
Bioinformatic Characterization of the Functional and Structural Effect of Single Nucleotide Mutations in Patients with High-Grade Glioma

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Samples for Sequencing
2.3. Molecular Mutation Panel
2.4. Prediction of Pathogenic SNPs
2.5. Prediction of Deleterious SNPs for the Protein
2.6. Predicting Protein Stability for Functionally Deleterious Glioma-Associated SNPs
2.7. Prediction of Protein Structural Alteration and Loss of Activity
2.8. Observation of Amino Acid Change
2.9. Structural Comparison between Normal and Mutated Residues
2.10. Group of Mutations
2.11. Protein–Protein Interaction
3. Results
3.1. Distribution of SNPs
3.2. Unique Non-Synonymous Polymorphisms Predicted Using SIFT and PolyPhen-2
3.3. Disease-Related Single Nucleotide Polymorphisms PhD-SNP SNPs and GO
3.4. Prediction of Protein Stability Alteration Using I-Mutant 3.0 and MUpro Software
3.5. Mutations Group
3.6. Impact of Single Nucleotide Polymorphisms on Protein Structural Activity
3.7. Alteration and Loss of Activity
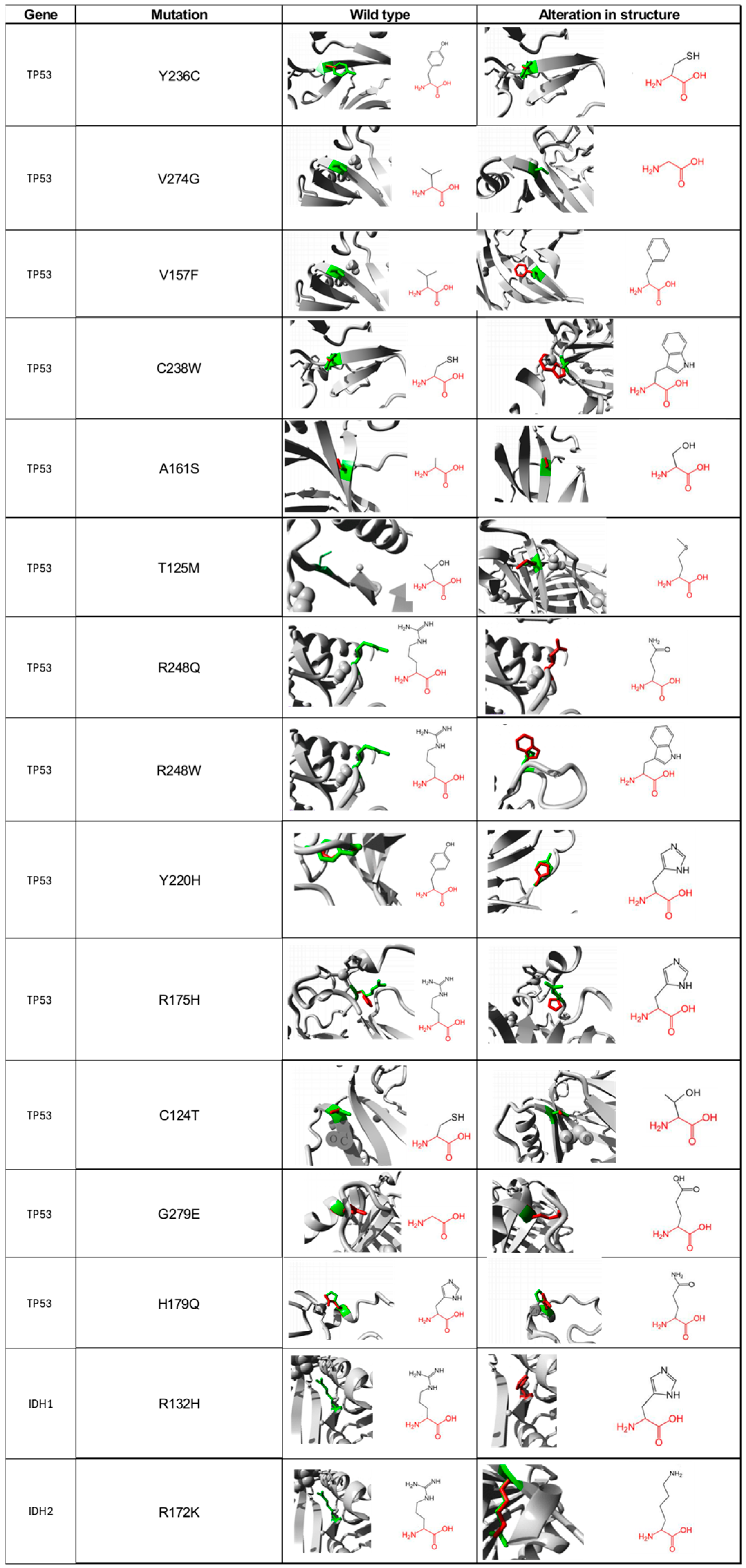
3.8. Structural Differences between Normal and Mutated Residues Using TM-Aling and Ramachandran Plots
3.9. Protein–Protein Interaction
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Komori, T. The 2021 WHO classification of tumors, 5th edition, central nervous system tumors: The 10 basic principles. Brain Tumor Pathol. 2022, 39, 47–50. [Google Scholar] [CrossRef]
- Weller, M.; Wick, W.; Aldape, K.; Brada, M.; Berger, M.; Pfister, S.M.; Nishikawa, R.; Rosenthal, M.; Wen, P.Y.; Stupp, R.; et al. Glioma. Nat. Rev. Dis. Primers 2015, 1, 15017. [Google Scholar] [CrossRef] [PubMed]
- Dekker, L.J.M.; Verheul, C.; Wensveen, N.; Leenders, W.; Lamfers, M.L.M.; Leenstra, S.; Luider, T.M. Effects of the IDH1 R132H Mutation on the Energy Metabolism: A Comparison between Tissue and Corresponding Primary Glioma Cell Cultures. ACS Omega 2022, 7, 3568–3578. [Google Scholar] [CrossRef] [PubMed]
- Palacios Paredes, L.F.; Silva, C.; García Matamoros, E.K. Gliomas de Alto Grado del Adulto: Biología Molecular (Parte I): Revisión Narrativa. Oncol. Ecuad. 2020, 30, 249–279. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Junaid, M.; Hamid, N.; Duan, J.-J.; Yang, X.; Pei, D.-S. Current understanding of gliomagenesis: From model to mechanism. Int. J. Med. Sci. 2022, 19, 2071–2079. [Google Scholar] [CrossRef]
- Zhang, Y.; Dube, C.; Gibert, M.; Cruickshanks, N.; Wang, B.; Coughlan, M.; Yang, Y.; Setiady, I.; Deveau, C.; Saoud, K.; et al. The p53 Pathway in Glioblastoma. Cancers 2018, 10, 297. [Google Scholar] [CrossRef]
- Krex, D.; Mohr, B.; Appelt, H.; Schackert, H.K.; Schackert, G. Genetic Analysis of a Multifocal Glioblastoma Multiforme: A Suitable Tool to Gain New Aspects in Glioma Development. Neurosurgery 2003, 53, 1377–1384. [Google Scholar] [CrossRef]
