
Deferasirox’s Anti-Chemoresistance and Anti-Metastatic Effect on Non-Small Cell Lung Carcinoma

 ,
,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Stocks and Dilutions
2.3. Cell Culture Conditions
2.4. Cell Viability Assay
2.5. Confocal Microscopy
2.6. Iron Binding Assay
2.7. Total Glutathione (GSH) Assay
2.8. Total Oxidative Stress Assay
2.9. Caspase 3 Activity Assay
2.10. Flow Cytometry
2.10.1. Cell Cycle Arrest
2.10.2. DNA Damage
2.10.3. Mitochondrial Membrane Potential
2.10.4. EGFR Protein Expression and Activation
2.11. Scratch Wound Healing Migration Assay
2.12. mRNA Gene Expression
2.13. In Vivo Assays
2.14. Statistical Analyses
3. Results
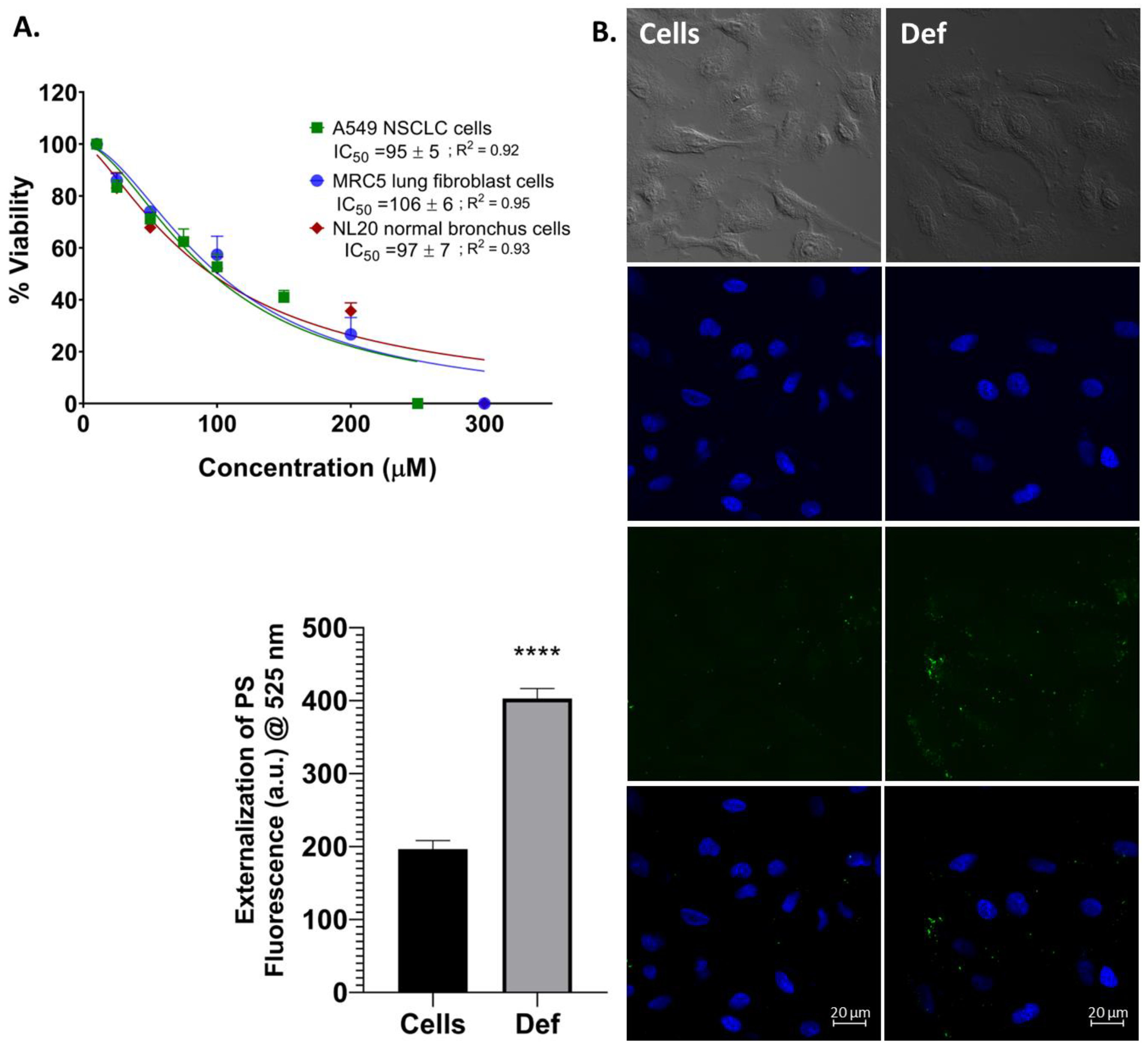
3.1. Def Induces Cell Death and Apoptosis in NSCLC Cells
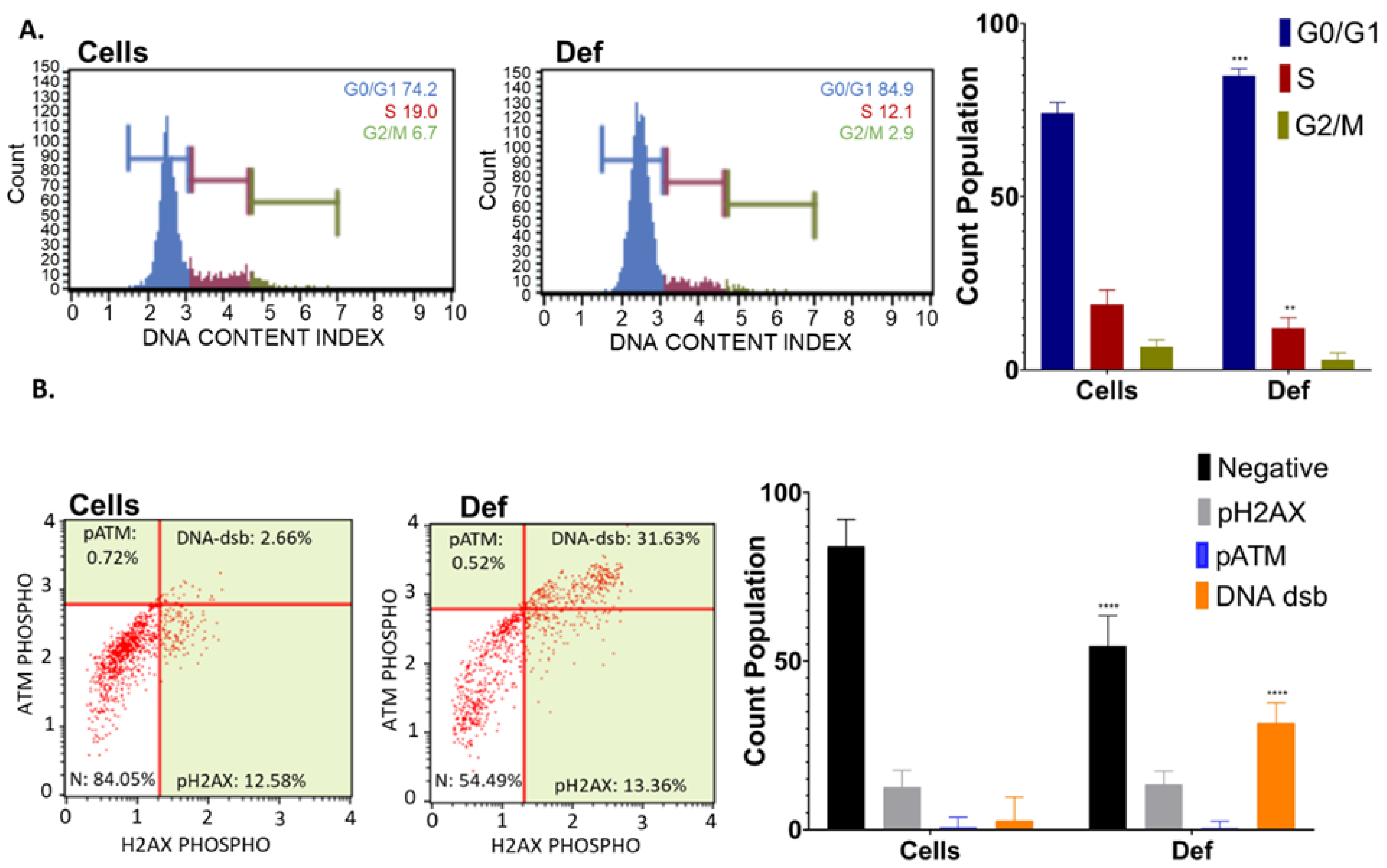
3.2. Def Promotes G0/G1 Cycle Arrest and DNA dsb
3.3. Def Induces Mitochondria Depolarization and Activation of Caspase 3/7
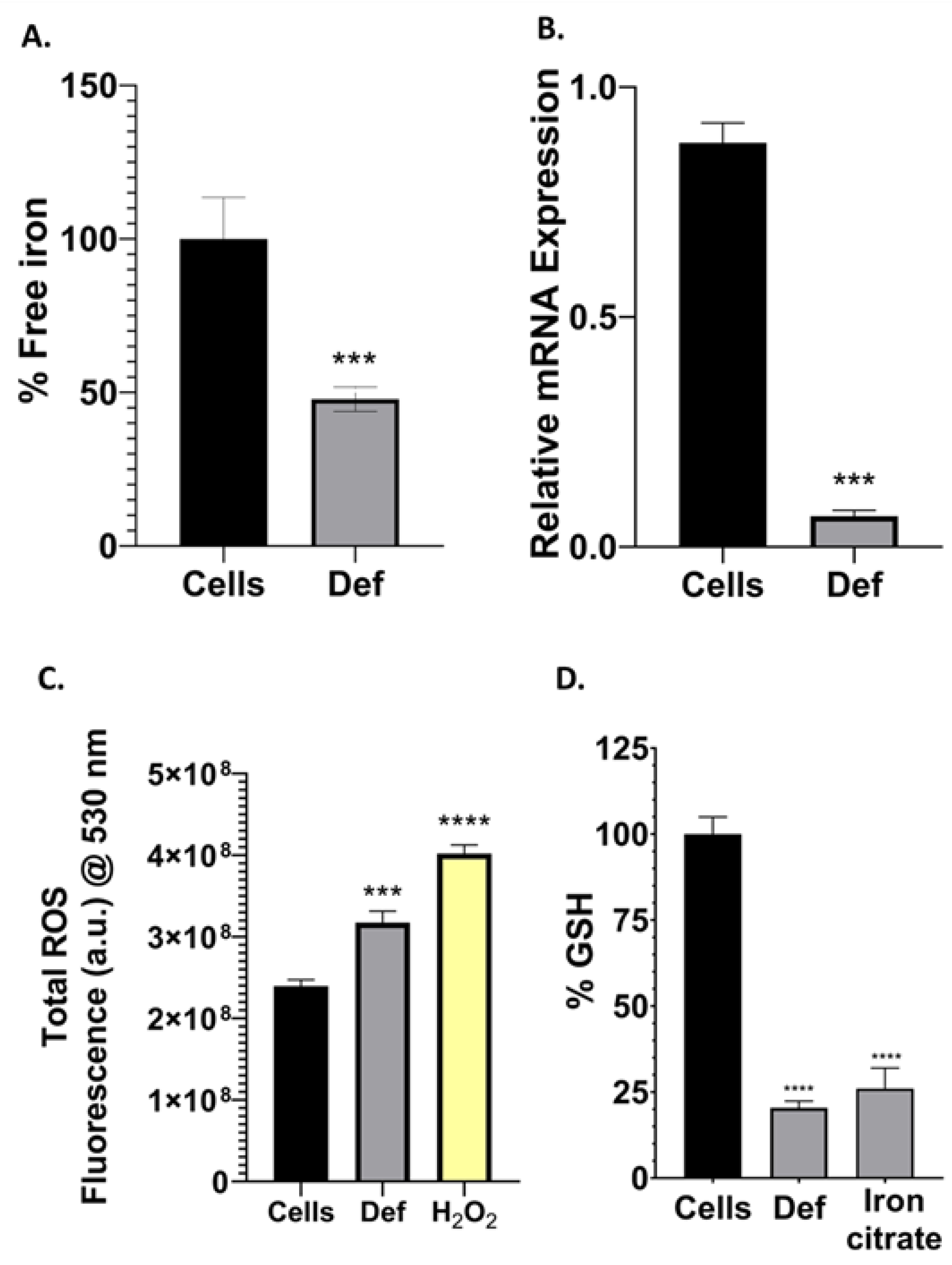
3.4. Def Chelates Iron Impairing Iron-Containing Proteins, GSH and Producing ROS
3.5. Def Decreases Cell Migration, the Expression of ABCB1 Gene and EGFR Protein, and Upregulates NDRG1 Gene
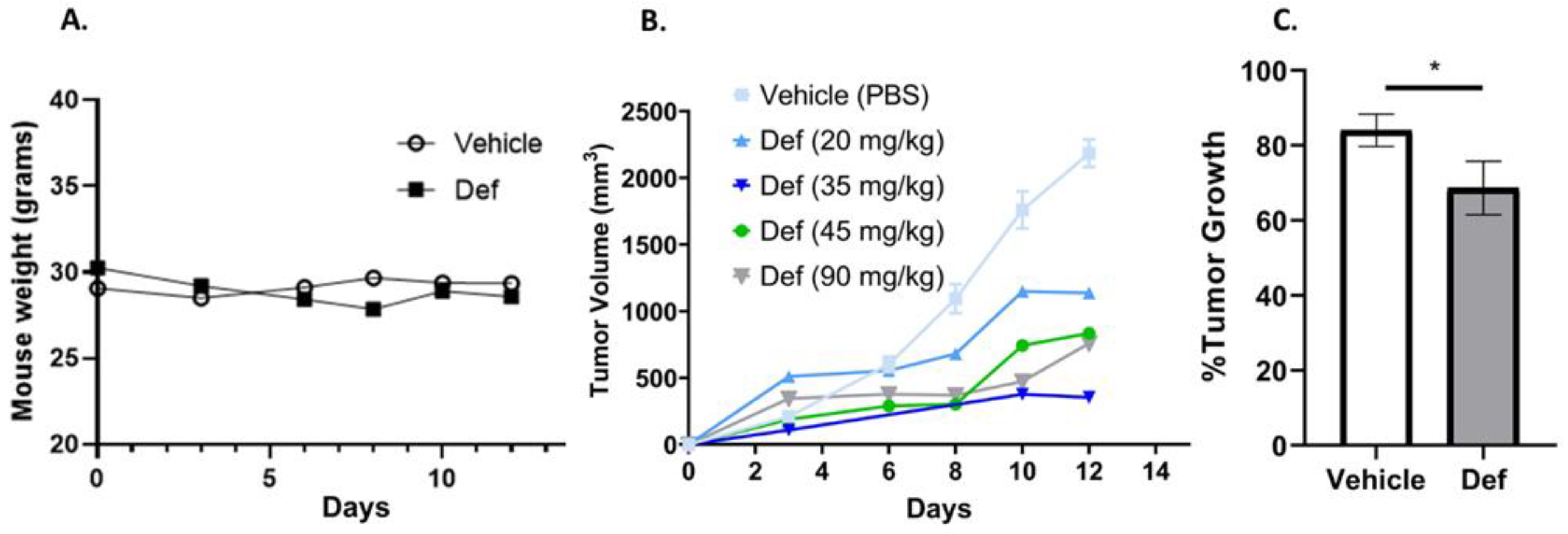
3.6. Def Is Safe and Diminishes Carcinoma Tumor Growth in Mice
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hsu, M.Y.; Mina, E.; Roetto, A.; Porporato, P.E. Iron: An Essential Element of Cancer Metabolism. Cells 2020, 9, 2591. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.A.M.; Richardson, K.L.; Kabir, T.D.; Trinder, D.; Ganss, R.; Leedman, P.J. Altered Iron Metabolism and Impact in Cancer Biology, Metastasis, and Immunology. Front. Oncol. 2020, 10, 476. [Google Scholar] [CrossRef]
- Morales, M.; Xue, X. Targeting Iron Metabolism in Cancer Therapy. Theranostics 2021, 11, 8412–8429. [Google Scholar] [CrossRef]
- Guo, Q.; Li, L.; Hou, S.; Yuan, Z.; Li, C.; Zhang, W.; Zheng, L.; Li, X. The Role of Iron in Cancer Progression. Front. Oncol. 2021, 11, 778492. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Schabath, M.B.; Cote, M.L. Cancer Progress and Priorities: Lung Cancer. Cancer Epidemiol. Biomark. Prev. 2019, 28, 1563–1579. [Google Scholar] [CrossRef] [PubMed]
