

Regulation of Airway Epithelial-Derived Alarmins in Asthma: Perspectives for Therapeutic Targets

Abstract
:1. Introduction
2. The Airway Epithelium: Structure and Function
3. The Cell-Derived Alarmins IL-25, IL-33 and TSLP and Their Role in Asthma Pathology
3.1. IL-25, IL-33 and TSLP Alarmin Expression and Signaling
3.2. Alarmin Cytokines Drive Type 2 Inflammation
4. Epithelial-Derived Cytokines HMGB1, IL-1α, IL-36 and TL1A in Asthma Pathology
4.1. HMGB1
4.2. IL-1α
4.3. IL-36
4.4. TL1A
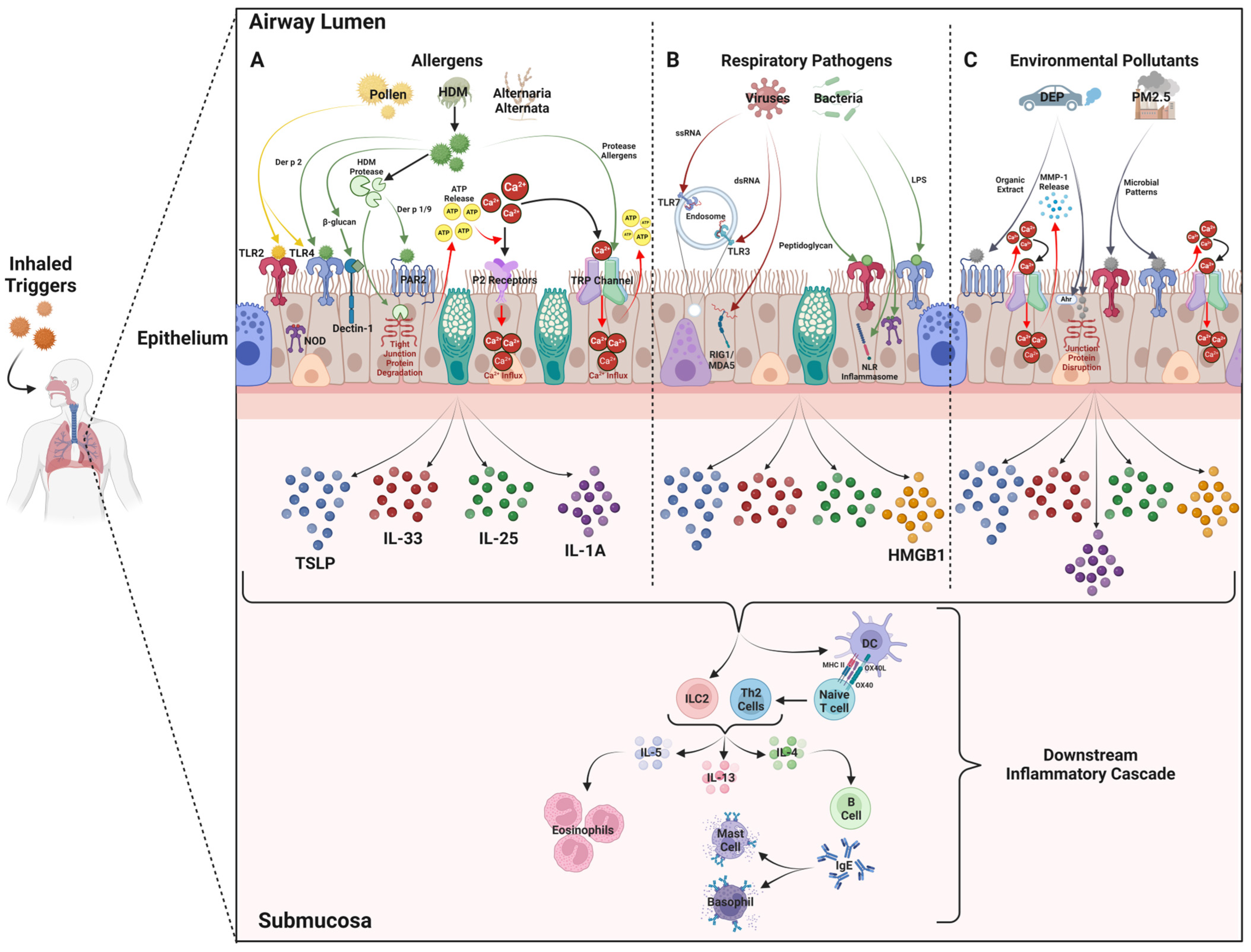
5. Triggers That Mediate the Release of Alarmin Cytokines from Airway Epithelial Cells
5.1. Aeroallergens
5.2. Respiratory Infections
5.3. Environmental Pollutants
6. Effect of Alarmin Blockade in Asthma—Review of Clinical Trials to Date
6.1. Anti-IL-25 Biologics
6.2. Anti-TSLP Biologics
6.3. Anti-IL-33 Biologics
6.4. Anti-ST2 Biologics
6.5. Personalized Medicine for Treatment of Asthma
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cecchi, L.; Vaghi, A.; Bini, F.; Martini, M.; Musarra, A.; Bilò, M.B. From triggers to asthma: A narrative review on epithelium dysfunction. Eur. Ann. Allergy Clin. Immunol. 2022, 54, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Holgate, S.T. Innate and adaptive immune responses in asthma. Nat. Med. 2012, 18, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekara, S.; Wark, P. Biologic therapies for severe asthma with persistent type 2 inflammation. Aust. Prescr. 2024, 47, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Dunican, E.M.; Fahy, J.V. The Role of Type 2 Inflammation in the Pathogenesis of Asthma Exacerbations. Ann. Am. Thorac. Soc. 2015, 12 (Suppl. S2), S144–S149. [Google Scholar] [CrossRef] [PubMed]
- Vatrella, A.; Maglio, A.; Pelaia, C.; Ciampo, L.; Pelaia, G.; Vitale, C. Eosinophilic inflammation: An Appealing Target for Pharmacologic Treatments in Severe Asthma. Biomedicines 2022, 10, 2181. [Google Scholar] [CrossRef]
- Inman, M.D.; Watson, R.M.; Rerecich, T.; Gauvreau, G.M.; Lutsky, B.N.; Stryszak, P.; O’Byrne, P.M. Dose-dependent effects of inhaled mometasone furoate on airway function and inflammation after allergen inhalation challenge. Am. J. Respir. Crit. Care Med. 2001, 164, 569–574. [Google Scholar] [CrossRef]
- Calvén, J.; Ax, E.; Rådinger, M. The Airway Epithelium-A Central Player in Asthma Pathogenesis. Int. J. Mol. Sci. 2020, 21, 8907. [Google Scholar] [CrossRef]
- Gauvreau, G.M.; Bergeron, C.; Boulet, L.P.; Cockcroft, D.W.; Côté, A.; Davis, B.E.; Leigh, R.; Myers, I.; O’Byrne, P.M.; Sehmi, R. Sounding the alarmins-The role of alarmin cytokines in asthma. Allergy 2023, 78, 402–417. [Google Scholar] [CrossRef]
- Kayalar, Ö.; Rajabi, H.; Konyalilar, N.; Mortazavi, D.; Aksoy, G.T.; Wang, J.; Bayram, H. Impact of particulate air pollution on airway injury and epithelial plasticity; underlying mechanisms. Front. Immunol. 2024, 15, 1324552. [Google Scholar] [CrossRef]
- Heijink, I.H.; Kuchibhotla, V.N.S.; Roffel, M.P.; Maes, T.; Knight, D.A.; Sayers, I.; Nawijn, M.C. Epithelial cell dysfunction, a major driver of asthma development. Allergy 2020, 75, 1902–1917. [Google Scholar] [CrossRef]
- Whetstone, C.E.; Ranjbar, M.; Omer, H.; Cusack, R.P.; Gauvreau, G.M. The Role of Airway Epithelial Cell Alarmins in Asthma. Cells 2022, 11, 1105. [Google Scholar] [CrossRef] [PubMed]
- Zakeri, A.; Russo, M. Dual Role of Toll-like Receptors in Human and Experimental Asthma Models. Front. Immunol. 2018, 9, 1027. [Google Scholar] [CrossRef] [PubMed]
- Peters, T.; Henry, P.J. Protease-activated receptors and prostaglandins in inflammatory lung disease. Br. J. Pharmacol. 2009, 158, 1017–1033. [Google Scholar] [CrossRef] [PubMed]
- Angelina, A.; Martín-Cruz, L.; de la Rocha-Muñoz, A.; Lavín-Plaza, B.; Palomares, O. C-Type Lectin Receptor Mediated Modulation of T2 Immune Responses to Allergens. Curr. Allergy Asthma Rep. 2023, 23, 141–151. [Google Scholar] [CrossRef]
- Zheng, J.; Shi, W.; Yang, Z.; Chen, J.; Qi, A.; Yang, Y.; Deng, Y.; Yang, D.; Song, N.; Song, B.; et al. RIG-I-like receptors: Molecular mechanism of activation and signaling. Adv. Immunol. 2023, 158, 1–74. [Google Scholar] [CrossRef]
- Fournier, B.; Philpott, D.J. Recognition of Staphylococcus aureus by the innate immune system. Clin. Microbiol. Rev. 2005, 18, 521–540. [Google Scholar] [CrossRef]
- Denney, L.; Ho, L.P. The role of respiratory epithelium in host defence against influenza virus infection. Biomed. J. 2018, 41, 218–233. [Google Scholar] [CrossRef]
- Bucheimer, R.E.; Linden, J. Purinergic regulation of epithelial transport. J. Physiol. 2004, 555, 311–321. [Google Scholar] [CrossRef]
- Brister, D.L.; Omer, H.; Whetstone, C.E.; Ranjbar, M.; Gauvreau, G.M. Multifactorial Causes and Consequences of TLSP Production, Function, and Release in the Asthmatic Airway. Biomolecules 2024, 14, 401. [Google Scholar] [CrossRef]
- Ko, H.K.; Cheng, S.L.; Lin, C.H.; Lin, S.H.; Hsiao, Y.H.; Su, K.C.; Yu, C.J.; Wang, H.C.; Sheu, C.C.; Chiu, K.C.; et al. Blood tryptase and thymic stromal lymphopoietin levels predict the risk of exacerbation in severe asthma. Sci. Rep. 2021, 11, 8425. [Google Scholar] [CrossRef]
- Furci, F.; Murdaca, G.; Pelaia, C.; Imbalzano, E.; Pelaia, G.; Caminati, M.; Allegra, A.; Senna, G.; Gangemi, S. TSLP and HMGB1: Inflammatory Targets and Potential Biomarkers for Precision Medicine in Asthma and COPD. Biomedicines 2023, 11, 437. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Hammel, M.; He, Y.; Tainer, J.A.; Jeng, U.S.; Zhang, L.; Wang, S.; Wang, X. Structural insights into the interaction of IL-33 with its receptors. Proc. Natl. Acad. Sci. USA 2013, 110, 14918–14923. [Google Scholar] [CrossRef]
- Liu, F.; Wu, J.X.; Zhao, J.P.; Li, H.J.; Liu, W.; Bi, W.X.; Dong, L. [IL-25 derived from epithelial cells has the potential to promote airway remodeling in asthma]. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi 2012, 28, 633–636. [Google Scholar] [PubMed]
- Préfontaine, D.; Lajoie-Kadoch, S.; Foley, S.; Audusseau, S.; Olivenstein, R.; Halayko, A.J.; Lemière, C.; Martin, J.G.; Hamid, Q. Increased expression of IL-33 in severe asthma: Evidence of expression by airway smooth muscle cells. J. Immunol. 2009, 183, 5094–5103. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Li, Y.; Lv, Z.; Chen, Y.; Li, Y.; Huang, K.; Corrigan, C.J.; Ying, S. Bronchial Allergen Challenge of Patients with Atopic Asthma Triggers an Alarmin (IL-33, TSLP, and IL-25) Response in the Airways Epithelium and Submucosa. J. Immunol. 2018, 201, 2221–2231. [Google Scholar] [CrossRef] [PubMed]
- Asaka, D.; Yoshikawa, M.; Nakayama, T.; Yoshimura, T.; Moriyama, H.; Otori, N. Elevated levels of interleukin-33 in the nasal secretions of patients with allergic rhinitis. Int. Arch. Allergy Immunol. 2012, 158 (Suppl. S1), 47–50. [Google Scholar] [CrossRef]
- Kim, D.W.; Kim, D.K.; Eun, K.M.; Bae, J.S.; Chung, Y.J.; Xu, J.; Kim, Y.M.; Mo, J.H. IL-25 Could Be Involved in the Development of Allergic Rhinitis Sensitized to House Dust Mite. Mediat. Inflamm. 2017, 2017, 3908049. [Google Scholar] [CrossRef]
- Mou, Z.; Xia, J.; Tan, Y.; Wang, X.; Zhang, Y.; Zhou, B.; Li, H.; Han, D. Overexpression of thymic stromal lymphopoietin in allergic rhinitis. Acta Otolaryngol. 2009, 129, 297–301. [Google Scholar] [CrossRef]
- Ranjbar, M.; Whetstone, C.E.; Cusack, R.P.; Al-Sajee, D.; Omer, H.; Alsaji, N.; Ho, T.; Duong, M.; Mitchell, P.; Satia, I.; et al. Comparison of upper and lower airway expression of SARS-CoV-2 receptors in allergic asthma. Allergy 2024, 79, 2856–2858. [Google Scholar] [CrossRef]
- Whetstone, C.; Ranjbar, M.; Alsaji, N.; Al-Sajee, D.; Wiltshire, L.; Wattie, J.; O’Byrne, P.; Cusack, R.; Sehmi, R.; Gauvreau, G. Alarmin Expression in the Upper and Lower Airways of Asthmatics with Allergic Rhinitis. Eur. Respir. J. 2022, 60, 3735. [Google Scholar] [CrossRef]
- Gurram, R.K.; Zhu, J. Orchestration between ILC2s and Th2 cells in shaping type 2 immune responses. Cell Mol. Immunol. 2019, 16, 225–235. [Google Scholar] [CrossRef]
- Camelo, A.; Rosignoli, G.; Ohne, Y.; Stewart, R.A.; Overed-Sayer, C.; Sleeman, M.A.; May, R.D. IL-33, IL-25, and TSLP induce a distinct phenotypic and activation profile in human type 2 innate lymphoid cells. Blood Adv. 2017, 1, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Barrett, N.A.; Kanaoka, Y.; Yoshimoto, E.; Garofalo, D.; Cirka, H.; Feng, C.; Boyce, J.A. Type 2 Cysteinyl Leukotriene Receptors Drive IL-33–Dependent Type 2 Immunopathology and Aspirin Sensitivity. J. Immunol. 2018, 200, 915–927. [Google Scholar] [CrossRef] [PubMed]
