Hanna, G. The landscape of the mesenchymal signature in brain tumours. Brain 2019, 142, 847–866. [Google Scholar] [CrossRef] [PubMed]
- Coller, H.A. Is cancer a metabolic disease? Am. J. Pathol. 2014, 184, 4–17. [Google Scholar] [CrossRef]
- Agnihotri, S.; Zadeh, G. Metabolic reprogramming in glioblastoma: The influence of cancer metabolism on epigenetics and unanswered questions. Neuro Oncol. 2016, 18, 160–172. [Google Scholar] [CrossRef]
- Ozcan, E.; Cakir, T. Reconstructed Metabolic Network Models Predict Flux-Level Metabolic Reprogramming in Glioblastoma. Front. Neurosci. 2016, 10, 156. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Mardinoglu, A.; Boren, J.; Smith, U.; Uhlen, M.; Nielsen, J. Systems biology in hepatology: Approaches and applications. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 365–377. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, J. Systems Biology of Metabolism. Annu. Rev. Biochem. 2017, 86, 245–275. [Google Scholar] [CrossRef] [PubMed]
- Mardinoglu, A.; Palsson, B.Ø. Genome-scale models in human metabologenomics. Nat. Rev. Genet. 2024, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Larsson, I.; Uhlen, M.; Zhang, C.; Mardinoglu, A. Genome-Scale Metabolic Modeling of Glioblastoma Reveals Promising Targets for Drug Development. Front. Genet. 2020, 11, 381. [Google Scholar] [CrossRef]
- Grossman, R.L.; Heath, A.P.; Ferretti, V.; Varmus, H.E.; Lowy, D.R.; Kibbe, W.A.; Staudt, L.M. Toward a Shared Vision for Cancer Genomic Data. N. Engl. J. Med. 2016, 375, 1109–1112. [Google Scholar] [CrossRef]
- RStudio Team. RStudio: Integrated Development for R; RStudio, PBC: Boston, MA, USA, 2020. [Google Scholar]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Howe, K.L.; Achuthan, P.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; Bhai, J.; et al. Ensembl 2021. Nucleic Acids Res 2021, 49, D884–D891. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.L.; Kocabas, P.; Wang, H.; Cholley, P.E.; Cook, D.; Nilsson, A.; Anton, M.; Ferreira, R.; Domenzain, I.; Billa, V.; et al. An atlas of human metabolism. Sci. Signal 2020, 13, eaaz1482. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Marcisauskas, S.; Sanchez, B.J.; Domenzain, I.; Hermansson, D.; Agren, R.; Nielsen, J.; Kerkhoven, E.J. RAVEN 2.0: A versatile toolbox for metabolic network reconstruction and a case study on Streptomyces coelicolor. PLoS Comput. Biol. 2018, 14, e1006541. [Google Scholar] [CrossRef]
- Agren, R.; Mardinoglu, A.; Asplund, A.; Kampf, C.; Uhlen, M.; Nielsen, J. Identification of anticancer drugs for hepatocellular carcinoma through personalized genome-scale metabolic modeling. Mol. Syst. Biol. 2014, 10, 721. [Google Scholar] [CrossRef]
- Inc, T.M. Optimization Toolbox Version: 9.14.0 (R2023a); The MathWorks Inc.: Natick, MA, USA, 2023. [Google Scholar]
- Franz, M.; Lopes, C.T.; Fong, D.; Kucera, M.; Cheung, M.; Siper, M.C.; Huck, G.; Dong, Y.; Sumer, O.; Bader, G.D. Cytoscape.js 2023 update: A graph theory library for visualization and analysis. Bioinformatics 2023, 39, btad031. [Google Scholar] [CrossRef]