- Ettinger, D.S.; Wood, D.E.; Aisner, D.L.; Akerley, W.; Bauman, J.R.; Bharat, A.; Bruno, D.S.; Chang, J.Y.; Chirieac, L.R.; D’Amico, T.A.; et al. Non-Small Cell Lung Cancer, Version 3.2022, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Canc. Netw. 2022, 20, 497–530. [Google Scholar] [CrossRef]
- Padinharayil, H.; Varghese, J.; John, M.C.; Rajanikant, G.K.; Wilson, C.M.; Al-Yozbaki, M.; Renu, K.; Dewanjee, S.; Sanyal, R.; Dey, A.; et al. Non-Small Cell Lung Carcinoma (NSCLC): Implications on Molecular Pathology and Advances in Early Diagnostics and Therapeutics. Genes Dis. 2023, 10, 960–989. [Google Scholar] [CrossRef]
- Mobarra, N.; Shanaki, M.; Ehteram, H.; Nasiri, H.; Sahmani, M.; Saeidi, M.; Goudarzi, M.; Pourkarim, H.; Azad, M. A Review on Iron Chelators in Treatment of Iron Overload Syndromes. Int. J. Hematol. Oncol. Stem Cell Res. 2016, 10, 239–247. [Google Scholar] [PubMed]
- Abdelaal, G.; Veuger, S. Reversing Oncogenic Transformation with Iron Chelation. Oncotarget 2021, 12, 106–124. [Google Scholar] [CrossRef]
- Islam, S.; Hoque, N.; Nasrin, N.; Hossain, M.; Rizwan, F.; Biswas, K.; Asaduzzaman, M.; Rahman, S.; Hoskin, D.W.; Sultana, S.; et al. Iron Overload and Breast Cancer: Iron Chelation as a Potential Therapeutic Approach. Life 2022, 12, 963. [Google Scholar] [CrossRef] [PubMed]
- Salem, A.; Desai, P.; Elgebaly, A. Efficacy and Safety of Combined Deferiprone and Deferasirox in Iron-Overloaded Patients: A Systematic Review. Cureus 2023, 15, e48276. [Google Scholar] [CrossRef] [PubMed]
- Samara, A.; Shapira, S.; Lubin, I.; Shpilberg, O.; Avigad, S.; Granot, G.; Raanani, P. Deferasirox Induces Cyclin D1 Degradation and Apoptosis in Mantle Cell Lymphoma in a Reactive Oxygen Species- and GSK3β-Dependent Mechanism. Br. J. Haematol. 2021, 192, 747–760. [Google Scholar] [CrossRef] [PubMed]
- Petronek, M.S.; Spitz, D.R.; Allen, B.G. Iron-Sulfur Cluster Biogenesis as a Critical Target in Cancer. Antioxidants 2021, 10, 1458. [Google Scholar] [CrossRef]
- Kontoghiorghes, G.J.; Kontoghiorghe, C.N. Iron and Chelation in Biochemistry and Medicine: New Approaches to Controlling Iron Metabolism and Treating Related Diseases. Cells 2020, 9, 1456. [Google Scholar] [CrossRef]