- Halim, T.Y.; Rana, B.M.; Walker, J.A.; Kerscher, B.; Knolle, M.D.; Jolin, H.E.; Serrao, E.M.; Haim-Vilmovsky, L.; Teichmann, S.A.; Rodewald, H.-R. Tissue-restricted adaptive type 2 immunity is orchestrated by expression of the costimulatory molecule OX40L on group 2 innate lymphoid cells. Immunity 2018, 48, 1195–1207.e6. [Google Scholar] [CrossRef]
- Chen, R.; Smith, S.G.; Salter, B.; El-Gammal, A.; Oliveria, J.P.; Obminski, C.; Watson, R.; O’Byrne, P.M.; Gauvreau, G.M.; Sehmi, R. Allergen-induced increases in sputum levels of group 2 innate lymphoid cells in subjects with asthma. Am. J. Respir. Crit. Care Med. 2017, 196, 700–712. [Google Scholar] [CrossRef]
- Corrigan, C.J.; Wang, W.; Meng, Q.; Fang, C.; Wu, H.; Reay, V.; Lv, Z.; Fan, Y.; An, Y.; Wang, Y.-H. T-helper cell type 2 (Th2) memory T cell-potentiating cytokine IL-25 has the potential to promote angiogenesis in asthma. Proc. Natl. Acad. Sci. USA 2011, 108, 1579–1584. [Google Scholar] [CrossRef]
- Furusawa, J.-i.; Moro, K.; Motomura, Y.; Okamoto, K.; Zhu, J.; Takayanagi, H.; Kubo, M.; Koyasu, S. Critical role of p38 and GATA3 in natural helper cell function. J. Immunol. 2013, 191, 1818–1826. [Google Scholar] [CrossRef]
- Seder, R.A.; Paul, W.E.; Davis, M.M.; Fazekas de St Groth, B. The presence of interleukin 4 during in vitro priming determines the lymphokine-producing potential of CD4+ T cells from T cell receptor transgenic mice. J. Exp. Med. 1992, 176, 1091–1098. [Google Scholar] [CrossRef] [PubMed]
- Jutel, M.; Pichler, W.J.; Skrbic, D.; Urwyler, A.; Dahinden, C.; Müller, U. Bee venom immunotherapy results in decrease of IL-4 and IL-5 and increase of IFN-gamma secretion in specific allergen-stimulated T cell cultures. J. Immunol 1995, 154, 4187–4194. [Google Scholar] [CrossRef]
- Schulz-Kuhnt, A.; Greif, V.; Hildner, K.; Knipfer, L.; Döbrönti, M.; Zirlik, S.; Fuchs, F.; Atreya, R.; Zundler, S.; López-Posadas, R. ILC2 lung-homing in cystic fibrosis patients: Functional involvement of CCR6 and impact on respiratory failure. Front. Immunol. 2020, 11, 691. [Google Scholar] [CrossRef]
- Krempski, J.W.; Iijima, K.; Kobayashi, T.; Kita, H. ILC2-derived IL-13 likely promotes development of peanut allergy. J. Immunol. 2018, 200, 46.12. [Google Scholar] [CrossRef]
- Macheleidt, O.; Sandhoff, K.; Kaiser, H.W. Deficiency of epidermal protein-bound ω-hydroxyceramides in atopic dermatitis. J. Investig. Dermatol. 2002, 119, 166–173. [Google Scholar] [CrossRef]
- Turner, J.-E.; Morrison, P.J.; Wilhelm, C.; Wilson, M.; Ahlfors, H.; Renauld, J.-C.; Panzer, U.; Helmby, H.; Stockinger, B. IL-9–mediated survival of type 2 innate lymphoid cells promotes damage control in helminth-induced lung inflammation. J. Exp. Med. 2013, 210, 2951–2965. [Google Scholar] [CrossRef] [PubMed]
- Smithgall, M.D.; Comeau, M.R.; Park Yoon, B.-R.; Kaufman, D.; Armitage, R.; Smith, D.E. IL-33 amplifies both Th1-and Th2-type responses through its activity on human basophils, allergen-reactive Th2 cells, iNKT and NK cells. Int. Immunol. 2008, 20, 1019–1030. [Google Scholar] [CrossRef]
- Wu, W.H.; Park, C.O.; Oh, S.H.; Kim, H.J.; Kwon, Y.S.; Bae, B.G.; Noh, J.Y.; Lee, K.H. Thymic stromal lymphopoietin–activated invariant natural killer T cells trigger an innate allergic immune response in atopic dermatitis. J. Allergy Clin. Immunol. 2010, 126, 290–299.e1-4. [Google Scholar] [CrossRef]
- Duchesne, M.; Okoye, I.; Lacy, P. Epithelial cell alarmin cytokines: Frontline mediators of the asthma inflammatory response. Front. Immunol. 2022, 13, 975914. [Google Scholar] [CrossRef]
- Allinne, J.; Scott, G.; Lim, W.K.; Birchard, D.; Erjefält, J.S.; Sandén, C.; Ben, L.H.; Agrawal, A.; Kaur, N.; Kim, J.H.; et al. IL-33 blockade affects mediators of persistence and exacerbation in a model of chronic airway inflammation. J. Allergy Clin. Immunol. 2019, 144, 1624–1637.e10. [Google Scholar] [CrossRef] [PubMed]
- Vannella, K.M.; Ramalingam, T.R.; Borthwick, L.A.; Barron, L.; Hart, K.M.; Thompson, R.W.; Kindrachuk, K.N.; Cheever, A.W.; White, S.; Budelsky, A.L.; et al. Combinatorial targeting of TSLP, IL-25, and IL-33 in type 2 cytokine-driven inflammation and fibrosis. Sci. Transl. Med. 2016, 8, 337ra365. [Google Scholar] [CrossRef]
- Cheung, P.F.; Wong, C.K.; Ip, W.K.; Lam, C.W. IL-25 regulates the expression of adhesion molecules on eosinophils: Mechanism of eosinophilia in allergic inflammation. Allergy 2006, 61, 878–885. [Google Scholar] [CrossRef]
- Tang, W.; Smith, S.G.; Du, W.; Gugilla, A.; Du, J.; Oliveria, J.P.; Howie, K.; Salter, B.M.; Gauvreau, G.M.; O’Byrne, P.M.; et al. Interleukin-25 and eosinophils progenitor cell mobilization in allergic asthma. Clin. Transl. Allergy 2018, 8, 5. [Google Scholar] [CrossRef]
- Tang, W.; Smith, S.G.; Salter, B.; Oliveria, J.P.; Mitchell, P.; Nusca, G.M.; Howie, K.; Gauvreau, G.M.; O’Byrne, P.M.; Sehmi, R. Allergen-Induced Increases in Interleukin-25 and Interleukin-25 Receptor Expression in Mature Eosinophils from Atopic Asthmatics. Int. Arch. Allergy Immunol. 2016, 170, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.K.; Cheung, P.F.; Ip, W.K.; Lam, C.W. Interleukin-25-induced chemokines and interleukin-6 release from eosinophils is mediated by p38 mitogen-activated protein kinase, c-Jun N-terminal kinase, and nuclear factor-kappaB. Am. J. Respir. Cell Mol. Biol. 2005, 33, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Bal, S.M.; Bernink, J.H.; Nagasawa, M.; Groot, J.; Shikhagaie, M.M.; Golebski, K.; Van Drunen, C.M.; Lutter, R.; Jonkers, R.E.; Hombrink, P. IL-1β, IL-4 and IL-12 control the fate of group 2 innate lymphoid cells in human airway inflammation in the lungs. Nat. Immunol. 2016, 17, 636–645. [Google Scholar] [CrossRef] [PubMed]
- Suzukawa, M.; Koketsu, R.; Iikura, M.; Nakae, S.; Matsumoto, K.; Nagase, H.; Saito, H.; Matsushima, K.; Ohta, K.; Yamamoto, K. Interleukin-33 enhances adhesion, CD11b expression and survival in human eosinophils. Lab. Investig. 2008, 88, 1245–1253. [Google Scholar] [CrossRef]
- Bouffi, C.; Rochman, M.; Zust, C.B.; Stucke, E.M.; Kartashov, A.; Fulkerson, P.C.; Barski, A.; Rothenberg, M.E. IL-33 markedly activates murine eosinophils by an NF-κB–dependent mechanism differentially dependent upon an IL-4–driven autoinflammatory loop. J. Immunol. 2013, 191, 4317–4325. [Google Scholar] [CrossRef]
- Mitchell, P.D.; Salter, B.M.; Oliveria, J.P.; El-Gammal, A.; Tworek, D.; Smith, S.G.; Sehmi, R.; Gauvreau, G.M.; PM, O.A.B. IL-33 and Its Receptor ST2 after Inhaled Allergen Challenge in Allergic Asthmatics. Int. Arch. Allergy Immunol. 2018, 176, 133–142. [Google Scholar] [CrossRef]
- Hui, C.C.; Asher, I.; Heroux, D.; Allakhverdi, Z.; Delespesse, G.; Denburg, J.A. Effects of thymic stromal lymphopoietin on cord blood progenitor cell differentiation and hemopoietic cytokine receptors expression. Allergy Asthma Clin. Immunol. Off. J. Can. Soc. Allergy Clin. Immunol. 2011, 7, A24. [Google Scholar]
- Allakhverdi, Z.; Comeau, M.R.; Smith, D.E.; Toy, D.; Endam, L.M.; Desrosiers, M.; Liu, Y.-J.; Howie, K.J.; Denburg, J.A.; Gauvreau, G.M. CD34+ hemopoietic progenitor cells are potent effectors of allergic inflammation. J. Allergy Clin. Immunol. 2009, 123, 472–478.e1. [Google Scholar] [CrossRef]
- Wong, C.K.; Hu, S.; Cheung, P.F.; Lam, C.W. Thymic stromal lymphopoietin induces chemotactic and prosurvival effects in eosinophils: Implications in allergic inflammation. Am. J. Respir. Cell Mol. Biol. 2010, 43, 305–315. [Google Scholar] [CrossRef]
- Wang, H.; Mobini, R.; Fang, Y.; Barrenäs, F.; Zhang, H.; Xiang, Z.; Benson, M. Allergen challenge of peripheral blood mononuclear cells from patients with seasonal allergic rhinitis increases IL-17RB, which regulates basophil apoptosis and degranulation. Clin. Exp. Allergy 2010, 40, 1194–1202. [Google Scholar] [CrossRef]
- Pecaric-Petkovic, T.; Didichenko, S.A.; Kaempfer, S.; Spiegl, N.; Dahinden, C.A. Human basophils and eosinophils are the direct target leukocytes of the novel IL-1 family member IL-33. Blood J. Am. Soc. Hematol. 2009, 113, 1526–1534. [Google Scholar] [CrossRef] [PubMed]
- Salter, B.M.; Oliveria, J.P.; Nusca, G.; Smith, S.G.; Tworek, D.; Mitchell, P.D.; Watson, R.M.; Sehmi, R.; Gauvreau, G.M. IL-25 and IL-33 induce Type 2 inflammation in basophils from subjects with allergic asthma. Respir. Res. 2016, 17, 5. [Google Scholar] [CrossRef] [PubMed]
- Blom, L.; Poulsen, B.C.; Jensen, B.M.; Hansen, A.; Poulsen, L.K. IL-33 induces IL-9 production in human CD4+ T cells and basophils. PLoS ONE 2011, 6, e21695. [Google Scholar] [CrossRef] [PubMed]