- Djuzenova, C.S.; Fiedler, V.; Memmel, S.; Katzer, A.; Hartmann, S.; Krohne, G.; Zimmermann, H.; Scholz, C.-J.; Polat, B.; Flentje, M.; et al. Actin cytoskeleton organization, cell surface modification and invasion rate of 5 glioblastoma cell lines differing in PTEN and p53 status. Exp. Cell Res. 2015, 330, 346–357. [Google Scholar] [CrossRef]
- Park, C.-M.; Park, M.-J.; Kwak, H.-J.; Moon, S.-I.; Yoo, D.-H.; Lee, H.-C.; Park, I.-C.; Rhee, C.H.; Hong, S.-I. Induction of p53-mediated apoptosis and recovery of chemosensitivity through p53 transduction in human glioblastoma cells by cisplatin. Int. J. Oncol. 2006, 28, 119–125. Available online: http://www.spandidos-publications.com/10.3892/ijo.28.1.119 (accessed on 20 September 2024). [CrossRef]
- Zheng, H.; Ying, H.; Yan, H.; Kimmelman, A.C.; Hiller, D.J.; Chen, A.-J.; Perry, S.R.; Tonon, G.; Chu, G.C.; Ding, Z.; et al. p53 and Pten control neural and glioma stem/progenitor cell renewal and differentiation. Nature 2008, 455, 1129–1133. [Google Scholar] [CrossRef]
- Chen, X.; Liu, J.; Li, Y.; Zeng, Y.; Wang, F.; Cheng, Z.; Duan, H.; Pan, G.; Yang, S.; Chen, Y.; et al. IDH1 mutation impairs antiviral response and potentiates oncolytic virotherapy in glioma. Nat. Commun. 2023, 14, 6781. Available online: https://www.nature.com/articles/s41467-023-42545-3 (accessed on 20 September 2024). [CrossRef] [PubMed]
- Grimi, A.; Bono, B.C.; Lazzarin, S.M.; Marcheselli, S.; Pessina, F.; Riva, M. Gliomagenesis, Epileptogenesis, and Remodeling of Neural Circuits: Relevance for Novel Treatment Strategies in Low- and High-Grade Gliomas. Int. J. Mol. Sci. 2024, 25, 8953. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Liu, Y.; Cai, S.J.; Qian, M.; Ding, J.; Larion, M.; Gilbert, M.R.; Yang, C. IDH mutation in glioma: Molecular mechanisms and potential therapeutic targets. Br. J. Cancer 2020, 122, 1580–1589. [Google Scholar] [CrossRef]
- Sim, N.-L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef]
- Capriotti, E.; Calabrese, R.; Casadio, R. Predicting the insurgence of human genetic diseases associated to single point protein mutations with support vector machines and evolutionary information. Bioinformatics 2006, 22, 2729–2734. [Google Scholar] [CrossRef]
- Calabrese, R.; Capriotti, E.; Fariselli, P.; Martelli, P.L.; Casadio, R. Functional annotations improve the predictive score of human disease-related mutations in proteins. Hum. Mutat. 2009, 30, 1237–1244. [Google Scholar] [CrossRef]
- Bava, K.A. ProTherm, version 4.0: Thermodynamic database for proteins and mutants. Nucleic Acids Res. 2004, 32, D120–D121. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Randall, A.; Baldi, P. Prediction of protein stability changes for single-site mutations using support vector machines. Proteins Struct. Funct. Bioinform. 2006, 62, 1125–1132. [Google Scholar] [CrossRef]
- Pejaver, V.; Urresti, J.; Lugo-Martinez, J.; Pagel, K.A.; Lin, G.N.; Nam, H.-J.; Mort, M.; Cooper, D.N.; Sebat, J.; Iakoucheva, L.M.; et al. Inferring the molecular and phenotypic impact of amino acid variants with MutPred2. Nat. Commun. 2020, 11, 5918. [Google Scholar] [CrossRef]