- Huff, S.E.; Winter, J.M.; Dealwis, C.G. Inhibitors of the Cancer Target Ribonucleotide Reductase, Past and Present. Biomolecules 2022, 12, 815. [Google Scholar] [CrossRef]
- Morganti, C.; Bonora, M.; Ito, K.; Ito, K. Electron Transport Chain Complex II Sustains High Mitochondrial Membrane Potential in Hematopoietic Stem and Progenitor Cells. Stem Cell Res. 2019, 40, 101573. [Google Scholar] [CrossRef]
- Abdelaal, G.; Carter, A.; Panayiotides, M.I.; Tetard, D.; Veuger, S. Novel Iron Chelator SK4 Demonstrates Cytotoxicity in a Range of Tumour Derived Cell Lines. Front. Mol. Biosci. 2022, 9, 1005092. [Google Scholar] [CrossRef]
- Zuo, Z.; Zhou, Z.; Chang, Y.; Liu, Y.; Shen, Y.; Li, Q.; Zhang, L. Ribonucleotide Reductase M2 (RRM2): Regulation, Function and Targeting Strategy in Human Cancer. Genes Dis. 2022, 11, 218–233. [Google Scholar] [CrossRef]
- Levitz, T.S.; Drennan, C.L. Starting a New Chapter on Class Ia Ribonucleotide Reductases. Curr. Opin. Struct. Biol. 2022, 77, 102489. [Google Scholar] [CrossRef]
- Jeleń, A.M.; Strehl, B.; Szmajda-Krygier, D.; Pązik, M.; Balcerczak, E. Bioinformatics-Based Characterization of ATP-Binding Cassette Subfamily B Member 1 (ABCB1) Gene Expression in Non-Small-Cell Lung Cancer (NSCLC). Appl. Sci. 2023, 13, 6576. [Google Scholar] [CrossRef]
- Hu, F.; Li, C.; Zheng, X.; Zhang, H.; Shen, Y.; Zhou, L.; Yang, X.; Han, B.; Zhang, X. Lung Adenocarcinoma Resistance to Therapy with EGFR Tyrosine Kinase Inhibitors Is Related to Increased Expression of Cancer Stem Cell Markers SOX2, OCT4 and NANOG. Oncol. Rep. 2020, 43, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Omori, M.; Noro, R.; Seike, M.; Matsuda, K.; Hirao, M.; Fukuizumi, A.; Takano, N.; Miyanaga, A.; Gemma, A. Inhibitors of ABCB1 and ABCG2 Overcame Resistance to Topoisomerase Inhibitors in Small Cell Lung Cancer. Thorac. Cancer 2022, 13, 2142–2151. [Google Scholar] [CrossRef] [PubMed]