- Suzukawa, M.; Iikura, M.; Koketsu, R.; Nagase, H.; Tamura, C.; Komiya, A.; Nakae, S.; Matsushima, K.; Ohta, K.; Yamamoto, K. An IL-1 cytokine member, IL-33, induces human basophil activation via its ST2 receptor. J. Immunol. 2008, 181, 5981–5989. [Google Scholar] [CrossRef]
- Salter, B.M.; Oliveria, J.P.; Nusca, G.; Smith, S.G.; Watson, R.M.; Comeau, M.; Sehmi, R.; Gauvreau, G.M. Thymic stromal lymphopoietin activation of basophils in patients with allergic asthma is IL-3 dependent. J. Allergy Clin. Immunol. 2015, 136, 1636–1644. [Google Scholar] [CrossRef]
- Allakhverdi, Z.; Comeau, M.R.; Jessup, H.K.; Yoon, B.-R.P.; Brewer, A.; Chartier, S.; Paquette, N.; Ziegler, S.F.; Sarfati, M.; Delespesse, G. Thymic stromal lymphopoietin is released by human epithelial cells in response to microbes, trauma, or inflammation and potently activates mast cells. J. Exp. Med. 2007, 204, 253–258. [Google Scholar] [CrossRef]
- Miyata, M.; Hatsushika, K.; Ando, T.; Shimokawa, N.; Ohnuma, Y.; Katoh, R.; Suto, H.; Ogawa, H.; Masuyama, K.; Nakao, A. Mast cell regulation of epithelial TSLP expression plays an important role in the development of allergic rhinitis. Eur. J. Immunol. 2008, 38, 1487–1492. [Google Scholar] [CrossRef]
- Corrigan, C.J.; Wang, W.; Meng, Q.; Fang, C.; Eid, G.; Caballero, M.R.; Lv, Z.; An, Y.; Wang, Y.-H.; Liu, Y.-J. Allergen-induced expression of IL-25 and IL-25 receptor in atopic asthmatic airways and late-phase cutaneous responses. J. Allergy Clin. Immunol. 2011, 128, 116–124. [Google Scholar] [CrossRef]
- Kim, B.S.; Wang, K.; Siracusa, M.C.; Saenz, S.A.; Brestoff, J.R.; Monticelli, L.A.; Noti, M.; Wojno, E.D.T.; Fung, T.C.; Kubo, M. Basophils promote innate lymphoid cell responses in inflamed skin. J. Immunol. 2014, 193, 3717–3725. [Google Scholar] [CrossRef]
- Morita, H.; Arae, K.; Unno, H.; Miyauchi, K.; Toyama, S.; Nambu, A.; Oboki, K.; Ohno, T.; Motomura, K.; Matsuda, A. An interleukin-33-mast cell-interleukin-2 axis suppresses papain-induced allergic inflammation by promoting regulatory T cell numbers. Immunity 2015, 43, 175–186. [Google Scholar] [CrossRef]
- Pajulas, A.; Fu, Y.; Cheung, C.C.; Chu, M.; Cannon, A.; Alakhras, N.; Zhang, J.; Ulrich, B.J.; Nelson, A.S.; Zhou, B. Interleukin-9 promotes mast cell progenitor proliferation and CCR2-dependent mast cell migration in allergic airway inflammation. Mucosal Immunol. 2023, 16, 432–445. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.-Y.; Smrž, D.; Desai, A.; Bandara, G.; Ito, T.; Iwaki, S.; Kang, J.-H.; Andrade, M.V.; Hilderbrand, S.C.; Brown, J.M. IL-33 induces a hyporesponsive phenotype in human and mouse mast cells. J. Immunol. 2013, 190, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Soumelis, V.; Reche, P.A.; Kanzler, H.; Yuan, W.; Edward, G.; Homey, B.; Gilliet, M.; Ho, S.; Antonenko, S.; Lauerma, A. Human epithelial cells trigger dendritic cell–mediated allergic inflammation by producing TSLP. Nat. Immunol. 2002, 3, 673–680. [Google Scholar] [CrossRef] [PubMed]
- Froidure, A.; Shen, C.; Gras, D.; Van Snick, J.; Chanez, P.; Pilette, C. Myeloid dendritic cells are primed in allergic asthma for thymic stromal lymphopoietin-mediated induction of Th2 and Th9 responses. Allergy 2014, 69, 1068–1076. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Wang, Y.-H.; Duramad, O.; Hori, T.; Delespesse, G.J.; Watanabe, N.; Qin, F.; Yao, Z.; Cao, W.; Liu, Y.-J. TSLP-activated dendritic cells induce an inflammatory T helper type 2 cell response through OX40 ligand. J. Exp. Med. 2005, 202, 1213–1223. [Google Scholar] [CrossRef]
- Reche, P.A.; Soumelis, V.; Gorman, D.M.; Clifford, T.; Liu, M.-r.; Travis, M.; Zurawski, S.M.; Johnston, J.; Liu, Y.-J.; Spits, H. Human thymic stromal lymphopoietin preferentially stimulates myeloid cells. J. Immunol. 2001, 167, 336–343. [Google Scholar] [CrossRef]
- Schmitz, J.; Owyang, A.; Oldham, E.; Song, Y.; Murphy, E.; McClanahan, T.K.; Zurawski, G.; Moshrefi, M.; Qin, J.; Li, X. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 2005, 23, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-H.; Angkasekwinai, P.; Lu, N.; Voo, K.S.; Arima, K.; Hanabuchi, S.; Hippe, A.; Corrigan, C.J.; Dong, C.; Homey, B. IL-25 augments type 2 immune responses by enhancing the expansion and functions of TSLP-DC–activated Th2 memory cells. J. Exp. Med. 2007, 204, 1837–1847. [Google Scholar] [CrossRef]
- Yamamoto, T.; Endo, Y.; Onodera, A.; Hirahara, K.; Asou, H.K.; Nakajima, T.; Kanno, T.; Ouchi, Y.; Uematsu, S.; Nishimasu, H. DUSP10 constrains innate IL-33-mediated cytokine production in ST2 hi memory-type pathogenic Th2 cells. Nat. Commun. 2018, 9, 4231. [Google Scholar] [CrossRef]
- Li, Z.-G.; Scott, M.J.; Brzoska, T.; Sundd, P.; Li, Y.-H.; Billiar, T.R.; Wilson, M.A.; Wang, P.; Fan, J. Lung epithelial cell-derived IL-25 negatively regulates LPS-induced exosome release from macrophages. Mil. Med. Res. 2018, 5, 24. [Google Scholar] [CrossRef]
- Han, H.; Headley, M.B.; Xu, W.; Comeau, M.R.; Zhou, B.; Ziegler, S.F. Thymic stromal lymphopoietin amplifies the differentiation of alternatively activated macrophages. J. Immunol. 2013, 190, 904–912. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.D.; Oak, S.R.; Hartigan, A.J.; Finn, W.G.; Kunkel, S.L.; Duffy, K.E.; Das, A.; Hogaboam, C.M. Interleukin-33 contributes to both M1 and M2 chemokine marker expression in human macrophages. BMC Immunol. 2010, 11, 52. [Google Scholar] [CrossRef] [PubMed]
- Kurowska-Stolarska, M.; Stolarski, B.; Kewin, P.; Murphy, G.; Corrigan, C.J.; Ying, S.; Pitman, N.; Mirchandani, A.; Rana, B.; van Rooijen, N. IL-33 amplifies the polarization of alternatively activated macrophages that contribute to airway inflammation. J. Immunol. 2009, 183, 6469–6477. [Google Scholar] [CrossRef] [PubMed]
- Rochman, I.; Watanabe, N.; Arima, K.; Liu, Y.-J.; Leonard, W.J. Cutting edge: Direct action of thymic stromal lymphopoietin on activated human CD4+ T cells. J. Immunol. 2007, 178, 6720–6724. [Google Scholar] [CrossRef] [PubMed]
- Omori, M.; Ziegler, S. Induction of IL-4 expression in CD4+ T cells by thymic stromal lymphopoietin. J. Immunol. 2007, 178, 1396–1404. [Google Scholar] [CrossRef]
- Kitajima, M.; Lee, H.C.; Nakayama, T.; Ziegler, S.F. TSLP enhances the function of helper type 2 cells. Eur. J. Immunol. 2011, 41, 1862–1871. [Google Scholar] [CrossRef]
- Li, H.; Zhao, H.; Yu, J.; Su, Y.; Cao, S.; An, X.; Ren, X. Increased prevalence of regulatory T cells in the lung cancer microenvironment: A role of thymic stromal lymphopoietin. Cancer Immunol. Immunother. 2011, 60, 1587–1596. [Google Scholar] [CrossRef]
- Watanabe, N.; Wang, Y.-H.; Lee, H.K.; Ito, T.; Wang, Y.-H.; Cao, W.; Liu, Y.-J. Hassall’s corpuscles instruct dendritic cells to induce CD4+ CD25+ regulatory T cells in human thymus. Nature 2005, 436, 1181–1185. [Google Scholar] [CrossRef]
- Akamatsu, T.; Watanabe, N.; Kido, M.; Saga, K.; Tanaka, J.; Kuzushima, K.; Nishio, A.; Chiba, T. Human TSLP directly enhances expansion of CD8+ T cells. Clin. Exp. Immunol. 2008, 154, 98–106. [Google Scholar] [CrossRef]
- Friend, S.L.; Hosier, S.; Nelson, A.; Foxworthe, D.; Williams, D.; Farr, A. A thymic stromal cell line supports in vitro development of surface IgM+ B cells and produces a novel growth factor affecting B and T lineage cells. Exp. Hematol. 1994, 22, 321–328. [Google Scholar]
- Milford, T.A.M.; Su, R.J.; Francis, O.L.; Baez, I.; Martinez, S.R.; Coats, J.S.; Weldon, A.J.; Calderon, M.N.; Nwosu, M.C.; Botimer, A.R. TSLP or IL-7 provide an IL-7Rα signal that is critical for human B lymphopoiesis. Eur. J. Immunol. 2016, 46, 2155–2161. [Google Scholar] [CrossRef] [PubMed]
- Moser, R.; Fehr, J.; Bruijnzeel, P. IL-4 controls the selective endothelium-driven transmigration of eosinophils from allergic individuals. J. Immunol. 1992, 149, 1432–1438. [Google Scholar] [CrossRef] [PubMed]
- Vu, A.T.; Baba, T.; Chen, X.; Le, T.A.; Kinoshita, H.; Xie, Y.; Kamijo, S.; Hiramatsu, K.; Ikeda, S.; Ogawa, H. Staphylococcus aureus membrane and diacylated lipopeptide induce thymic stromal lymphopoietin in keratinocytes through the Toll-like receptor 2–Toll-like receptor 6 pathway. J. Allergy Clin. Immunol. 2010, 126, 985–993.e3. [Google Scholar] [CrossRef]
- Pawankar, R.; Okuda, M.; Yssel, H.; Okumura, K.; Ra, C. Nasal mast cells in perennial allergic rhinitics exhibit increased expression of the Fc epsilonRI, CD40L, IL-4, and IL-13, and can induce IgE synthesis in B cells. J. Clin. Investig. 1997, 99, 1492–1499. [Google Scholar] [CrossRef]