- Venselaar, H.; Beek, T.A.T.; Kuipers, R.K.; Hekkelman, M.L.; Vriend, G. Protein structure analysis of mutations causing inheritable diseases. An e-Science approach with life scientist friendly interfaces. BMC Bioinform. 2010, 11, 548. [Google Scholar] [CrossRef]
- Csaba, G.; Birzele, F.; Zimmer, R. Systematic comparison of SCOP and CATH: A new gold standard for protein structure analysis. BMC Struct. Biol. 2009, 9, 23. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Skolnick, J. TM-Align: A protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005, 33, 2302–2309. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; De Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Meyer, M.J.; Lapcevic, R.; Romero, A.E.; Yoon, M.; Das, J.; Beltrán, J.F.; Mort, M.; Stenson, P.D.; Cooper, D.N.; Paccanaro, A.; et al. mutation3D: Cancer Gene Prediction Through Atomic Clustering of Coding Variants in the Structural Proteome. Hum. Mutat. 2016, 37, 447–456. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Chiang, Y.-T.; Chien, Y.-C.; Lin, Y.-H.; Wu, H.-H.; Lee, D.-F.; Yu, Y.-L. The Function of the Mutant p53-R175H in Cancer. Cancers 2021, 13, 4088. [Google Scholar] [CrossRef]
- Baugh, E.H.; Ke, H.; Levine, A.J.; Bonneau, R.A.; Chan, C.S. Why are there hotspot mutations in the TP53 gene in human cancers? Cell Death Differ. 2018, 25, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Cheng, H.; Yu, L.; Zhang, J.; Wang, Y.; Liang, Y.; Lou, F.; Wang, H.; Cao, S. Mutation patterns and evolutionary action score of TP53 enable identification of a patient population with poor prognosis in advanced non-small cell lung cancer. Cancer Med. 2023, 12, 6649–6658. [Google Scholar] [CrossRef]
- Yokouchi, H.; Nishihara, H.; Harada, T.; Yamazaki, S.; Kikuchi, H.; Oizumi, S.; Uramoto, H.; Tanaka, F.; Harada, M.; Akie, K.; et al. Detection of somatic TP53 mutation in surgically resected small-cell lung cancer by targeted exome sequencing: Association with longer relapse-free survival. Heliyon 2020, 6, e04439. [Google Scholar] [CrossRef]
- Song, P.; Zhang, F.; Li, Y.; Yang, G.; Li, W.; Ying, J.; Gao, S. Concomitant TP53 mutations with response to crizotinib treatment in patients with ALK-rearranged non-small-cell lung cancer. Cancer Med. 2019, 8, 1551–1557. [Google Scholar] [CrossRef] [PubMed]
- Barta, J.A.; Pauley, K.; Kossenkov, A.V.; McMahon, S.B. The lung-enriched p53 mutants V157F and R158L/P regulate a gain of function transcriptome in lung cancer. Carcinogenesis 2020, 41, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, A.; Hoshida, Y. Depicting the role of TP53 in hepatocellular carcinoma progression. J. Hepatol. 2011, 55, 724–725. [Google Scholar] [CrossRef]
- Mena, A.C.; Pulido, E.G.; Guillen-Ponce, C. Understanding the molecular-based mechanism of action of the tyrosine kinase inhibitor: Sunitinib. Anticancer. Drugs 2010, 21 (Suppl. S1), S3–S11. [Google Scholar] [CrossRef]