- Menezes, S.V.; Fouani, L.; Huang, M.L.H.; Geleta, B.; Maleki, S.; Richardson, A.; Richardson, D.R.; Kovacevic, Z. The Metastasis Suppressor, NDRG1, Attenuates Oncogenic TGF-β and NF-κB Signaling to Enhance Membrane E-Cadherin Expression in Pancreatic Cancer Cells. Carcinogenesis 2019, 40, 805–818. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.C.; Geleta, B.; Maleki, S.; Richardson, D.R.; Kovačević, Ž. The Metastasis Suppressor NDRG1 Directly Regulates Androgen Receptor Signaling in Prostate Cancer. J. Biol. Chem. 2021, 297, 101414. [Google Scholar] [CrossRef] [PubMed]
- Kontoghiorghes, G.J. New Iron Metabolic Pathways and Chelation Targeting Strategies Affecting the Treatment of All Types and Stages of Cancer. Int. J. Mol. Sci. 2022, 23, 13990. [Google Scholar] [CrossRef]
- Uribe, M.L.; Marrocco, I.; Yarden, Y. EGFR in Cancer: Signaling Mechanisms, Drugs, and Acquired Resistance. Cancers 2021, 13, 2748. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Zhang, G.; Zhou, S.; Wang, S.; Lv, H.; Zhou, L.; Shang, P. Iron Chelator Induces Apoptosis in Osteosarcoma Cells by Disrupting Intracellular Iron Homeostasis and Activating the MAPK Pathway. Int. J. Mol. Sci. 2021, 22, 7168. [Google Scholar] [CrossRef]
- Zhou, N.; Cui, Y.; Zhu, R.; Kuang, Y.; Ma, W.; Hou, J.; Zhu, Y.; Chen, S.; Xu, X.; Tan, K.; et al. Deferasirox Shows Inhibition Activity against Cervical Cancer in Vitro and in Vivo. Gynecol. Oncol. 2022, 166, 126–137. [Google Scholar] [CrossRef]
- Torres-Sanchez, A.; Torres, G.; Estrada, S.; Perez, D.; Garcia, C.; Milian, M.; Velazquez, E.; Molina, V.; Delgado, Y. Unraveling the Impact of Six Pentacyclic Triterpenes Regulating Metabolic Pathways on Lung Carcinoma Cells. Pharmaceuticals 2024, 17, 694. [Google Scholar] [CrossRef]
- Torres-Martinez, Z.; Pérez, D.; Torres, G.; Estrada, S.; Correa, C.; Mederos, N.; Velazquez, K.; Castillo, B.; Griebenow, K.; Delgado, Y. A Synergistic pH-Responsive Serum Albumin-Based Drug Delivery System Loaded with Doxorubicin and Pentacyclic Triterpene Betulinic Acid for Potential Treatment of NSCLC. BioTech 2023, 12, 13. [Google Scholar] [CrossRef] [PubMed]