- Hoontrakoon, R.; Kailey, J.; Bratton, D. IL-4 and TNF alpha synergize to enhance eosinophil survival. J. Allergy Clin. Immunol. 1999, 103, S239. [Google Scholar]
- Dabbagh, K.; Takeyama, K.; Lee, H.-M.; Ueki, I.F.; Lausier, J.A.; Nadel, J.A. IL-4 induces mucin gene expression and goblet cell metaplasia in vitro and in vivo. J. Immunol. 1999, 162, 6233–6237. [Google Scholar] [CrossRef] [PubMed]
- Doucet, C.; Brouty-Boyé, D.; Pottin-Clemenceau, C.; Jasmin, C.; Canonica, G.W.; Azzarone, B. IL-4 and IL-13 specifically increase adhesion molecule and inflammatory cytokine expression in human lung fibroblasts. Int. Immunol. 1998, 10, 1421–1433. [Google Scholar] [CrossRef]
- Oettgen, H.C.; Geha, R.S. IgE regulation and roles in asthma pathogenesis. J. Allergy Clin. Immunol. 2001, 107, 429–441. [Google Scholar] [CrossRef]
- Ortega, H.G.; Liu, M.C.; Pavord, I.D.; Brusselle, G.G.; FitzGerald, J.M.; Chetta, A.; Humbert, M.; Katz, L.E.; Keene, O.N.; Yancey, S.W. Mepolizumab treatment in patients with severe eosinophilic asthma. N. Engl. J. Med. 2014, 371, 1198–1207. [Google Scholar] [CrossRef]
- Panettieri, R.A.; Sjöbring, U.; Péterffy, A.; Wessman, P.; Bowen, K.; Piper, E.; Colice, G.; Brightling, C.E. Tralokinumab for severe, uncontrolled asthma (STRATOS 1 and STRATOS 2): Two randomised, double-blind, placebo-controlled, phase 3 clinical trials. Lancet Respir. Med. 2018, 6, 511–525. [Google Scholar] [CrossRef]
- Luttmann, W.; Knoechel, B.; Foerster, M.; Matthys, H.; Virchow, J.C., Jr.; Kroegel, C. Activation of human eosinophils by IL-13. Induction of CD69 surface antigen, its relationship to messenger RNA expression, and promotion of cellular viability. J. Immunol. 1996, 157, 1678–1683. [Google Scholar] [CrossRef] [PubMed]
- Pope, S.M.; Brandt, E.B.; Mishra, A.; Hogan, S.P.; Zimmermann, N.; Matthaei, K.I.; Foster, P.S.; Rothenberg, M.E. IL-13 induces eosinophil recruitment into the lung by an IL-5–and eotaxin-dependent mechanism. J. Allergy Clin. Immunol. 2001, 108, 594–601. [Google Scholar] [CrossRef]
- Bochner, B.S.; Klunk, D.A.; Sterbinsky, S.A.; Coffman, R.L.; Schleimer, R.P. IL-13 selectively induces vascular cell adhesion molecule-1 expression in human endothelial cells. J. Immunol. 1995, 154, 799–803. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Xia, Y.; Nguyen, A.; Lai, Y.H.; Feng, L.; Mosmann, T.R.; Lo, D. Effects of Th2 cytokines on chemokine expression in the lung: IL-13 potently induces eotaxin expression by airway epithelial cells. J. Immunol. 1999, 162, 2477–2487. [Google Scholar] [CrossRef]
- LAPORTE, J.C.; Moore, P.E.; Baraldo, S.; Jouvin, M.-H.; Church, T.L.; Schwartzman, I.N.; Panettieri, R.A., Jr.; Kinet, J.-P.; Shore, S.A. Direct effects of interleukin-13 on signaling pathways for physiological responses in cultured human airway smooth muscle cells. Am. J. Respir. Crit. Care Med. 2001, 164, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Wills-Karp, M. IL-12/IL-13 axis in allergic asthma. J. Allergy Clin. Immunol. 2001, 107, 9–18. [Google Scholar] [CrossRef]
- Kuperman, D.A.; Huang, X.; Koth, L.L.; Chang, G.H.; Dolganov, G.M.; Zhu, Z.; Elias, J.A.; Sheppard, D.; Erle, D.J. Direct effects of interleukin-13 on epithelial cells cause airway hyperreactivity and mucus overproduction in asthma. Nat. Med. 2002, 8, 885–889. [Google Scholar] [CrossRef]
- Kondo, M.; Tamaoki, J.; Takeyama, K.; Isono, K.; Kawatani, K.; Izumo, T.; Nagai, A. Elimination of IL-13 reverses established goblet cell metaplasia into ciliated epithelia in airway epithelial cell culture. Allergol. Int. 2006, 55, 329–336. [Google Scholar] [CrossRef]
- Kanoh, S.; Tanabe, T.; Rubin, B.K. IL-13-induced MUC5AC production and goblet cell differentiation is steroid resistant in human airway cells. Clin. Exp. Allergy 2011, 41, 1747–1756. [Google Scholar] [CrossRef]
- Venegas Garrido, C.; Mukherjee, M.; Svenningsen, S.; Nair, P. Eosinophil-mucus interplay in severe asthma: Implications for treatment with biologicals. Allergol. Int. 2024, 73, 351–361. [Google Scholar] [CrossRef]
- Stirling, R.G.; Van Rensen, E.L.; Barnes, P.J.; Fan Chung, K. Interleukin-5 induces CD34+ eosinophil progenitor mobilization and eosinophil CCR3 expression in asthma. Am. J. Respir. Crit. Care Med. 2001, 164, 1403–1409. [Google Scholar] [CrossRef] [PubMed]
- Dorman, S.C.; Efthimiadis, A.; Babirad, I.; Watson, R.M.; Denburg, J.A.; Hargreave, F.E.; O’Byrne, P.M.; Sehmi, R. Sputum CD34+ IL-5Rα+ cells increase after allergen: Evidence for in situ eosinophilopoiesis. Am. J. Respir. Crit. Care Med. 2004, 169, 573–577. [Google Scholar] [CrossRef] [PubMed]
- Wood, L.J.; Sehmi, R.; Dorman, S.; Hamid, Q.; Tulic, M.K.; Watson, R.M.; Foley, R.; Wasi, P.; Denburg, J.A.; Gauvreau, G. Allergen-induced increases in bone marrow T lymphocytes and interleukin-5 expression in subjects with asthma. Am. J. Respir. Crit. Care Med. 2002, 166, 883–889. [Google Scholar] [CrossRef]
- Park, S.W.; Kim, D.J.; Chang, H.S.; Park, S.J.; Lee, Y.M.; Park, J.S.; Chung, I.Y.; Lee, J.H.; Park, C.-S. Association of interleukin-5 and eotaxin with acute exacerbation of asthma. Int. Arch. Allergy Immunol. 2003, 131, 283–290. [Google Scholar] [CrossRef]
- Kolbeck, R.; Kozhich, A.; Koike, M.; Peng, L.; Andersson, C.K.; Damschroder, M.M.; Reed, J.L.; Woods, R.; Dall’acqua, W.W.; Stephens, G.L.; et al. MEDI-563, a humanized anti-IL-5 receptor alpha mAb with enhanced antibody-dependent cell-mediated cytotoxicity function. J. Allergy Clin. Immunol. 2010, 125, 1344–1353.e2. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.G.; Teague, W.G.; Feng, X.; Welch, C.; Etter, E.; Negri, J.; Spano, M.; Wavell, K.; Braciale, T.; Steinke, J.W.; et al. Interleukin-5 receptor alpha (CD125) expression on human blood and lung neutrophils. Ann. Allergy Asthma Immunol. 2022, 128, 53–60.e3. [Google Scholar] [CrossRef]
- Whetstone, C.E.; Cusack, R.P.; Price, E.; Howie, K.; Stevens, C.; Al-Sajee, D.; Beaudin, S.; Wattie, J.; Alsaji, N.; Schlatman, A.; et al. Effect of benralizumab on inflammation in skin after intradermal allergen challenge in patients with moderate-to-severe atopic dermatitis. J. Allergy Clin. Immunol. Glob. 2024, 3, 100310. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, R. HMGB1 is a promising therapeutic target for asthma. Cytokine 2023, 165, 156171. [Google Scholar] [CrossRef] [PubMed]
- Hou, C.; Zhao, H.; Liu, L.; Li, W.; Zhou, X.; Lv, Y.; Shen, X.; Liang, Z.; Cai, S.; Zou, F. High mobility group protein B1 (HMGB1) in Asthma: Comparison of patients with chronic obstructive pulmonary disease and healthy controls. Mol. Med. 2011, 17, 807–815. [Google Scholar] [CrossRef]
- Watanabe, T.; Asai, K.; Fujimoto, H.; Tanaka, H.; Kanazawa, H.; Hirata, K. Increased levels of HMGB-1 and endogenous secretory RAGE in induced sputum from asthmatic patients. Respir. Med. 2011, 105, 519–525. [Google Scholar] [CrossRef]
- Shim, E.J.; Chun, E.; Lee, H.S.; Bang, B.R.; Kim, T.W.; Cho, S.H.; Min, K.U.; Park, H.W. The role of high-mobility group box-1 (HMGB1) in the pathogenesis of asthma. Clin. Exp. Allergy 2012, 42, 958–965. [Google Scholar] [CrossRef]
- Hou, C.; Kong, J.; Liang, Y.; Huang, H.; Wen, H.; Zheng, X.; Wu, L.; Chen, Y. HMGB1 contributes to allergen-induced airway remodeling in a murine model of chronic asthma by modulating airway inflammation and activating lung fibroblasts. Cell Mol. Immunol. 2015, 12, 409–423. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Wang, J.; Zhu, F.; Li, R.; Liu, B.; Xu, W.; He, G.; Cao, H.; Wang, Y.; Yang, J. HMGB1 regulates T helper 2 and T helper17 cell differentiation both directly and indirectly in asthmatic mice. Mol. Immunol. 2018, 97, 45–55. [Google Scholar] [CrossRef]
- Matarazzo, L.; Hernandez Santana, Y.E.; Walsh, P.T.; Fallon, P.G. The IL-1 cytokine family as custodians of barrier immunity. Cytokine 2022, 154, 155890. [Google Scholar] [CrossRef] [PubMed]
- Godwin, M.S.; Reeder, K.M.; Garth, J.M.; Blackburn, J.P.; Jones, M.; Yu, Z.; Matalon, S.; Hastie, A.T.; Meyers, D.A.; Steele, C. IL-1RA regulates immunopathogenesis during fungal-associated allergic airway inflammation. JCI Insight 2019, 4, e129055. [Google Scholar] [CrossRef] [PubMed]
- Osei, E.T.; Mostaço-Guidolin, L.B.; Hsieh, A.; Warner, S.M.; Al-Fouadi, M.; Wang, M.; Cole, D.J.; Maksym, G.N.; Hallstrand, T.S.; Timens, W.; et al. Epithelial-interleukin-1 inhibits collagen formation by airway fibroblasts: Implications for asthma. Sci. Rep. 2020, 10, 8721. [Google Scholar] [CrossRef] [PubMed]
- Willart, M.A.; Deswarte, K.; Pouliot, P.; Braun, H.; Beyaert, R.; Lambrecht, B.N.; Hammad, H. Interleukin-1α controls allergic sensitization to inhaled house dust mite via the epithelial release of GM-CSF and IL-33. J. Exp. Med. 2012, 209, 1505–1517. [Google Scholar] [CrossRef]