- Bougeard, G.; Renaux-Petel, M.; Flaman, J.-M.; Charbonnier, C.; Fermey, P.; Belotti, M.; Gauthier-Villars, M.; Stoppa-Lyonnet, D.; Consolino, E.; Brugières, L.; et al. Revisiting Li-Fraumeni Syndrome From TP53 Mutation Carriers. J. Clin. Oncol. 2015, 33, 2345–2352. [Google Scholar] [CrossRef]
- Lai, Z.-Y.; Tsai, K.-Y.; Chang, S.-J.; Chuang, Y.-J. Gain-of-Function Mutant TP53 R248Q Overexpressed in Epithelial Ovarian Carcinoma Alters AKT-Dependent Regulation of Intercellular Trafficking in Responses to EGFR/MDM2 Inhibitor. Int. J. Mol. Sci. 2021, 22, 8784. [Google Scholar] [CrossRef] [PubMed]
- Allende-Vega, N.; Villalba, M. Metabolic stress controls mutant p53 R248Q stability in acute myeloid leukemia cells. Sci. Rep. 2019, 9, 5637. [Google Scholar] [CrossRef]
- Klemke, L.; Fehlau, C.F.; Winkler, N.; Toboll, F.; Singh, S.K.; Moll, U.M.; Schulz-Heddergott, R. The Gain-of-Function p53 R248W Mutant Promotes Migration by STAT3 Deregulation in Human Pancreatic Cancer Cells. Front. Oncol. 2021, 11, 642603. [Google Scholar] [CrossRef]
- Yoshikawa, K.; Hamada, J.-I.; Tada, M.; Kameyama, T.; Nakagawa, K.; Suzuki, Y.; Ikawa, M.; Hassan, N.M.M.; Kitagawa, Y.; Moriuchi, T. Mutant p53 R248Q but not R248W enhances in vitro invasiveness of human lung cancer NCI-H1299 cells. Biomed. Res. 2010, 31, 401–411. [Google Scholar] [CrossRef]
- Bauer, M.R.; Krämer, A.; Settanni, G.; Jones, R.N.; Ni, X.; Tareque, R.K.; Fersht, A.R.; Spencer, J.; Joerger, A.C. Targeting Cavity-Creating p53 Cancer Mutations with Small-Molecule Stabilizers: The Y220X Paradigm. ACS Chem. Biol. 2020, 15, 657–668. [Google Scholar] [CrossRef]
- Gomes, A.S.; Ramos, H.; Inga, A.; Sousa, E.; Saraiva, L. Structural and Drug Targeting Insights on Mutant p53. Cancers 2021, 13, 3344. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Gao, S.-J.; Soubise, B.; Douet-Guilbert, N.; Liu, Z.-L.; Troadec, M.-B. TP53 in Myelodysplastic Syndromes. Cancers 2021, 13, 5392. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, X.; Tang, Y.; Lao, Z.; Lei, J.; Wei, G. Common cancer mutations R175H and R273H drive the p53 DNA-binding domain towards aggregation-prone conformations. Phys. Chem. Chem. Phys. 2020, 22, 9225–9232. [Google Scholar] [CrossRef] [PubMed]
- Sarma, P.P.; Dutta, D.; Mirza, Z.; Saikia, K.K.; Baishya, B.K. Point mutations in the DNA binding domain of p53 contribute to glioma progression and poor prognosis. Mol. Biol. 2017, 51, 293–299. [Google Scholar] [CrossRef]
- Schaefer, K.N.; Geil, W.M.; Sweredoski, M.J.; Moradian, A.; Hess, S.; Barton, J.K. Oxidation of p53 through DNA Charge Transport Involves a Network of Disulfides within the DNA-Binding Domain. Biochemistry 2015, 54, 932–941. [Google Scholar] [CrossRef]
- Synoradzki, K.J.; Bartnik, E.; Czarnecka, A.M.; Fiedorowicz, M.; Firlej, W.; Brodziak, A.; Stasinska, A.; Rutkowski, P.; Grieb, P. TP53 in Biology and Treatment of Osteosarcoma. Cancers 2021, 13, 4284. [Google Scholar] [CrossRef]
- Carson, C.; Omolo, B.; Chu, H.; Zhou, Y.; Sambade, M.J.; Peters, E.C.; Tompkins, P.; Simpson, D.A.; Thomas, N.E.; Fan, C.; et al. A prognostic signature of defective p53-dependent G1 checkpoint function in melanoma cell lines. Pigment. Cell Melanoma Res. 2012, 25, 514–526. [Google Scholar] [CrossRef]