- Barcelo-Bovea, V.; Dominguez-Martinez, I.; Joaquin-Ovalle, F.; Amador, L.A.; Castro-Rivera, E.; Medina-Álvarez, K.; McGoron, A.; Griebenow, K.; Ferrer-Acosta, Y. Optimization and Characterization of Protein Nanoparticles for the Targeted and Smart Delivery of Cytochrome c to Non-Small Cell Lung Carcinoma. Cancers 2020, 12, 1215. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Martinez, I.; Joaquin-Ovalle, F.; Ferrer-Acosta, Y.; Griebenow, K.H. Folate-Decorated Cross-Linked Cytochrome c Nanoparticles for Active Targeting of Non-Small Cell Lung Carcinoma (NSCLC). Pharmaceutics 2022, 14, 490. [Google Scholar] [CrossRef] [PubMed]
- Entezari, S.; Haghi, S.M.; Norouzkhani, N.; Sahebnazar, B.; Vosoughian, F.; Akbarzadeh, D.; Islampanah, M.; Naghsh, N.; Abbasalizadeh, M.; Deravi, N. Iron Chelators in Treatment of Iron Overload. J. Toxicol. 2022, 2022, 4911205. [Google Scholar] [CrossRef]
- Morana, O.; Wood, W.; Gregory, C.D. The Apoptosis Paradox in Cancer. Int. J. Mol. Sci. 2022, 23, 1328. [Google Scholar] [CrossRef]
- Ding, S.M.; Lu, J.F.; Edoo, M.I.A.; Zhou, L.; Xie, H.Y.; Zheng, S.S.; Li, Q.Y. MRC-5 Cancer-Associated Fibroblasts Influence Production of Cancer Stem Cell Markers and Inflammation-Associated Cell Surface Molecules, in Liver Cancer Cell Lines. Int. J. Med. Sci. 2019, 16, 1157–1170. [Google Scholar] [CrossRef]
- Li, N.; Chen, Q.; Gu, J.; Li, S.; Zhao, G.; Wang, W.; Wang, Z.; Wang, X. Synergistic Inhibitory Effects of Deferasirox in Combination with Decitabine on Leukemia Cell Lines SKM-1, THP-1, and K-562. Oncotarget 2017, 8, 36517–36530. [Google Scholar] [CrossRef]
- Chantrel-Groussard, K.; Gaboriau, F.; Pasdeloup, N.; Havouis, R.; Nick, H.; Pierre, J.L.; Brissot, P.; Lescoat, G. The New Orally Active Iron Chelator ICL670A Exhibits a Higher Antiproliferative Effect in Human Hepatocyte Cultures than O-Trensox. Eur. J. Pharmacol. 2006, 541, 129–137. [Google Scholar] [CrossRef]
- Khan, A.; Singh, P.; Srivastava, A. Iron: Key Player in Cancer and Cell Cycle? J. Trace Elem. Med. Biol. 2020, 62, 126582. [Google Scholar] [CrossRef]
- Gillyard, T.; Davis, J. DNA Double-Strand Break Repair in Cancer: A Path to Achieving Precision Medicine. Int. Rev. Cell Mol. Biol. 2021, 364, 111–137. [Google Scholar] [CrossRef]