- Gu, J.; Qin, G.; Jiang, L.; Xu, W.; Wang, Y.; Liao, J.; Pan, H.; Liang, Z. Correlations between IL-36 family cytokines in peripheral blood and subjective and objective assessment results in patients with allergic rhinitis. Allergy Asthma Clin. Immunol. 2023, 19, 79. [Google Scholar] [CrossRef]
- Li, J.; Wang, Z.; Dong, H.; Hao, Y.; Gao, P.; Li, W. Different expression levels of interleukin-36 in asthma phenotypes. Allergy Asthma Clin. Immunol. 2024, 20, 3. [Google Scholar] [CrossRef]
- Qin, X.; Liu, M.; Zhang, S.; Wang, C.; Zhang, T. The role of IL-36γ and its regulation in eosinophilic inflammation in allergic rhinitis. Cytokine 2019, 117, 84–90. [Google Scholar] [CrossRef]
- Murphy, R.; Gharib, S.; Sehmi, R.; Gauvreau, G.; Hallstrand, T. Kinetics of Airway Gene Expression in Atopic Asthmatics Following Inhaled Allergen Challenge. In B101. Varied Omics Techniques Applied to Allergic and Respiratory Traits; American Thoracic Society: New York, NY, USA, 2022; p. A3477. [Google Scholar]
- Pires, S.; Longman, R.S. Sounding the alarm in the lung with TL1A. J. Exp. Med. 2024, 221, e20240389. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, P.; Duval, A.; Camus, M.; Lefrançais, E.; Roga, S.; Dedieu, C.; Ortega, N.; Bellard, E.; Mirey, E.; Mouton-Barbosa, E.; et al. TL1A is an epithelial alarmin that cooperates with IL-33 for initiation of allergic airway inflammation. J. Exp. Med. 2024, 221, e20231236. [Google Scholar] [CrossRef] [PubMed]
- Machida, K.; Aw, M.; Salter, B.M.A.; Ju, X.; Mukherjee, M.; Gauvreau, G.M.; O’Byrne, P.M.; Nair, P.; Sehmi, R. The Role of the TL1A/DR3 Axis in the Activation of Group 2 Innate Lymphoid Cells in Subjects with Eosinophilic Asthma. Am. J. Respir. Crit. Care Med. 2020, 202, 1105–1114. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Yang, H.; Dong, X.L.; Zhang, J.T.; Liu, X.F.; Pan, Y.; Zhang, J.; Xu, J.W.; Wang, Z.H.; Cui, W.J.; et al. TL1A/DR3 Axis, A Key Target of TNF-a, Augments the Epithelial-Mesenchymal Transformation of Epithelial Cells in OVA-Induced Asthma. Front. Immunol. 2022, 13, 854995. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, D.; Pan, Y.; Liu, X.; Xu, J.; Qiao, X.; Cui, W.; Dong, L. The TL1A-DR3 Axis in Asthma: Membrane-Bound and Secreted TL1A Co-Determined the Development of Airway Remodeling. Allergy Asthma Immunol. Res. 2022, 14, 233–253. [Google Scholar] [CrossRef]
- Lambrecht, B.N.; Hammad, H. Allergens and the airway epithelium response: Gateway to allergic sensitization. J. Allergy Clin. Immunol. 2014, 134, 499–507. [Google Scholar] [CrossRef]
- Fuchs, B.; Knothe, S.; Rochlitzer, S.; Nassimi, M.; Greweling, M.; Lauenstein, H.D.; Nassenstein, C.; Müller, M.; Ebensen, T.; Dittrich, A.M.; et al. A Toll-like receptor 2/6 agonist reduces allergic airway inflammation in chronic respiratory sensitisation to Timothy grass pollen antigens. Int. Arch. Allergy Immunol. 2010, 152, 131–139. [Google Scholar] [CrossRef]
- Mueller, G.A.; Pedersen, L.C.; Lih, F.B.; Glesner, J.; Moon, A.F.; Chapman, M.D.; Tomer, K.B.; London, R.E.; Pomés, A. The novel structure of the cockroach allergen Bla g 1 has implications for allergenicity and exposure assessment. J. Allergy Clin. Immunol. 2013, 132, 1420–1426. [Google Scholar] [CrossRef]
- Ait Yahia, S.; Audousset, C.; Alvarez-Simon, D.; Vorng, H.; Togbe, D.; Marquillies, P.; Delacre, M.; Rose, S.; Bouscayrol, H.; Rifflet, A.; et al. NOD1 sensing of house dust mite-derived microbiota promotes allergic experimental asthma. J. Allergy Clin. Immunol. 2021, 148, 394–406. [Google Scholar] [CrossRef]
- Bergougnan, C.; Dittlein, D.C.; Hümmer, E.; Riepl, R.; Eisenbart, S.; Böck, D.; Griesbaum, L.; Weigl, A.; Damialis, A.; Hartwig, A.; et al. Physical and immunological barrier of human primary nasal epithelial cells from non-allergic and allergic donors. World Allergy Organ. J. 2020, 13, 100109. [Google Scholar] [CrossRef]
- Do, D.C.; Zhao, Y.; Gao, P. Cockroach allergen exposure and risk of asthma. Allergy 2016, 71, 463–474. [Google Scholar] [CrossRef] [PubMed]
- Jacquet, A.; Robinson, C. Proteolytic, lipidergic and polysaccharide molecular recognition shape innate responses to house dust mite allergens. Allergy 2020, 75, 33–53. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Zou, Z.; Meng, F.; Raz, E.; Huang, Y.; Tao, A.; Ai, Y. Dust mite-derived Der f 3 activates a pro-inflammatory program in airway epithelial cells via PAR-1 and PAR-2. Mol. Immunol. 2019, 109, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Finkelman, M.A.; Lempitski, S.J.; Slater, J.E. beta-Glucans in standardized allergen extracts. J. Endotoxin Res. 2006, 12, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Nathan, A.T.; Peterson, E.A.; Chakir, J.; Wills-Karp, M. Innate immune responses of airway epithelium to house dust mite are mediated through beta-glucan-dependent pathways. J. Allergy Clin. Immunol. 2009, 123, 612–618. [Google Scholar] [CrossRef]
- Kouzaki, H.; Iijima, K.; Kobayashi, T.; O’Grady, S.M.; Kita, H. The danger signal, extracellular ATP, is a sensor for an airborne allergen and triggers IL-33 release and innate Th2-type responses. J. Immunol. 2011, 186, 4375–4387. [Google Scholar] [CrossRef]
- Schiffers, C.; Hristova, M.; Habibovic, A.; Dustin, C.M.; Danyal, K.; Reynaert, N.L.; Wouters, E.F.M.; van der Vliet, A. The Transient Receptor Potential Channel Vanilloid 1 Is Critical in Innate Airway Epithelial Responses to Protease Allergens. Am. J. Respir. Cell Mol. Biol. 2020, 63, 198–208. [Google Scholar] [CrossRef]
- Ouyang, X.; Reihill, J.A.; Douglas, L.E.J.; Dunne, O.M.; Sergeant, G.P.; Martin, S.L. House dust mite allergens induce Ca2+ signalling and alarmin responses in asthma airway epithelial cells. Biochim. Biophys. Acta Mol. Basis Dis. 2024, 1870, 167079. [Google Scholar] [CrossRef]
- Kouzaki, H.; O’Grady, S.M.; Lawrence, C.B.; Kita, H. Proteases induce production of thymic stromal lymphopoietin by airway epithelial cells through protease-activated receptor-2. J. Immunol. 2009, 183, 1427–1434. [Google Scholar] [CrossRef]
- Kouzaki, H.; Tojima, I.; Kita, H.; Shimizu, T. Transcription of interleukin-25 and extracellular release of the protein is regulated by allergen proteases in airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 2013, 49, 741–750. [Google Scholar] [CrossRef]
- Snelgrove, R.J.; Gregory, L.G.; Peiró, T.; Akthar, S.; Campbell, G.A.; Walker, S.A.; Lloyd, C.M. Alternaria-derived serine protease activity drives IL-33-mediated asthma exacerbations. J. Allergy Clin. Immunol. 2014, 134, 583–592.e6. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Kouzaki, H.; Matsumoto, K.; Hosoi, J.; Shimizu, T. The effect of calprotectin on TSLP and IL-25 production from airway epithelial cells. Allergol. Int. 2017, 66, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Nakagome, K.; Nagata, M. Innate Immune Responses by Respiratory Viruses, Including Rhinovirus, During Asthma Exacerbation. Front. Immunol. 2022, 13, 865973. [Google Scholar] [CrossRef] [PubMed]
- Eisele, N.A.; Anderson, D.M. Host Defense and the Airway Epithelium: Frontline Responses That Protect against Bacterial Invasion and Pneumonia. J. Pathog. 2011, 2011, 249802. [Google Scholar] [CrossRef]
- Brown, M.A.; Morgan, S.B.; Donachie, G.E.; Horton, K.L.; Pavord, I.D.; Arancibia-Cárcamo, C.V.; Hinks, T.S.C. Epithelial immune activation and intracellular invasion by non-typeable Haemophilus influenzae. Front. Cell Infect. Microbiol. 2023, 13, 1141798. [Google Scholar] [CrossRef]
- Mahmutovic Persson, I.; Akbarshahi, H.; Menzel, M.; Brandelius, A.; Uller, L. Increased expression of upstream TH2-cytokines in a mouse model of viral-induced asthma exacerbation. J. Transl. Med. 2016, 14, 52. [Google Scholar] [CrossRef]
- Ravanetti, L.; Dijkhuis, A.; Dekker, T.; Sabogal Pineros, Y.S.; Ravi, A.; Dierdorp, B.S.; Erjefält, J.S.; Mori, M.; Pavlidis, S.; Adcock, I.M.; et al. IL-33 drives influenza-induced asthma exacerbations by halting innate and adaptive antiviral immunity. J. Allergy Clin. Immunol. 2019, 143, 1355–1370.e16. [Google Scholar] [CrossRef]
- Norlander, A.E.; Peebles, R.S., Jr. Innate Type 2 Responses to Respiratory Syncytial Virus Infection. Viruses 2020, 12, 521. [Google Scholar] [CrossRef]
- Chen, S.; Yu, G.; Xie, J.; Tang, W.; Gao, L.; Long, X.; Ren, L.; Xie, X.; Deng, Y.; Fu, Z.; et al. High-mobility group box-1 protein from CC10+ club cells promotes type 2 response in the later stage of respiratory syncytial virus infection. Am. J. Physiol. Lung Cell Mol. Physiol. 2019, 316, L280–L290. [Google Scholar] [CrossRef]