- Gousias, K.; Theocharous, T.; Simon, M. Mechanisms of Cell Cycle Arrest and Apoptosis in Glioblastoma. Biomedicines 2022, 10, 564. [Google Scholar] [CrossRef] [PubMed]
- Pei, Z.; Lee, K.C.; Khan, A.; Erisnor, G.; Wang, H.Y. Pathway analysis of glutamate-mediated, calcium-related signaling in glioma progression. Biochem. Pharmacol. 2020, 176, 113814. [Google Scholar] [CrossRef]
- Sami, A.; Karsy, M. Targeting the PI3K/AKT/mTOR signaling pathway in glioblastoma: Novel therapeutic agents and advances in understanding. Tumour Biol. 2013, 34, 1991–2002. [Google Scholar] [CrossRef]
- Daniel, P.M.; Filiz, G.; Mantamadiotis, T. Sensitivity of GBM cells to cAMP agonist-mediated apoptosis correlates with CD44 expression and agonist resistance with MAPK signaling. Cell Death Dis. 2016, 7, e2494. [Google Scholar] [CrossRef] [PubMed]
- Morandi, F.; Horenstein, A.L.; Malavasi, F. The Key Role of NAD(+) in Anti-Tumor Immune Response: An Update. Front. Immunol. 2021, 12, 658263. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, S.K.; Venneti, S. Glutamine Metabolism in Brain Tumors. Cancers 2019, 11, 1628. [Google Scholar] [CrossRef]
- Kośliński, P.; Pluskota, R.; Koba, M.; Siedlecki, Z.; Śniegocki, M. Comparative Analysis of Amino Acid Profiles in Patients with Glioblastoma and Meningioma Using Liquid Chromatography Electrospray Ionization Tandem Mass Spectrometry (LC-ESI-MS/MS). Molecules 2023, 28, 7699. [Google Scholar] [CrossRef]
- Gouasmi, R.; Ferraro-Peyret, C.; Nancey, S.; Coste, I.; Renno, T.; Chaveroux, C.; Aznar, N.; Ansieau, S. The Kynurenine Pathway and Cancer: Why Keep It Simple When You Can Make It Complicated. Cancers 2022, 14, 2793. [Google Scholar] [CrossRef] [PubMed]
- Navas, L.E.; Carnero, A. NAD+ metabolism, stemness, the immune response, and cancer. Signal Transduct. Target. Ther. 2021, 6, 2. [Google Scholar] [CrossRef] [PubMed]
- Stine, Z.E.; Schug, Z.T.; Salvino, J.M.; Dang, C.V. Targeting cancer metabolism in the era of precision oncology. Nat. Rev. Drug Discov. 2022, 21, 141–162. [Google Scholar] [CrossRef] [PubMed]
- Seyfried, T.N.; Kiebish, M.A.; Marsh, J.; Shelton, L.M.; Huysentruyt, L.C.; Mukherjee, P. Metabolic management of brain cancer. Biochim. Biophys. Acta (BBA) Bioenerg. 2011, 1807, 577–594. [Google Scholar] [CrossRef]
- Shakya, S.; Gromovsky, A.D.; Hale, J.S.; Knudsen, A.M.; Prager, B.; Wallace, L.C.; Penalva, L.O.F.; Brown, H.A.; Kristensen, B.W.; Rich, J.N.; et al. Altered lipid metabolism marks glioblastoma stem and non-stem cells in separate tumor niches. Acta Neuropathol. Commun. 2021, 9, 101. [Google Scholar] [CrossRef]
- Pridham, K.J.; Hutchings, K.R.; Beck, P.; Liu, M.; Xu, E.; Saechin, E.; Bui, V.; Patel, C.; Solis, J.; Huang, L.; et al. Selective regulation of chemosensitivity in glioblastoma by phosphatidylinositol 3-kinase beta. iScience 2024, 27, 109921. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Madrid, P.; Fluitt, A.; Stahl, A.; Xie, X.S. Development and validation of a high-throughput screening assay for human long-chain fatty acid transport proteins 4 and 5. J. Biomol. Screen. 