- Bendahou, M.A.; Arrouchi, H.; Lakhlili, W.; Allam, L.; Aanniz, T.; Cherradi, N.; Ibrahimi, A.; Boutarbouch, M. Computational Analysis of IDH1, IDH2, and TP53 Mutations in Low-Grade Gliomas Including Oligodendrogliomas and Astrocytomas. Cancer Inform. 2020, 19, 117693512091583. [Google Scholar] [CrossRef]
- Zhan, D.; Ma, D.; Wei, S.; Lal, B.; Fu, Y.; Eberhart, C.; Laterra, J.; Ying, M.; Li, Y.; Meeker, A.; et al. Monoallelic IDH1 R132H Mutation Mediates Glioma Cell Response to Anticancer Therapies via Induction of Senescence. Mol. Cancer Res. 2021, 19, 1878–1888. [Google Scholar] [CrossRef]
- Kayabolen, A.; Yilmaz, E.; Bagci-Onder, T. IDH Mutations in Glioma: Double-Edged Sword in Clinical Applications? Biomedicines 2021, 9, 799. [Google Scholar] [CrossRef]
- Ahmed, H.U.; Paul, A.; Mahmud, Z.; Rahman, T.; Hosen, I. Comprehensive characterization of the single nucleotide polymorphisms located in the isocitrate dehydrogenase isoform 1 and 2 genes using in silico approach. Gene Rep. 2021, 24, 101259. [Google Scholar] [CrossRef]
- Najafi, S.; Esmaeili, S.; Zhaleh, H.; Rahmati, Y. The role of IDH1 mutation on gene expression in glioblastoma. Inform. Med. Unlocked 2022, 28, 100812. [Google Scholar] [CrossRef]
- Lemonnier, F.; Cairns, R.A.; Inoue, S.; Li, W.Y.; Dupuy, A.; Broutin, S.; Martin, N.; Fataccioli, V.; Pelletier, R.; Wakeham, A.; et al. The IDH2 R172K mutation associated with angioimmunoblastic T-cell lymphoma produces 2HG in T cells and impacts lymphoid development. Proc. Natl. Acad. Sci. USA 2016, 113, 15084–15089. [Google Scholar] [CrossRef] [PubMed]
- Kotredes, K.P.; Razmpour, R.; Lutton, E.; Alfonso-Prieto, M.; Ramirez, S.H.; Gamero, A.M. Characterization of cancer-associated IDH2 mutations that differ in tumorigenicity, chemosensitivity and 2-hydroxyglutarate production. Oncotarget 2019, 10, 2675–2692. [Google Scholar] [CrossRef]
- Sporikova, Z.; Slavkovsky, R.; Tuckova, L.; Kalita, O.; Houdova, M.M.; Ehrmann, J.; Hajduch, M.; Hrabalek, L.; Vaverka, M. IDH1/2 Mutations in Patients With Diffuse Gliomas: A Single Centre Retrospective Massively Parallel Sequencing Analysis. Appl. Immunohistochem. Mol. Morphol. 2022, 30, 178–183. [Google Scholar] [CrossRef]
- Alfadul, S.M.; Matnurov, E.M.; Varakutin, A.E.; Babak, M.V. Metal-Based Anticancer Complexes and p53: How Much Do We Know? Cancers 2023, 15, 2834. [Google Scholar] [CrossRef]
- Cho, Y.; Gorina, S.; Jeffrey, P.D.; Pavletich, N.P. Crystal Structure of a p53 Tumor Suppressor-DNA Complex: Understanding Tumorigenic Mutations. Science 1994, 265, 346–355. [Google Scholar] [CrossRef]
- Joerger, A.C.; Fersht, A.R. Structural Biology of the Tumor Suppressor p53 and Cancer-Associated Mutants. In Advances in Cancer Research [Internet]; Elsevier: Amsterdam, The Netherlands, 2007; pp. 1–23. Available online: https://linkinghub.elsevier.com/retrieve/pii/S0065230X06970018 (accessed on 17 September 2024).