- Lan, F.; Zhang, N.; Holtappels, G.; De Ruyck, N.; Krysko, O.; Van Crombruggen, K.; Braun, H.; Johnston, S.L.; Papadopoulos, N.G.; Zhang, L.; et al. Staphylococcus aureus Induces a Mucosal Type 2 Immune Response via Epithelial Cell-derived Cytokines. Am. J. Respir. Crit. Care Med. 2018, 198, 452–463. [Google Scholar] [CrossRef]
- Li, J.; Kanju, P.; Patterson, M.; Chew, W.L.; Cho, S.H.; Gilmour, I.; Oliver, T.; Yasuda, R.; Ghio, A.; Simon, S.A.; et al. TRPV4-mediated calcium influx into human bronchial epithelia upon exposure to diesel exhaust particles. Environ. Health Perspect. 2011, 119, 784–793. [Google Scholar] [CrossRef] [PubMed]
- Park, E.; Kim, B.Y.; Lee, S.; Son, K.H.; Bang, J.; Hong, S.H.; Lee, J.W.; Uhm, K.O.; Kwak, H.J.; Lim, H.J. Diesel exhaust particle exposure exacerbates ciliary and epithelial barrier dysfunction in the multiciliated bronchial epithelium models. Ecotoxicol. Environ. Saf. 2024, 273, 116090. [Google Scholar] [CrossRef]
- Becker, S.; Dailey, L.; Soukup, J.M.; Silbajoris, R.; Devlin, R.B. TLR-2 is involved in airway epithelial cell response to air pollution particles. Toxicol. Appl. Pharmacol. 2005, 203, 45–52. [Google Scholar] [CrossRef]
- Chen, X.; Deng, T.; Huo, T.; Dong, F.; Deng, J. MiR-140-5p/TLR4/NF-κB signaling pathway: Crucial role in inflammatory response in 16HBE cells induced by dust fall PM2.5. Ecotoxicol. Environ. Saf. 2021, 208, 111414. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Zhang, Y.; Wang, M.; Zhang, H.; Chen, Y.; Adcock, I.M.; Chung, K.F.; Mo, J.; Zhang, Y.; Li, F. TRPV1 and TRPA1 in Lung Inflammation and Airway Hyperresponsiveness Induced by Fine Particulate Matter (PM2.5). Oxidative Med. Cell. Longev. 2019, 2019, 7450151. [Google Scholar] [CrossRef] [PubMed]
- Bao, Z.J.; Fan, Y.M.; Cui, Y.F.; Sheng, Y.F.; Zhu, M. Effect of PM2.5 mediated oxidative stress on the innate immune cellular response of Der p1 treated human bronchial epithelial cells. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 2907–2912. [Google Scholar]
- Watterson, T.L.; Hamilton, B.; Martin, R.S.; Coulombe, R.A., Jr. Urban particulate matter activates Akt in human lung cells. Arch. Toxicol. 2012, 86, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Ye, D.; Liu, S.; Hu, J.; Zhu, T.; He, F.; Ran, P. GSK-3β Inhibitors Attenuate the PM2.5-Induced Inflammatory Response in Bronchial Epithelial Cells. Int. J. Chron. Obs. Pulmon Dis. 2021, 16, 2845–2856. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Sunusi, S.; Lu, H. Group 2 innate lymphoid cells (ILC2s) are important in typical type 2 immune-mediated diseases and an essential therapeutic target. J. Int. Med. Res. 2022, 50, 3000605211053156. [Google Scholar] [CrossRef]
- Abu Khweek, A.; Kim, E.; Joldrichsen, M.R.; Amer, A.O.; Boyaka, P.N. Insights Into Mucosal Innate Immune Responses in House Dust Mite-Mediated Allergic Asthma. Front. Immunol. 2020, 11, 534501. [Google Scholar] [CrossRef]
- Figueiredo, I.A.D.; Ferreira, S.R.D.; Fernandes, J.M.; Silva, B.A.D.; Vasconcelos, L.H.C.; Cavalcante, F.A. A review of the pathophysiology and the role of ion channels on bronchial asthma. Front. Pharmacol. 2023, 14, 1236550. [Google Scholar] [CrossRef] [PubMed]
- Rossi, G.A.; Ballarini, S.; Salvati, P.; Sacco, O.; Colin, A.A. Alarmins and innate lymphoid cells 2 activation: A common pathogenetic link connecting respiratory syncytial virus bronchiolitis and later wheezing/asthma? Pediatr. Allergy Immunol. 2022, 33, e13803. [Google Scholar] [CrossRef]
- Pelaia, G.; Vatrella, A.; Gallelli, L.; Renda, T.; Cazzola, M.; Maselli, R.; Marsico, S.A. Respiratory infections and asthma. Respir. Med. 2006, 100, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Schaunaman, N.; Sanchez, A.; Dimasuay, K.G.; Pavelka, N.; Numata, M.; Alam, R.; Martin, R.J.; Chu, H.W. Interleukin 1 Receptor-Like 1 (IL1RL1) Promotes Airway Bacterial and Viral Infection and Inflammation. Infect. Immun. 2019, 87, e00340-19. [Google Scholar] [CrossRef]
- Cattani-Cavalieri, I.; Trombetta-Lima, M.; Yan, H.; Manzano-Covarrubias, A.L.; Baarsma, H.A.; Oun, A.; van der Veen, M.M.; Oosterhout, E.; Dolga, A.M.; Ostrom, R.S.; et al. Diesel exhaust particles alter mitochondrial bioenergetics and cAMP producing capacity in human bronchial epithelial cells. Front. Toxicol. 2024, 6, 1412864. [Google Scholar] [CrossRef]
- Auger, F.; Gendron, M.C.; Chamot, C.; Marano, F.; Dazy, A.C. Responses of well-differentiated nasal epithelial cells exposed to particles: Role of the epithelium in airway inflammation. Toxicol. Appl. Pharmacol. 2006, 215, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Schwarze, P.E.; Totlandsdal, A.I.; Låg, M.; Refsnes, M.; Holme, J.A.; Øvrevik, J. Inflammation-related effects of diesel engine exhaust particles: Studies on lung cells in vitro. BioMed Res. Int. 2013, 2013, 685142. [Google Scholar] [CrossRef]
- Busse, W.W.; Holgate, S.; Kerwin, E.; Chon, Y.; Feng, J.; Lin, J.; Lin, S.L. Randomized, double-blind, placebo-controlled study of brodalumab, a human anti-IL-17 receptor monoclonal antibody, in moderate to severe asthma. Am. J. Respir. Crit. Care Med. 2013, 188, 1294–1302. [Google Scholar] [CrossRef]
- Gauvreau Gail, M.; O’Byrne Paul, M.; Boulet, L.-P.; Wang, Y.; Cockcroft, D.; Bigler, J.; FitzGerald, J.M.; Boedigheimer, M.; Davis Beth, E.; Dias, C.; et al. Effects of an Anti-TSLP Antibody on Allergen-Induced Asthmatic Responses. N. Engl. J. Med. 2014, 370, 2102–2110. [Google Scholar] [CrossRef]
- Corren, J.; Parnes, J.R.; Wang, L.; Mo, M.; Roseti, S.L.; Griffiths, J.M.; Merwe, R.v.d. Tezepelumab in Adults with Uncontrolled Asthma. N. Engl. J. Med. 2017, 377, 936–946. [Google Scholar] [CrossRef]
- Sverrild, A.; Hansen, S.; Hvidtfeldt, M.; Clausson, C.-M.; Cozzolino, O.; Cerps, S.; Uller, L.; Backer, V.; Erjefält, J.; Porsbjerg, C. The effect of tezepelumab on airway hyperresponsiveness to mannitol in asthma (UPSTREAM). Eur. Respir. J. 2022, 59, 2101296. [Google Scholar] [CrossRef] [PubMed]
- Emson, C.; Diver, S.; Chachi, L.; Megally, A.; Small, C.; Downie, J.; Parnes, J.R.; Bowen, K.; Colice, G.; Brightling, C.E. CASCADE: A phase 2, randomized, double-blind, placebo-controlled, parallel-group trial to evaluate the effect of tezepelumab on airway inflammation in patients with uncontrolled asthma. Respir. Res. 2020, 21, 265. [Google Scholar] [CrossRef] [PubMed]
- Menzies-Gow, A.; Corren, J.; Bourdin, A.; Chupp, G.; Israel, E.; Wechsler, M.E.; Brightling, C.E.; Griffiths, J.M.; Hellqvist, Å.; Bowen, K.; et al. Tezepelumab in Adults and Adolescents with Severe, Uncontrolled Asthma. N. Engl. J. Med. 2021, 384, 1800–1809. [Google Scholar] [CrossRef]
- Shinkai, M.; Ebisawa, M.; Fukushima, Y.; Takeuchi, S.; Okada, H.; Tokiyo, T.; Hayashi, N.; Takikawa, M.; Colice, G.; Almqvist, G. One-year safety and tolerability of tezepelumab in Japanese patients with severe uncontrolled asthma: Results of the NOZOMI study. J. Asthma 2023, 60, 616–624. [Google Scholar] [CrossRef] [PubMed]
- Wechsler, M.E.; Colice, G.; Griffiths, J.M.; Almqvist, G.; Skärby, T.; Piechowiak, T.; Kaur, P.; Bowen, K.; Hellqvist, Å.; Mo, M.; et al. SOURCE: A phase 3, multicentre, randomized, double-blind, placebo-controlled, parallel group trial to evaluate the efficacy and safety of tezepelumab in reducing oral corticosteroid use in adults with oral corticosteroid dependent asthma. Respir. Res. 2020, 21, 264. [Google Scholar] [CrossRef] [PubMed]
- Menzies-Gow, A.; Ponnarambil, S.; Downie, J.; Bowen, K.; Hellqvist, Å.; Colice, G. DESTINATION: A phase 3, multicentre, randomized, double-blind, placebo-controlled, parallel-group trial to evaluate the long-term safety and tolerability of tezepelumab in adults and adolescents with severe, uncontrolled asthma. Respir. Res. 2020, 21, 279. [Google Scholar] [CrossRef]
- A Phase 2 Multi-Centre Randomized Double-Blind Placebo-Controlled Parallel Group Study to Examine the Effects of 24 Weeks Tezepelumab 210 mg sc q4wks on Methacholine Airway Hyperresponsiveness in Participants with Mild Allergic Asthma. Available online: https://clinicaltrials.gov/study/NCT05740748 (accessed on 16 September 2024).
- Tezepelumab in the Treatment of Co-Morbid Allergic Rhinitis and Allergic Asthma Study (TEZARS)—An Open-Label Exploratory Mechanistic Pilot Study to Evaluate Tezepelumab Efficacy in Asthma and Allergic Rhinitis. Available online: https://clinicaltrials.gov/study/NCT06189742 (accessed on 16 September 2024).