2010, 15, 488–497. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.Q.; De Marchi, T.; Timmermans, A.; Trapman-Jansen, A.M.; Foekens, R.; Look, M.P.; Smid, M.; van Deurzen, C.H.; Span, P.N.; Sweep, F.C.; et al. Prognostic significance of nuclear expression of UMP-CMP kinase in triple negative breast cancer patients. Sci. Rep. 2016, 6, 32027. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Li, L.; Wang, Z.; Pang, J.; Guan, X.; Yuan, Y.; Xia, Z.; Yi, W. A comprehensive analysis of the role of QPRT in breast cancer. Sci. Rep. 2023, 13, 15414. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Feng, X.; Lai, J.; Yi, W.; Yang, J.; Du, T.; Long, X.; Zhang, Y.; Xiao, Y. A novel role of kynureninase in the growth control of breast cancer cells and its relationships with breast cancer. J. Cell Mol. Med. 2019, 23, 6700–6707. [Google Scholar] [CrossRef] [PubMed]
- Cali, J.J.; Russell, D.W. Characterization of human sterol 27-hydroxylase. A mitochondrial cytochrome P-450 that catalyzes multiple oxidation reaction in bile acid biosynthesis. J. Biol. Chem. 1991, 266, 7774–7778. [Google Scholar] [CrossRef] [PubMed]
- Mast, N.; Anderson, K.W.; Lin, J.B.; Li, Y.; Turko, I.V.; Tatsuoka, C.; Bjorkhem, I.; Pikuleva, I.A. Cytochrome P450 27A1 Deficiency and Regional Differences in Brain Sterol Metabolism Cause Preferential Cholestanol Accumulation in the Cerebellum. J. Biol. Chem. 2017, 292, 4913–4924. [Google Scholar] [CrossRef] [PubMed]
- Pikuleva, I.A.; Babiker, A.; Waterman, M.R.; Bjorkhem, I. Activities of recombinant human cytochrome P450c27 (CYP27) which produce intermediates of alternative bile acid biosynthetic pathways. J. Biol. Chem. 1998, 273, 18153–18160. [Google Scholar] [CrossRef]
- Naz, H.; Islam, A.; Waheed, A.; Sly, W.S.; Ahmad, F.; Hassan, M.I. Human β-glucuronidase: Structure, function, and application in enzyme replacement therapy. Rejuvenation Res. 2013, 16, 352–363. [Google Scholar] [CrossRef]
- Erbs, E.; Brasen, C.L.; Lund, A.M.; Rasmussen, M. Adult patient diagnosed with NADSYN1 associated congenital NAD deficiency and analysis of NAD levels to be published in: European Journal of Medical Genetics. Eur. J. Med. Genet. 2023, 66, 104698. [Google Scholar] [CrossRef] [PubMed]
- Zhai, L.; Ladomersky, E.; Lenzen, A.; Nguyen, B.; Patel, R.; Lauing, K.L.; Wu, M.; Wainwright, D.A. IDO1 in cancer: A Gemini of immune checkpoints. Cell Mol. Immunol. 2018, 15, 447–457. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING database in 2023: Protein–protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2022, 51, D638–D646. [Google Scholar] [CrossRef]
- Digre, A.; Lindskog, C. The human protein atlas—Integrated omics for single cell mapping of the human proteome. Protein Sci. 2023, 32, e4562. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Zhang, C.; von Feilitzen, K.; Shi, M.; Li, X.; Yang, H.; Song, X.; Turkez, H.; Uhlén, M.; Mardinoglu, A. The Human Pathology Atlas for deciphering the prognostic features of human cancers. Res. Sq. 2024; preprint. [Google Scholar] [CrossRef]