- Butler, J.S.; Loh, S.N. Structure, Function, and Aggregation of the Zinc-Free Form of the p53 DNA Binding Domain. Biochemistry 2003, 42, 2396–2403. [Google Scholar] [CrossRef]
- Bullock, A.N.; Henckel, J.; Fersht, A.R. Quantitative analysis of residual folding and DNA binding in mutant p53 core domain: Definition of mutant states for rescue in cancer therapy. Oncogene 2000, 19, 1245–1256. [Google Scholar] [CrossRef]
- Loh, S.N. The missing Zinc: p53 misfolding and cancer. Metallomics 2010, 2, 442–449. [Google Scholar] [CrossRef]
- Pavletich, N.P.; Chambers, K.A.; Pabo, C.O. The DNA-binding domain of p53 contains the four conserved regions and the major mutation hot spots. Genes. Dev. 1993, 7, 2556–2564. [Google Scholar] [CrossRef] [PubMed]
- Joerger, A.; Fersht, A.R. Structure–function–rescue: The diverse nature of common p53 cancer mutants. Oncogene 2007, 26, 2226–2242. [Google Scholar] [CrossRef] [PubMed]
- Freed-Pastor, W.A.; Prives, C. Mutant p53: One name, many proteins. Genes. Dev. 2012, 26, 1268–1286. [Google Scholar] [CrossRef]
- Kogan, S.; Carpizo, D.R. Zinc Metallochaperones as Mutant p53 Reactivators: A New Paradigm in Cancer Therapeutics. Cancers 2018, 10, 166. [Google Scholar] [CrossRef]
- Blanden, A.R.; Yu, X.; Wolfe, A.J.; Gilleran, J.A.; Augeri, D.J.; O’dell, R.S.; Olson, E.C.; Kimball, S.D.; Emge, T.J.; Movileanu, L.; et al. Synthetic Metallochaperone ZMC1 Rescues Mutant p53 Conformation by Transporting Zinc into Cells as an Ionophore. Mol. Pharmacol. 2015, 87, 825–831. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Gao, T.; Wang, Y.; Yang, G. Investigating the Influence of Magnesium Ions on p53–DNA Binding Using Atomic Force Microscopy. Int. J. Mol. Sci. 2017, 18, 1585. [Google Scholar] [CrossRef]
- Méplan, C.; Richard, M.-J.; Hainaut, P. Metalloregulation of the tumor suppressor protein p53: Zinc mediates the renaturation of p53 after exposure to metal chelators in vitro and in intact cells. Oncogene 2000, 19, 5227–5236. [Google Scholar] [CrossRef]
- Butler, J.S.; Loh, S.N. Zn2+-Dependent Misfolding of the p53 DNA Binding Domain. Biochemistry 2007, 46, 2630–2639. [Google Scholar] [CrossRef]
- Hainaut, P.; Milner, J. A structural role for metal ions in the “wild-type” conformation of the tumor suppressor protein p53. Cancer Res. 1993, 53, 1739–1742. [Google Scholar]
- Liu, S.; Abboud, M.I.; John, T.; Mikhailov, V.; Hvinden, I.; Walsby-Tickle, J.; Liu, X.; Pettinati, I.; Cadoux-Hudson, T.; McCullagh, J.S.O.; et al. Roles of metal ions in the selective inhibition of oncogenic variants of isocitrate dehydrogenase 1. Commun. Biol. 2021, 4, 1243. Available online: https://www.nature.com/articles/s42003-021-02743-5 (accessed on 20 September 2024). [CrossRef]
- Murnyak, B.; Huang, L.E. Association of TP53 Alteration with Tissue Specificity and Patient Outcome of IDH1-Mutant Glioma. Cells 2021, 10, 2116. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Chromosome | Mutation Position | RS | Gene | Amino Acid Alteration | Classification According to ClinVar | MSC/ PolyPhen Score | Prediction PolyPhen | MSC/SIFT Score | Prediction | PhD SNP | SNP and GO |
|---|---|---|---|---|---|---|---|---|---|---|---|
| SIFT | |||||||||||
| 17 | 7577574 | rs730882026 | TP53 | p.Tyr236Cys | Conflict of interpretation | 0.033/1 | Pathogenic | 0.133/0 | Pathogenic | Deleterious to protein | Deleterious to protein |
| 17 | 7577117 | rs1057520006 | TP53 | p.Val274Gly | Conflict of interpretation | 0.033/1 | Pathogenic | 0.133/0 | Pathogenic | Deleterious to protein | Deleterious to protein |
| 17 | 7578461 | rs121912654 | TP53 | p.Val157Phe | Conflict of interpretation | 0.033/0.999 | Pathogenic | 0.133/0.01 | Pathogenic | Deleterious to protein | Deleterious to protein |