- Gauvreau, G.M.; Hohlfeld, J.M.; FitzGerald, J.M.; Boulet, L.P.; Cockcroft, D.W.; Davis, B.E.; Korn, S.; Kornmann, O.; Leigh, R.; Mayers, I.; et al. Inhaled anti-TSLP antibody fragment, ecleralimab, blocks responses to allergen in mild asthma. Eur. Respir. J. 2023, 61, 2201193. [Google Scholar] [CrossRef]
- Doffman, S.; Dosanjh, D.; Sadiq, M.; Asimus, S.; Cooper, J.; Zhou, X.-H.; Seth, H.; Pandya, H.; Saralaya, D.; Beier, J. Phase 1 Safety and Efficacy of AZD8630/AMG 104 Inhaled Anti-TSLP in Healthy Volunteers and Patients With Asthma on Medium-High Dose Inhaled Corticosteroid (ICS) and Long-Acting Beta-agonist (LABA) With Elevated Baseline Fractional Exhaled Nitric Oxide (FeNO). In A34. Late Breaking Abstracts in Asthma And Allergic Inflammation; American Thoracic Society: New York, NY, USA, 2024; p. A1386. [Google Scholar]
- Deiteren, A.; Bontinck, L.; Conickx, G.; Vigan, M.; Dervaux, N.; Gassiot, M.; Bas, S.; Suratt, B.; Staudinger, H.; Krupka, E. A first-in-human, single and multiple dose study of lunsekimig, a novel anti-TSLP/anti-IL-13 NANOBODY® compound, in healthy volunteers. Clin. Transl. Sci. 2024, 17, e13864. [Google Scholar] [CrossRef]
- Fei, Y.; Li, N.; Qian, W.; Fan, Y.; Shen, Y.; Wang, Q.; McLendon, K.; Shen, K. A phase 1, randomized, double-blind, placebo-controlled, dose escalation study to evaluate the safety, tolerability, pharmacokinetics and immunogenicity of SHR-1905, a long-acting anti-thymic stromal lymphopoietin antibody, in healthy subjects. Front. Pharmacol. 2024, 15, 1400696. [Google Scholar] [CrossRef]
- Singh, D.; Deykin, A.; Lloyd, P.; Nestorov, I.; Kalra, A.; Biswas, S.; Sinha, A.; Brickman, C.; Becker, O. A Multiple Ascending-dose Study With Verekitug, a Novel Antibody to the Human Thymic Stromal Lymphopoietin Receptor, in Adults with Asthma. In D21. Terminator: Control of Airway Inflammation and Immune Response in Asthma; American Thoracic Society: New York, NY, USA, 2024; p. A6996. [Google Scholar]
- Wechsler, M.E.; Ruddy, M.K.; Pavord, I.D.; Israel, E.; Rabe, K.F.; Ford, L.B.; Maspero, J.F.; Abdulai, R.M.; Hu, C.-C.; Martincova, R. Efficacy and safety of itepekimab in patients with moderate-to-severe asthma. N. Engl. J. Med. 2021, 385, 1656–1668. [Google Scholar] [CrossRef]
- A Randomized, Double-Blind, Placebo-Controlled, Parallel-Group, 12-Week Proof-of-Concept (PoC) Study to Assess the Efficacy, Safety, and Tolerability of SAR440340 and the Coadministration of SAR440340 and Dupilumab in Patients with Moderate-to-Severe Asthma Who Are Not Well Controlled on Inhaled Corticosteroid (ICS) Plus Long-acting β2 Adrenergic Agonist (LABA) Therapy. 2017. Available online: https://clinicaltrials.gov/study/NCT03387852 (accessed on 23 September 2024).
- A Randomized, Placebo-Controlled, Parallel Panel Study to Assess the Effects of REGN3500, Dupilumab, and Combination of REGN3500 Plus Dupilumab on Markers of Inflammation After Bronchial Allergen Challenge in Patients with Allergic Asthma. Available online: https://clinicaltrials.gov/study/NCT03112577 (accessed on 16 September 2024).
- Placebo-Controlled Proof of Concept Study to Investigate ANB020 Activity in Adult Patients with Severe Eosinophilic Asthma. 2018. Available online: https://www.clinicaltrialsregister.eu/ctr-search/trial/2017-000647-40/results (accessed on 23 September 2024).
- A Phase II, Randomised, Double-blind, Placebo-Controlled Study to Assess the Efficacy and Safety of MEDI3506 in Adult Participants with Uncontrolled Moderate-to-severe Asthma. 2020. Available online: https://clinicaltrials.gov/study/NCT04570657 (accessed on 23 September 2024).
- A Randomized, Double-Blind, Parallel Group, Multicenter, Stratified Study Evaluating the Efficacy and Safety of Repeat Doses of GSK3772847 Compared with Placebo in Participants with Moderately Severe Asthma. 2017. Available online: https://clinicaltrials.gov/study/NCT03207243 (accessed on 23 September 2024).
- A Double Blind (Sponsor Open) Placebo-Controlled, Stratified, Parallel Group Study to Evaluate the Efficacy and Safety of Repeat Doses of GSK3772847 in Participants with Moderate to Severe Asthma with Allergic Fungal Airway Disease (AFAD). 2018. Available online: https://clinicaltrials.gov/study/NCT03393806 (accessed on 23 September 2024).
- Kelsen, S.G.; Agache, I.O.; Soong, W.; Israel, E.; Chupp, G.L.; Cheung, D.S.; Theess, W.; Yang, X.; Staton, T.L.; Choy, D.F.; et al. Astegolimab (anti-ST2) efficacy and safety in adults with severe asthma: A randomized clinical trial. J. Allergy Clin. Immunol. 2021, 148, 790–798. [Google Scholar] [CrossRef] [PubMed]
- Alvarado, D.; Vitale, L.; Murphy, M.B.; Wasiuk, A.; Widger, J.; Crocker, A.; Patterson, C.; Mills-Chen, L.; O’Neill, T.; Baronas, A.R.; et al. Dual targeting of mast cells and TSLP with a bispecific antibody. J. Immunol. 2023, 210, 246.07. [Google Scholar] [CrossRef]
- Liu, X.; Han, J.; Wang, Q.; Wang, P.; Li, L.; Du, K.; Jiang, F.; Zhang, P.; Liu, H.; Huang, J. Development of a novel humanized anti-TSLP monoclonal antibody HZ-1127 with anti-allergic diseases and cancer potential. Antib. Ther. 2024, 7, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Gai, J.; Zhu, M.; Li, G.; Qiao, P. First-in-class Inhalable Anti-TSLP Single Domain Antibody with Therapeutic Potential in Asthma Treatment. In A33. Late Breaking Advances in Asthma and Immunology; American Thoracic Society: New York, NY, USA, 2023; p. A1378. [Google Scholar]
- A Phase Ib Randomized, Double-Blinded, Placebo-Controlled Multiple Rising Dose Clinical Trial to Evaluate the Safety, Efficacy, Pharmacokinetics, Pharmacodynamics, and Immunogenicity of Intravenous MK-8226 in Patients with Moderate to Severe Atopic Dermatitis. Available online: https://clinicaltrials.gov/study/NCT01732510 (accessed on 16 September 2024).
- Mayawala, K.; Aliprantis, A.; Hernandez Escalante, J.; Han, H.; Ullas, S.; Bai, M.; Sinclair, C.; Dworakowski, W.; Goyal, L.; Amiri, K.; et al. A Long-Acting High Affinity Anti-TSLP Antibody (GB-0895) for Severe Asthma Identified Leveraging a Proprietary Machine Learning Platform. Available online: https://delta.larvol.com/Products/?ProductId=0ee1870b-0207-4cca-9616-494fe95968d5 (accessed on 23 September 2024).
- A Randomized, Double-Blind, Placebo-Controlled Phase II Clinical Study to Evaluate the Efficacy and Safety of CM326 in Subjects with Moderate to Severe Asthma. Available online: https://clinicaltrials.gov/study/NCT05774340 (accessed on 16 September 2024).
- Tian, X.; Zhang, X. Safety, tolerability, pharmacokinetics, and immunogenicity of a human monoclonal antibody TQC2731 targeting thymic stromal lymphopoietin in healthy adults: A first-in-human, randomized, placebo-controlled, double-blind, phase 1 study. J. Allergy Clin. Immunol. 2023, 151, AB20. [Google Scholar] [CrossRef]
- Adhikary, P.P.; Idowu, T.; Tan, Z.; Hoang, C.; Shanta, S.; Dumbani, M.; Mappalakayil, L.; Awasthi, B.; Bermudez, M.; Weiner, J.; et al. Disrupting TSLP-TSLP receptor interactions via putative small molecule inhibitors yields a novel and efficient treatment option for atopic diseases. EMBO Mol. Med. 2024, 16, 1630–1656. [Google Scholar] [CrossRef] [PubMed]
- Osbourn, M.; Soares, D.C.; Vacca, F.; Cohen, E.S.; Scott, I.C.; Gregory, W.F.; Smyth, D.J.; Toivakka, M.; Kemter, A.M.; le Bihan, T.; et al. HpARI Protein Secreted by a Helminth Parasite Suppresses Interleukin-33. Immunity 2017, 47, 739–751.e5. [Google Scholar] [CrossRef] [PubMed]
- Holgado, A.; Braun, H.; Van Nuffel, E.; Detry, S.; Schuijs, M.J.; Deswarte, K.; Vergote, K.; Haegman, M.; Baudelet, G.; Haustraete, J.; et al. IL-33trap is a novel IL-33-neutralizing biologic that inhibits allergic airway inflammation. J. Allergy Clin. Immunol. 2019, 144, 204–215. [Google Scholar] [CrossRef]
- Duan, S.; Wang, J.; Lou, X.; Chen, D.; Shi, P.; Jiang, H.; Wang, Z.; Li, W.; Qian, F. A novel anti-IL-33 antibody recognizes an epitope FVLHN of IL-33 and has a therapeutic effect on inflammatory diseases. Int. Immunopharmacol. 2023, 122, 110578. [Google Scholar] [CrossRef]
- Vacca, F.; Chauché, C.; Jamwal, A.; Hinchy, E.C.; Heieis, G.; Webster, H.; Ogunkanbi, A.; Sekne, Z.; Gregory, W.F.; Wear, M.; et al. A helminth-derived suppressor of ST2 blocks allergic responses. Elife 2020, 9, e54017. [Google Scholar] [CrossRef]
- Højen, J.F.; Kristensen, M.L.V.; McKee, A.S.; Wade, M.T.; Azam, T.; Lunding, L.P.; de Graaf, D.M.; Swartzwelter, B.J.; Wegmann, M.; Tolstrup, M.; et al. IL-1R3 blockade broadly attenuates the functions of six members of the IL-1 family, revealing their contribution to models of disease. Nat. Immunol. 2019, 20, 1138–1149. [Google Scholar] [CrossRef]
- Williams, T.C.; Loo, S.L.; Nichol, K.S.; Reid, A.T.; Veerati, P.C.; Esneau, C.; Wark, P.A.B.; Grainge, C.L.; Knight, D.A.; Vincent, T.; et al. IL-25 blockade augments antiviral immunity during respiratory virus infection. Commun. Biol. 2022, 5, 415. [Google Scholar] [CrossRef] [PubMed]
- Bone, R.; Fennell, B.J.; Tam, A.; Sheldon, R.; Nocka, K.; Varghese, S.; Chang, C.S.; Hawerkamp, H.C.; Yeow, A.; Saunders, S.P.; et al. Discovery and multi-parametric optimization of a high-affinity antibody against interleukin-25 with neutralizing activity in a mouse model of skin inflammation. Antib. Ther. 2022, 5, 258–267. [Google Scholar] [CrossRef] [PubMed]
- A Randomized, Double-Blind, Placebo-Controlled, Phase I Single Ascending Dose Study to Evaluate the Safety, Tolerability, and Pharmacokinetics of XKH001Injection in Healthy Adults. Available online: https://clinicaltrials.gov/study/NCT05128409 (accessed on 16 September 2024).