- Samara, A.; Shapira, S.; Lubin, I.; Avigad, S.; Raanani, P.; Granot, G. Deferasirox Induces Apoptosis in Mantle Cell Lymphoma in a Mechanism Involving Cyclin-D1, p53, and the Ribonucleotide Reductase. Blood 2017, 130, 1481. [Google Scholar] [CrossRef]
- Tait, S.W.; Green, D.R. Mitochondrial Regulation of Cell Death. Cold Spring Harb. Perspect. Biol. 2013, 5, a008706. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Y.; Chen, X.; Gueydan, C.; Han, J. Plasma Membrane Changes during Programmed Cell Deaths. Cell Res. 2018, 28, 9–21. [Google Scholar] [CrossRef]
- Martin-Sanchez, D.; Gallegos-Villalobos, A.; Fontecha-Barriuso, M.; Carrasco, S.; Sanchez-Niño, M.D.; Lopez-Hernandez, F.J.; Ruiz-Ortega, M.; Egido, J.; Ortiz, A.; Sanz, A.B. Deferasirox-Induced Iron Depletion Promotes BclxL Downregulation and Death of Proximal Tubular Cells. Sci. Rep. 2017, 7, 41510. [Google Scholar] [CrossRef]
- Ricci, J.E.; Gottlieb, R.A.; Green, D.R. Caspase-Mediated Loss of Mitochondrial Function and Generation of Reactive Oxygen Species during Apoptosis. J. Cell Biol. 2003, 160, 65–75. [Google Scholar] [CrossRef]
- Calabrese, C.; Panuzzo, C.; Stanga, S.; Andreani, G.; Ravera, S.; Maglione, A.; Pironi, L.; Petiti, J.; Shahzad Ali, M.S.; Scaravaglio, P.; et al. Deferasirox-Dependent Iron Chelation Enhances Mitochondrial Dysfunction and Restores p53 Signaling by Stabilization of p53 Family Members in Leukemic Cells. Int. J. Mol. Sci. 2020, 21, 7674. [Google Scholar] [CrossRef] [PubMed]
- Gottwald, E.M.; Schuh, C.D.; Drücker, P.; Haenni, D.; Pearson, A.; Ghazi, S.; Bugarski, M.; Polesel, M.; Duss, M.; Landau, E.M.; et al. The Iron Chelator Deferasirox Causes Severe Mitochondrial Swelling Without Depolarization Due to a Specific Effect on Inner Membrane Permeability. Sci. Rep. 2020, 10, 1577. [Google Scholar] [CrossRef]
- Ford, S.J.; Obeidy, P.; Lovejoy, D.B.; Bedford, M.; Nichols, L.; Chadwick, C.; Tucker, O.; Lui, G.Y.; Kalinowski, D.S.; Jansson, P.J.; et al. Deferasirox (ICL670A) Effectively Inhibits Oesophageal Cancer Growth In Vitro and In Vivo. Br. J. Pharmacol. 2013, 168, 1316–1328. [Google Scholar] [CrossRef]