- Patel, M.; Li, Y.; Anderson, J.; Castro-Pedrido, S.; Skinner, R.; Lei, S.; Finkel, Z.; Rodriguez, B.; Esteban, F.; Lee, K.B.; et al. Gsx1 promotes locomotor functional recovery after spinal cord injury. Mol. Ther. 2021, 29, 2469–2482. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, L.; Yao, Y.; Peng, L.; Cui, F.; Chen, C.; Zhang, Y.; Sun, L.; Yu, Q.; Lin, K. Silencing GS Homeobox 2 Alleviates Gemcitabine Resistance in Pancreatic Cancer Cells by Activating SHH/GLI1 Signaling Pathway. Dig. Dis. Sci. 2022, 67, 3773–3782. [Google Scholar] [CrossRef]
- Tsang, S.M.; Oliemuller, E.; Howard, B.A. Regulatory roles for SOX11 in development, stem cells and cancer. Semin. Cancer Biol. 2020, 67, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Z.; Yin, X.; Wei, L.; Peng, M.; Zhu, Q.; Lu, X.; Guo, J.; Zhang, J.; Li, X.; Zou, Y. LILRB4 Checkpoint for Immunotherapy: Structure, Mechanism and Disease Targets. Biomolecules 2024, 14, 187. [Google Scholar] [CrossRef]
- Myers, B.L.; Brayer, K.J.; Paez-Beltran, L.E.; Keith, M.S.; Suzuki, H.; Newville, J.; Anderson, R.H.; Lo, Y.; Mertz, C.M.; Kollipara, R.; et al. Glioblastoma initiation, migration, and cell types are regulated by core bHLH transcription factors ASCL1 and OLIG2. bioRxiv, 2023; preprint. [Google Scholar] [CrossRef]
- Vue, T.Y.; Kollipara, R.K.; Borromeo, M.D.; Smith, T.; Mashimo, T.; Burns, D.K.; Bachoo, R.M.; Johnson, J.E. ASCL1 regulates neurodevelopmental transcription factors and cell cycle genes in brain tumors of glioma mouse models. Glia 2020, 68, 2613–2630. [Google Scholar] [CrossRef]
- Yu, D.; Khan, O.F.; Suva, M.L.; Dong, B.; Panek, W.K.; Xiao, T.; Wu, M.; Han, Y.; Ahmed, A.U.; Balyasnikova, I.V.; et al. Multiplexed RNAi therapy against brain tumor-initiating cells via lipopolymeric nanoparticle infusion delays glioblastoma progression. Proc. Natl. Acad. Sci. USA 2017, 114, E6147–E6156. [Google Scholar] [CrossRef] [PubMed]
- Sowparani, S.; Mahalakshmi, P.; Sweety, J.P.; Francis, A.P.; Dhanalekshmi, U.M.; Selvasudha, N. Ubiquitous Neural Cell Adhesion Molecule (NCAM): Potential Mechanism and Valorisation in Cancer Pathophysiology, Drug Targeting and Molecular Transductions. Mol. Neurobiol. 2022, 59, 5902–5924. [Google Scholar] [CrossRef]
- Olechnowicz, A.; Oleksiewicz, U.; Machnik, M. KRAB-ZFPs and cancer stem cells identity. Genes. Dis. 2023, 10, 1820–1832. [Google Scholar] [CrossRef]
- Playfoot, C.J.; Duc, J.; Sheppard, S.; Dind, S.; Coudray, A.; Planet, E.; Trono, D. Transposable elements and their KZFP controllers are drivers of transcriptional innovation in the developing human brain. Genome Res. 2021, 31, 1531–1545. [Google Scholar] [CrossRef] [PubMed]
- Aguirre-Ducler, A.; Gianino, N.; Villarroel-Espindola, F.; Desai, S.; Tang, D.; Zhao, H.; Syrigos, K.; Trepicchio, W.L.; Kannan, K.; Gregory, R.C.; et al. Tumor cell SYK expression modulates the tumor immune microenvironment composition in human cancer via TNF-alpha dependent signaling. J. Immunother. Cancer 2022, 10, e005113. [Google Scholar] [CrossRef] [PubMed]