| 17 | 7577107 | rs193920789 | TP53 | p.Cys238Trp | Pathogenic | 0.033/1 | Pathogenic | 0.133/0 | Pathogenic | Deleterious to protein | Deleterious to protein |
| 17 | 7578449 | * | TP53 | p.Ala161Ser | New | 0.033/0.967 | Pathogenic | 0.133/0.08 | Tolerable | Deleterious to protein | Deleterious to protein |
| 17 | 7577086 | rs786201057 | TP53 | p.Thr125Met | Conflict of interpretation | 0.033/1 | Pathogenic | 0.133/0 | Pathogenic | Deleterious to protein | Deleterious to protein |
| 17 | 7577538 | rs11540652 | TP53 | p.Arg248Gln | Pathogenic | 0.033/1 | Pathogenic | 0.133/0 | Pathogenic | Deleterious to protein | Deleterious to protein |
| 17 | 7577539 | rs121912651 | TP53 | p.Arg248Trp | Pathogenic | 0.033/1 | Pathogenic | 0.133/0 | Pathogenic | Deleterious to protein | Deleterious to protein |
| 17 | 7578191 | rs530941076 | TP53 | p.Tyr220His | Pathogenic | 0.033/1 | Pathogenic | 0.133/0 | Pathogenic | Deleterious to protein | Deleterious to protein |
| 17 | 7578406 | rs28934578 | TP53 | p.Arg175His | Pathogenic | 0.033/0.881 | Pathogenic | 0.133/0 | Pathogenic | Deleterious to protein | Deleterious to protein |
| 17 | 7579315 | rs1555526478 | TP53 | p.Cys124Ter | Pathogenic | 0.033/0.694 | Pathogenic | 0.133/0.11 | Tolerable | Deleterious to protein | Deleterious to protein |
| 17 | 7577102 | rs1064793881 | TP53 | p.Gly279Glu | Conflict of interpretation | 0.033/1 | Pathogenic | 0.133/0 | Pathogenic | Deleterious to protein | Deleterious to protein |
| 17 | 7578393 | rs876660821 | TP53 | p.His179Gln | Pathogenic | 0.033/0.999 | Pathogenic | 0.133/0 | Pathogenic | Deleterious to protein | Deleterious to protein |
| 2 | 209113112 | rs121913500 | IDH1 | p.Arg132His | Pathogenic | 0.823/0.013 | benign | 0.995/0 | Pathogenic | Deleterious to protein | Deleterious to protein |
| 15 | 90631838 | rs121913503 | IDH2 | p.Arg172Lys | Pathogenic | 1.000/0.999 | Pathogenic | 0.995/0 | Pathogenic | Deleterious to protein | Deleterious to protein |
| Gene | Alteration | MUpro | I-Mutant 3.0 | ||
|---|---|---|---|---|---|
| Value DDG | Protein Stability | Reliability Index | Protein Stability | ||
| (Kcal/mol) | |||||
| TP53 | Y236C | −15,011.602 | Decreases | 3 | Decreases |
| TP53 | V274G | −25,962.627 | Decreases | 9 | Decreases |
| TP53 | V157F | −0.94054347 | Decreases | 9 | Decreases |
| TP53 | C238W | −14,075.569 | Decreases | 5 | Decreases |
| TP53 | A161S | −0.73476729 | Decreases | 9 | Decreases |
| TP53 | T125M | −0.22237917 | Decreases | 3 | Decreases |
| TP53 | R248Q | −18,402.017 | Decreases | 7 | Decreases |
| TP53 | R248W | −14,246.049 | Decreases | 5 | Decreases |
| TP53 | Y220H | −0.85387287 | Decreases | 7 | Decreases |
| TP53 | R175H | −29,105.592 | Decreases | 8 | Decreases |
| TP53 | C124T | −29,105.592 | Decreases | 1 | Decreases |
| TP53 | G279E | 0.062230681 | Increases | 3 | Decreases |
| TP53 | H179Q | −0.19938582 | Decreases | 7 | Decreases |
| IDH1 | R132H | −14,231.866 | Decreases | 7 | Decreases |
| IDH2 | R172K | −12,425.191 | Decreases | 5 | Decreases |
| Altered Properties Due to SNP | SNPs Gene TP53 | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Percentage Probability of Change in Protein Function | |||||||||
| Y236C | V274G | V157F | C238W | Y220H | R175H | C124T | G279E | H179Q | |
| Altered ordered interface | 28% | - | - | 27% | - | - | 28% | - | - |
| Loss of strand | 27% | - | - | - | 28% | - | - | 27% | - |
| Gain of disulfide bond at C238 | 22% | - | - | 26% | - | - | - | - | - |
| Altered stability | 16% | 73% | 11% | - | - | 51% | 13% | - | - |
| Loss of N-linked glycosylation at N239 | 3% | - | - | 6% | - | - | - | - | - |
| Gain of intrinsic disorder | - | 42% | - | - | - | - | - | - | - |
| Gain of strand | - | - | - | 26% | - | - | - | - | - |