- Loza-Rosas, S.A.; Vázquez-Salgado, A.M.; Rivero, K.I.; Negrón, L.J.; Delgado, Y.; Benjamín-Rivera, J.A.; Vázquez-Maldonado, A.L.; Parks, T.B.; Munet-Colón, C.; Tinoco, A.D. Expanding the Therapeutic Potential of the Iron Chelator Deferasirox in the Development of Aqueous Stable Ti(IV) Anticancer Complexes. Inorg. Chem. 2017, 56, 7788–7802. [Google Scholar] [CrossRef]
- Khan, F.H.; Dervan, E.; Bhattacharyya, D.D.; McAuliffe, J.D.; Miranda, K.M.; Glynn, S.A. The Role of Nitric Oxide in Cancer: Master Regulator or Not? Int. J. Mol. Sci. 2020, 21, 9393. [Google Scholar] [CrossRef]
- Tirichen, H.; Yaigoub, H.; Xu, W.; Wu, C.; Li, R.; Li, Y. Mitochondrial Reactive Oxygen Species and Their Contribution in Chronic Kidney Disease Progression through Oxidative Stress. Front. Physiol. 2021, 12, 627837. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.F.; Zou, T.; Tuo, Q.Z.; Xu, S.; Li, H.; Belaidi, A.A.; Lei, P. Ferroptosis: Mechanisms and Links with Diseases. Signal Transduct. Target. Ther. 2021, 6, 49. [Google Scholar] [CrossRef] [PubMed]
- Hsu, W.Y.; Wang, L.T.; Lin, P.C.; Liao, Y.M.; Hsu, S.H.; Chiou, S.S. Deferasirox Causes Leukaemia Cell Death through Nrf2-Induced Ferroptosis. Antioxidants 2024, 13, 424. [Google Scholar] [CrossRef] [PubMed]
- Meirson, T.; Gil-Henn, H.; Samson, A.O. Invasion and Metastasis: The Elusive Hallmark of Cancer. Oncogene 2020, 39, 2024–2026. [Google Scholar] [CrossRef]
- Vasan, N.; Baselga, J.; Hyman, D.M. A View on Drug Resistance in Cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef]
- Eslami, M.; Memarsadeghi, O.; Davarpanah, A.; Arti, A.; Nayernia, K.; Behnam, B. Overcoming Chemotherapy Resistance in Metastatic Cancer: A Comprehensive Review. Biomedicines 2024, 12, 183. [Google Scholar] [CrossRef]
- Nishitani, S.; Noma, K.; Ohara, T.; Tomono, Y.; Watanabe, S.; Tazawa, H.; Shirakawa, Y.; Fujiwara, T. Iron Depletion-Induced Downregulation of N-Cadherin Expression Inhibits Invasive Malignant Phenotypes in Human Esophageal Cancer. Int. J. Oncol. 2016, 49, 1351–1359. [Google Scholar] [CrossRef]
- Chen, Z.; Zhang, D.; Yue, F.; Zheng, M.; Kovacevic, Z.; Richardson, D.R. The Iron Chelators Dp44mT and DFO Inhibit TGF-β-Induced Epithelial-Mesenchymal Transition via Up-Regulation of N-Myc Downregulated Gene 1 (NDRG1). J. Biol. Chem. 2012, 287, 17016–17028. [Google Scholar] [CrossRef]