- Al-Moussawy, M.; Abdelsamed, H.A.; Lakkis, F.G. Immunoglobulin-like receptors and the generation of innate immune memory. Immunogenetics 2022, 74, 179–195. [Google Scholar] [CrossRef] [PubMed]
- Frazzi, R. BIRC3 and BIRC5: Multi-faceted inhibitors in cancer. Cell Biosci. 2021, 11, 8. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Weng, J. A comprehensive bioinformatic analysis of cyclin-dependent kinase 2 (CDK2) in glioma. Gene 2022, 822, 146325. [Google Scholar] [CrossRef]
- Zhang, Y.; Dube, C.; Gibert, M., Jr.; Cruickshanks, N.; Wang, B.; Coughlan, M.; Yang, Y.; Setiady, I.; Deveau, C.; Saoud, K.; et al. The p53 Pathway in Glioblastoma. Cancers 2018, 10, 297. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Ren, T.; Zhao, H.; Chen, S.; Zheng, M.; Gao, X.; Feng, D.; Yang, L.; Jin, X.; Zhuo, R. Suppression of PTTG1 inhibits cell angiogenesis, migration and invasion in glioma cells. Med. Oncol. 2020, 37, 73. [Google Scholar] [CrossRef] [PubMed]
- Cloughesy, T.F.; Mochizuki, A.Y.; Orpilla, J.R.; Hugo, W.; Lee, A.H.; Davidson, T.B.; Wang, A.C.; Ellingson, B.M.; Rytlewski, J.A.; Sanders, C.M.; et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat. Med. 2019, 25, 477–486. [Google Scholar] [CrossRef]
- Lv, Y.; Ma, X.; Ma, Y.; Du, Y.; Feng, J. A new emerging target in cancer immunotherapy: Galectin-9 (LGALS9). Genes. & Diseases 2023, 10, 2366–2382. [Google Scholar] [CrossRef]
- Kou, Y.; Geng, F.; Guo, D. Lipid Metabolism in Glioblastoma: From De Novo Synthesis to Storage. Biomedicines 2022, 10, 1943. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A.; Nadler, J.L.; Mirmira, R.G.; Casimiro, I. Regulation of Tissue Inflammation by 12-Lipoxygenases. Biomolecules 2021, 11, 717. [Google Scholar] [CrossRef]
- Ackermann, J.A.; Hofheinz, K.; Zaiss, M.M.; Kronke, G. The double-edged role of 12/15-lipoxygenase during inflammation and immunity. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Li, S. Survival regulation of leukemia stem cells. Cell Mol. Life Sci. 2016, 73, 1039–1050. [Google Scholar] [CrossRef] [PubMed]
- O‘Flaherty, J.T.; Rogers, L.C.; Paumi, C.M.; Hantgan, R.R.; Thomas, L.R.; Clay, C.E.; High, K.; Chen, Y.Q.; Willingham, M.C.; Smitherman, P.K.; et al. 5-Oxo-ETE analogs and the proliferation of cancer cells. Biochim. Biophys. Acta 2005, 1736, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Powell, W.S.; Rokach, J. The eosinophil chemoattractant 5-oxo-ETE and the OXE receptor. Prog. Lipid Res. 2013, 52, 651–665. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Dai, F.; Wei, J.; Chen, Z. Biological Roles of 5-Oxo-6,8,11,14-Eicosatetraenoic Acid and the OXE Receptor in Allergic Diseases: Collegium Internationale Allergologicum Update 2024. Int. Arch Allergy Immunol. 2024, 185, 301–310. [Google Scholar] [CrossRef]
- Castruccio Castracani, C.; Longhitano, L.; Distefano, A.; Di Rosa, M.; Pittala, V.; Lupo, G.; Caruso, M.; Corona, D.; Tibullo, D.; Li Volti, G. Heme Oxygenase-1 and Carbon Monoxide Regulate Growth and Progression in Glioblastoma Cells. Mol. Neurobiol. 2020, 57, 2436–2446. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Zhang, L.; Zhou, H.; Ye, W.; Luo, C.; Yang, L.; Fang, N.; Tang, A. HMOX1 promotes lung adenocarcinoma metastasis by affecting macrophages and mitochondrion complexes. Front. Oncol. 2022, 12, 978006. [Google Scholar] [CrossRef] [PubMed]