| Altered metal binding | - | - | - | - | 27% | 67% | - | - | 72% |
| Gain of factor B | - | - | - | - | 24% | - | - | - | - |
| Altered transmembrane protein | - | - | - | - | 14% | - | - | - | - |
| Loss of sulfation at Y220 | - | - | - | - | 1% | - | - | - | - |
| Loss of helix | - | - | - | - | - | 28% | - | - | - |
| Gain of ubiquitylation at K120 | - | - | - | - | - | - | 15% | - | - |
| Gain of helix | - | - | - | - | - | - | - | 30% | - |
| Gain of intrinsic disorder | - | - | - | - | - | - | - | 31% | - |
| Altered Properties Due to SNP | SNPs Gene IDH1 | ||||||||
| Percentage Probability of Change in Protein Function | |||||||||
| R132H | |||||||||
| Altered metal binding | 55% | ||||||||
| Loss of allosteric site at R132 | 36% | ||||||||
| Altered DNA binding | 34% | ||||||||
| Altered disordered interface | 29% | ||||||||
| Altered transmembrane protein | 27% | ||||||||
| Loss of relative solvent accessibility | 27% | ||||||||
| Altered ordered interface | 26% | ||||||||
| Gain of catalytic site at H133 | 26% | ||||||||
| Altered Properties Due to SNP | SNPs Gene IDH2 | ||||||||
| Percentage Probability of Change in Protein Function | |||||||||
| R172K | |||||||||
| Altered DNA binding | 40% | ||||||||
| Loss of allosteric site at H173 | 34% | ||||||||
| Altered metal binding | 32% | ||||||||
| Gain of relative solvent accessibility | 30% | ||||||||
| Altered ordered interface | 29% | ||||||||
| Loss of catalytic site at R172 | 26% | ||||||||
| Altered transmembrane protein | 22% | ||||||||
| Gene | nsSNP | TM-Aling | SWISS-MODEL | |
|---|---|---|---|---|
| TM-Score a | Value RMSD b | Ramachandran Graphic Score c | ||
| TP53 | Y236C | 1.00 | 0.003 | 95.40% |
| TP53 | V274G | 1.00 | 0.004 | 95.40% |
| TP53 | V157F | 1.00 | 0.041 | 95.40% |
| TP53 | C238W | 0.999 | 0.053 | 93.86% |
| TP53 | A161S | 1.00 | 0.001 | 95.40% |
| TP53 | T125M | 1.00 | 0.002 | 95.40% |
| TP53 | R248Q | 1.00 | 0.002 | 95.40% |
| TP53 | R248W | 1.00 | 0.001 | 95.40% |
| TP53 | Y220H | 1.00 | 0.002 | 95.40% |
| TP53 | R175H | 1.00 | 0.008 | 95.40% |
| TP53 | C124T | 1.00 | 0.006 | 95.40% |
| TP53 | H179Q | 1.00 | 0.002 | 95.40% |
| TP53 | G279E | 1.00 | 0.004 | 95.40% |
| IDH1 | R132H | 0.8382 | 3.730 | 96.33% |
| IDH2 | R172K | 1.00 | 0.004 | 96.44% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vélez Gómez, S.; Martínez Garro, J.M.; Ortiz Gómez, L.D.; Salazar Flórez, J.E.; Monroy, F.P.; Peláez Sánchez, R.G. Bioinformatic Characterization of the Functional and Structural Effect of Single Nucleotide Mutations in Patients with High-Grade Glioma. Biomedicines 2024, 12, 2287. https://doi.org/10.3390/biomedicines12102287
Vélez Gómez S, Martínez Garro JM, Ortiz Gómez LD, Salazar Flórez JE, Monroy FP, Peláez Sánchez RG. Bioinformatic Characterization of the Functional and Structural Effect of Single Nucleotide Mutations in Patients with High-Grade Glioma. Biomedicines. 2024; 12(10):2287. https://doi.org/10.3390/biomedicines12102287
Chicago/Turabian StyleVélez Gómez, Sara, Juliana María Martínez Garro, León Darío Ortiz Gómez, Jorge Emilio Salazar Flórez, Fernando P. Monroy, and Ronald Guillermo Peláez Sánchez. 2024. "Bioinformatic Characterization of the Functional and Structural Effect of Single Nucleotide Mutations in Patients with High-Grade Glioma" Biomedicines 12, no. 10: 2287. https://doi.org/10.3390/biomedicines12102287
APA StyleVélez Gómez, S., Martínez Garro, J. M., Ortiz Gómez, L. D., Salazar Flórez, J. E., Monroy, F. P., & Peláez Sánchez, R. G. (2024). Bioinformatic Characterization of the Functional and Structural Effect of Single Nucleotide Mutations in Patients with High-Grade Glioma. Biomedicines, 12(10), 2287. https://doi.org/10.3390/biomedicines12102287