- Sigismund, S.; Avanzato, D.; Lanzetti, L. Emerging Functions of the EGFR in Cancer. Mol. Oncol. 2018, 12, 3–20. [Google Scholar] [CrossRef]
- Sandoval, T.A.; Salvagno, C.; Chae, C.S.; Awasthi, D.; Giovanelli, P.; Marin Falco, M.; Hwang, S.M.; Teran-Cabanillas, E.; Suominen, L.; Yamazaki, T.; et al. Iron Chelation Therapy Elicits Innate Immune Control of Metastatic Ovarian Cancer. Cancer Discov. 2024, 14, 1901–1921. [Google Scholar] [CrossRef]
- Talbot, S.R.; Biernot, S.; Bleich, A.; van Dijk, R.M.; Ernst, L.; Häger, C.; Helgers, S.O.A.; Koegel, B.; Koska, I.; Kuhla, A. Defining Body-Weight Reduction as a Humane Endpoint: A Critical Appraisal. Lab. Anim. 2020, 54, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Lui, G.Y.; Obeidy, P.; Ford, S.J.; Tselepis, C.; Sharp, D.M.; Jansson, P.J.; Kalinowski, D.S.; Kovacevic, Z.; Lovejoy, D.B.; Richardson, D.R. The Iron Chelator, Deferasirox, as a Novel Strategy for Cancer Treatment: Oral Activity Against Human Lung Tumor Xenografts and Molecular Mechanism of Action. Mol. Pharmacol. 2013, 83, 179–190. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Delgado, Y.; Torres-Sanchez, A.; Perez, D.; Torres, G.; Estrada, S.; Ortiz Alvelo, N.; Vega, J.; Santos, L.; Torres, A.; Madera, B.A.; et al. Deferasirox’s Anti-Chemoresistance and Anti-Metastatic Effect on Non-Small Cell Lung Carcinoma. Biomedicines 2024, 12, 2272. https://doi.org/10.3390/biomedicines12102272
Delgado Y, Torres-Sanchez A, Perez D, Torres G, Estrada S, Ortiz Alvelo N, Vega J, Santos L, Torres A, Madera BA, et al. Deferasirox’s Anti-Chemoresistance and Anti-Metastatic Effect on Non-Small Cell Lung Carcinoma. Biomedicines. 2024; 12(10):2272. https://doi.org/10.3390/biomedicines12102272
Chicago/Turabian StyleDelgado, Yamixa, Anamaris Torres-Sanchez, Daraishka Perez, Grace Torres, Sthephanie Estrada, Natalia Ortiz Alvelo, Jaisy Vega, Laurie Santos, Aracelis Torres, Bismark A. Madera, and et al. 2024. "Deferasirox’s Anti-Chemoresistance and Anti-Metastatic Effect on Non-Small Cell Lung Carcinoma" Biomedicines 12, no. 10: 2272. https://doi.org/10.3390/biomedicines12102272