- Ye, W.; Liu, Z.; Liu, F.; Luo, C. Heme Oxygenase-1 Predicts Risk Stratification and Immunotherapy Efficacy in Lower Grade Gliomas. Front. Cell Dev. Biol. 2021, 9, 760800. [Google Scholar] [CrossRef] [PubMed]
- Sohoni, S.; Ghosh, P.; Wang, T.; Kalainayakan, S.P.; Vidal, C.; Dey, S.; Konduri, P.C.; Zhang, L. Elevated Heme Synthesis and Uptake Underpin Intensified Oxidative Metabolism and Tumorigenic Functions in Non-Small Cell Lung Cancer Cells. Cancer Res 2019, 79, 2511–2525. [Google Scholar] [CrossRef]
- Zhang, W.; Jiang, J.; Kang, X.; Wang, C.; Chen, F.; Zhang, B.; Li, S.; Huang, S.; Li, W. Dietary B vitamins and glioma: A case-control study based on Chinese population. Front. Nutr. 2023, 10, 1122540. [Google Scholar] [CrossRef]
- Li, J.; Li, Y.; Fan, Z.; Chen, S.; Yan, X.; Yue, Z.; Huang, G.; Liu, S.; Zhang, H.; Chen, S.; et al. Two Amphioxus ApeC-Containing Proteins Bind to Microbes and Inhibit the TRAF6 Pathway. Front. Immunol. 2021, 12, 715245. [Google Scholar] [CrossRef]
- Kaynar, A.; Ozcan, M.; Li, X.; Turkez, H.; Zhang, C.; Uhlén, M.; Shoaie, S.; Mardinoglu, A. Discovery of a therapeutic agent for Glioblastoma using a systems biology-based drug repositioning approach. Int. J. Mol. Sci. 2024, 25, 7868. [Google Scholar] [CrossRef] [PubMed]
- Chuang, T.D.; Chen, S.J.; Lin, F.F.; Veeramani, S.; Kumar, S.; Batra, S.K.; Tu, Y.; Lin, M.F. Human prostatic acid phosphatase, an authentic tyrosine phosphatase, dephosphorylates ErbB-2 and regulates prostate cancer cell growth. J. Biol. Chem. 2010, 285, 23598–23606. [Google Scholar] [CrossRef] [PubMed]
- Yuan, B.; Liu, G.; Dai, Z.; Wang, L.; Lin, B.; Zhang, J. CYP1B1: A Novel Molecular Biomarker Predicts Molecular Subtype, Tumor Microenvironment, and Immune Response in 33 Cancers. Cancers 2022, 14, 5641. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Upregulated Gene Affected Metabolites | Short Summary |
|---|---|
| Lipid Metabolism and Cholesterol-Related Metabolites | |
| 13(S)-HPODE | Associated with breast cancer cell proliferation and invasion. |
| 5(S)-HPETE | Involved in the arachidonic acid pathway. |
| Cholesterol-ester | Implicated in steroid hormone biosynthesis and lipid metabolism. |
| Dehydrocholic acid | Investigated for potential anticancer properties. |
| Taurodeoxycholate and lithocholate | Involved in cholesterol metabolism. |
| Nucleotide Synthesis and Purine Metabolism | |
| GAR (Glycinamide ribonucleotide) | Involved in purine biosynthesis and can affect cancer cell growth. |
| Adenine, threonine, tetrahydrofolate | Critical for nucleotide synthesis and DNA methylation. |
| Extracellular Matrix and Invasion | |
| Hyaluronate | A component of the extracellular matrix plays a key role in cancer cell invasion and metastasis. |
| Pentose Phosphate Pathway and Glycogen Metabolism | |
| Ribose-5-phosphate | A component of the pentose phosphate pathway, involved in nucleotide synthesis, impacts cancer cell proliferation. |
| Glucose-1-phosphate | Involved in glycogen, energy, and glycosylation metabolism. |
| Glycosphingolipid Metabolism | |
| Galactosylglycerol | Involved in glycosphingolipid metabolism, cell signalling, and adhesion. |
| Inflammatory Response and Cell Survival | |
| Hepoxilin A3 | Implicated in inflammatory responses, chemoattractant for neutrophils. |
| Downregulated Gene Affected Metabolites | Short Summary |
|---|---|
| Amino Acid Metabolism | |
| Glutamate and Glutamine | Key players in brain metabolism, including neurotransmission, potentially contribute to the disease pathophysiology and might indicate altered energy and biosynthetic needs in GBM cells [47]. |
| Phenylalanine and its derivatives Phenylpyruvate and Hydroxyphenylpyruvate | It may be an indicator of GBM’s metabolic adaptations, affecting cancer progression. Glioma cell consumes more Phenylalanine, a precursor for neurotransmitters. Hydroxyphenylpyruvate influences oxidative stress via tyrosine metabolism [48]. |
| Tyrosine | It plays a role in multiple signalling pathways. Its involvement could influence cell signalling, potentially contributing to disease development. Tyrosine de novo synthesis metabolic task defective in all GBM models (Figure 4B) [49,50,51]. |
| Ketone Metabolism | |
| Acetoacetate | An alternative energy source via ketone metabolism that may indicate the energy demands of rapidly proliferating cells [52]. |
| Lipid Signaling | |
| Phosphatidylinositol (PI) | Phosphatidylinositol (PI) and its derivatives, including phosphatidylinositol-4,5-bisphosphate (PIP2) and phosphatidylinositol-3,4,5-trisphosphate (PIP3), are involved in signalling cascades linked to cell growth and survival. Dysregulation of them may be indicative of potential disruptions in crucial signalling pathways, contributing to tumour growth and evasion of cell death [53,54]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaynar, A.; Kim, W.; Ceyhan, A.B.; Zhang, C.; Uhlén, M.; Turkez, H.; Shoaie, S.; Mardinoglu, A. Unveiling the Molecular Mechanisms of Glioblastoma through an Integrated Network-Based Approach. Biomedicines 2024, 12, 2237. https://doi.org/10.3390/biomedicines12102237
Kaynar A, Kim W, Ceyhan AB, Zhang C, Uhlén M, Turkez H, Shoaie S, Mardinoglu A. Unveiling the Molecular Mechanisms of Glioblastoma through an Integrated Network-Based Approach. Biomedicines. 2024; 12(10):2237. https://doi.org/10.3390/biomedicines12102237
Chicago/Turabian StyleKaynar, Ali, Woonghee Kim, Atakan Burak Ceyhan, Cheng Zhang, Mathias Uhlén, Hasan Turkez, Saeed Shoaie, and Adil Mardinoglu. 2024. "Unveiling the Molecular Mechanisms of Glioblastoma through an Integrated Network-Based Approach" Biomedicines 12, no. 10: 2237. https://doi.org/10.3390/biomedicines12102237
APA StyleKaynar, A., Kim, W., Ceyhan, A. B., Zhang, C., Uhlén, M., Turkez, H., Shoaie, S., & Mardinoglu, A. (2024). Unveiling the Molecular Mechanisms of Glioblastoma through an Integrated Network-Based Approach. Biomedicines, 12(10), 2237. https://doi.org/10.3390/biomedicines12102237