- Corren, J.; Karpefors, M.; Hellqvist, Å.; Parnes, J.R.; Colice, G. Tezepelumab reduces exacerbations across all seasons in patients with severe, uncontrolled asthma: A post hoc analysis of the PATHWAY phase 2b study. J. Asthma Allergy 2021, 14, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wechsler, M.E.; Menzies-Gow, A.; Brightling, C.E.; Kuna, P.; Korn, S.; Welte, T.; Griffiths, J.M.; Sałapa, K.; Hellqvist, Å.; Almqvist, G.; et al. Evaluation of the oral corticosteroid-sparing effect of tezepelumab in adults with oral corticosteroid-dependent asthma (SOURCE): A randomised, placebo-controlled, phase 3 study. Lancet Respir. Med. 2022, 10, 650–660. [Google Scholar] [CrossRef]
- A Multicentre, Single-Arm, Phase 3b Efficacy and Safety Study of Tezepelumab 210 mg Administered Subcutaneously to Reduce Oral Corticosteroid Use in Adult Participants with Severe Asthma on High-dose Inhaled Corticosteroid Plus Long-acting β2 Agonist and Long-Term Oral Corticosteroid Therapy (WAYFINDER). Available online: https://clinicaltrials.gov/study/NCT05274815 (accessed on 16 September 2024).
- A Randomised, Double-Blind, Parallel-Group, Placebo-Controlled 28-Week Phase 3 Efficacy and Safety Study of Tezepelumab in Reducing Oral Corticosteroid Use in Adults with Oral Corticosteroid Dependent Asthma (SUNRISE). Available online: https://clinicaltrials.gov/study/NCT05398263 (accessed on 16 September 2024).
- Jackson, D.; Lugogo, N.; Gurnell, M.; Llanos-Ackert, J.; Martin, N.; Keeling, N.; Salapa, K.; Cook, B. Tezepelumab reduced OCS use in OCS-dependent patients with severe asthma: PHASE 3B WAYFINDER interim results. Ann. Allergy Asthma Immunol. 2023, 131, S43. [Google Scholar] [CrossRef]
- Diver, S.; Khalfaoui, L.; Emson, C.; Wenzel, S.E.; Menzies-Gow, A.; Wechsler, M.E.; Johnston, J.; Molfino, N.; Parnes, J.R.; Megally, A.; et al. Effect of tezepelumab on airway inflammatory cells, remodelling, and hyperresponsiveness in patients with moderate-to-severe uncontrolled asthma (CASCADE): A double-blind, randomised, placebo-controlled, phase 2 trial. Lancet Respir. Med. 2021, 9, 1299–1312. [Google Scholar] [CrossRef]
- Shen, S.; Xian, M.; Yan, B.; Lan, F.; Wang, C.; Zhang, L. Anti-thymic stromal lymphopoietin monoclonal antibody in patients with chronic rhinosinusitis with nasal polyps (DUBHE): Rationale and design of a multicenter, randomized, double-blind, placebo-controlled study. Asia Pac. Allergy 2024, 14, 26–31. [Google Scholar] [CrossRef]
- A Randomized, Double-Blind, Placebo-Controlled, Parallel-Group, Dose Ranging Study to Assess the Efficacy, Safety, and Tolerability of Subcutaneous Lunsekimig in Adult Participants with Moderate-to-Severe Asthma. Available online: https://clinicaltrials.gov/study/NCT06102005 (accessed on 16 September 2024).
- Komai-Koma, M.; Xu, D.; Li, Y.; McKenzie, A.N.; McInnes, I.B.; Liew, F.Y. IL-33 is a chemoattractant for human Th2 cells. Eur. J. Immunol. 2007, 37, 2779–2786. [Google Scholar] [CrossRef]
- Baye, T.M.; Abebe, T.; Wilke, R.A. Genotype–Environment Interactions and Their Translational Implications. Pers. Med. 2011, 8, 59–70. [Google Scholar] [CrossRef]
- Cho, S.H.; Oh, S.Y.; Bahn, J.W.; Choi, J.Y.; Chang, Y.S.; Kim, Y.K.; Min, K.U.; Kim, Y.Y. Association between bronchodilating response to short-acting β-agonist and non-synonymous single-nucleotide polymorphisms of β2-adrenoceptor gene. Clin. Exp. Allergy 2005, 35, 1162–1167. [Google Scholar] [CrossRef]
- Lopert, A.; Rijavec, M.; Žavbi, M.; Korošec, P.; Fležar, M. Asthma treatment outcome in adults is associated with rs9910408 in TBX21 gene. Sci. Rep. 2013, 3, 2915. [Google Scholar] [CrossRef] [PubMed]
- Tantisira, K.G.; Lima, J.; Sylvia, J.; Klanderman, B.; Weiss, S.T. 5-lipoxygenase pharmacogenetics in asthma: Overlap with Cys-leukotriene receptor antagonist loci. Pharmacogenet. Genom. 2009, 19, 244–247. [Google Scholar] [CrossRef] [PubMed]
- de Lima, L.C.; Cruz, Á.A.; Costa, R.d.S.; Silva, H.d.S.; Coelho, R.S.; Teixeira, H.M.; Oliveira, P.R.; Barnes, K.C.; Figueiredo, C.A.; Carneiro, V.L. TSLP and IL25 variants are related to asthma and atopy. Gene Rep. 2023, 30, 101727. [Google Scholar] [CrossRef]
- Matloubi, M.; Ranjbar, M.; Assarehzadegan, M.A.; Fallahpour, M.; Sadeghi, F.; Soleyman-Jahi, S.; Janani, L. The Impact of Interleukin (IL)-33 Gene Polymorphisms and Environmental Factors on Risk of Asthma in the Iranian Population. Lung 2020, 198, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Ranjbar, M.; Matloubi, M.; Assarehzadegan, M.A.; Fallahpour, M.; Sadeghi, F.; Soleyman-Jahi, S.; Janani, L. Association between Two Single Nucleotide Polymorphisms of Thymic Stromal Lymphopoietin (TSLP) Gene and Asthma in Iranian Population. Iran. J. Allergy Asthma Immunol. 2020, 19, 362–372. [Google Scholar] [CrossRef]
- Slager, R.E.; Otulana, B.A.; Hawkins, G.A.; Yen, Y.P.; Peters, S.P.; Wenzel, S.E.; Meyers, D.A.; Bleecker, E.R. IL-4 receptor polymorphisms predict reduction in asthma exacerbations during response to an anti-IL-4 receptor α antagonist. J. Allergy Clin. Immunol. 2012, 130, 516–522.e4. [Google Scholar] [CrossRef]

{kind=link}
| Drug Name | Study Name | Clinical Trial ID | Characteristics | Study Population | Main Results/Endpoints | References |
|---|---|---|---|---|---|---|
| IL-25 | ||||||
| Brodalumab | - | NCT01199289 | Phase 2b, Randomized, Double-Blind, Placebo-Controlled Study | Moderate-to-severe uncontrolled asthmatic patients taking ICS | - Generally well tolerated with no unexpected safety concerns - No treatment differences observed between treated vs. placebo group | [179] |
| TSLP | ||||||
| Tezepelumab | - | NCT01405963 | Phase 1, Randomized, Double-Blind, Placebo-Controlled, Parallel Design, Multiple-Dose Study | Mild atopic asthma | - Reduced allergen-induced bronchoconstriction - Decreased FeNO - Decreased blood and sputum eosinophil counts | [180] |
| PATHWAY | NCT02054130 | Phase 2 Randomized, Double-Blind, Placebo-Controlled Study | Severe, uncontrolled asthmatic patients taking ICS | - Decreased asthma exacerbations - Reduced AAER - Improved the ACQ-6 score - Reduced serum IgE concentrations - Reduced blood eosinophil counts and FeNO levels. - Reduced blood levels of periostin, IL-5, IL-13, and TARC | [181] | |
| UPSTREAM | NCT02698501 | Phase 2 Randomized Double-Blind, Placebo-Controlled Trial | Adult asthmatic patients taking ICS with or without LABA | - Improved mannitol PD15, - Decreased eosinophil counts in airway tissue, BAL, sputum, and blood | [182] | |
| CASCADE | NCT03688074 | A Phase 2, Multicentre, Randomized, Double-Blind, Placebo-Controlled, Parallel-Group Study | Uncontrolled moderate-to-severe asthma | - Improved mannitol-induced AHR - Decreased airway eosinophil counts | [183] | |
| NAVIGATOR | NCT03347279 | A Phase 3 Multicenter, Randomized, Double-Blind, Placebo-Controlled, Parallel-Group Study | Uncontrolled moderate-to-severe asthma | - Reduced AAER - Improved ACQ-6 and the Asthma AQLQ scores - Improved lung function - Decreased serum IgE, FeNO, and blood eosinophil counts | [184] | |
| NOZOMI | NCT04048343 | A Phase 3, 52-Week, Open-Label, Multicenter Study | Uncontrolled moderate-to-severe asthma | - No new adverse events were observed | [185] | |
| SOURCE | NCT03406078 | A Phase 3 Multicenter, Randomized, Double-Blind, Placebo-Controlled Study | OCS-dependent severe asthma | - No significant effect on OCS use - Improved number of asthma exacerbations, lung function, blood eosinophil count, FeNO, and serum total IgE concentration at week 48 | [186] | |
| DESTINATION | NCT03706079 | A Phase 3 Multicenter, Double-Blind, Randomized, Placebo-Controlled, Parallel-Group, Safety Extension Study | Uncontrolled moderate-to-severe asthma | Ongoing | [187] | |
| - | NCT05740748 | Phase 2 Multicenter, Randomized, Double-Blind Placebo-Controlled Parallel-Group Study | Mild allergic asthma | Ongoing | [188] | |
| TEZARS | NCT06189742 | A Phase II Open-Label Exploratory Mechanistic Pilot Study | Patients with coexisting allergic asthma and allergic rhinitis | Ongoing | [189] | |
| Ecleralimab (CSJ117) | - | NCT03138811 | A Phase 1, Randomized, Subject- and Investigator-Blinded, Placebo-Controlled, Parallel Design, Bronchoprovocation Study | Mild atopic asthma | - Reduced allergen-induced bronchoconstriction | [190] |
| AZD8630/AMG104 | - | NCT05110976 | A Phase 1, Randomized, Blinded, Placebo-Controlled Safety Study | Healthy adults and asthmatic adults on medium-/high-dose ICS/LABA | -Significant reduction in FeNO in asthmatics evident from Day 7 until end of treatment (Day 28) - Lower levels of serum IL-5 and IL-13 in treated individuals compared to placebo | [191] |
| Lunsekimig | - | NCT06566764 | A Phase 1, Randomized, Double-Blind, Placebo-Controlled, Safety and Proof of Mechanism Study | Mild-to-moderate asthma | - Well tolerated with mild-to-moderate treatment-emerging adverse events that were similar to placebo group - Reduced FeNO and blood biomarkers (IL-5, eotaxin-3, TARC, IgE and eosinophils) - Pre-bronchodilator FEV1 improved by week 1 in treatment group and persisted through Day 29 | [192] |
| SHR-1905 | - | NCT04905602 | A Phase 1, Randomized, Double-blind, Placebo-Controlled Dose Escalation Study | Mild asthma | - Safe and well-tolerated mild-to-moderate treatment-related adverse effects - Treatment reduced eosinophil, FeNO, TARC, and eotaxin-3 levels - Significantly improved FEV1 | [193] |
| TSLP Receptor | ||||||
| Verekitug (UPB-101/ASP7266) | - | - | A Randomized, Double-Blind, Placebo-Controlled, Multiple Ascending Dose Study | Mild-to-moderate asthma | - Well tolerated and safe at all doses tested - 100 mg dose resulted in 54% and 54% mean percent change from baseline FeNO and eosinophils, respectively, at week 12 | [194] |
| IL-33 | ||||||
| Itepekimab (REGN3500, SAR440340) | - | NCT02999711 | A Phase 1, Randomized, Double-Blind, Placebo-controlled, Multiple Ascending Dose Study | Mild-to-moderate asthma | - Improved FEV1 and overall asthma control | [195] |
| - | NCT03387852 | A Phase 2, Randomized, Double-Blind, Placebo-Controlled, Parallel-Group, 12-Week Proof-of-Concept (PoC) Study to Assess the Efficacy, Safety, and Tolerability of SAR440340 and the Coadministration of SAR440340 and Dupilumab | Uncontrolled moderate-to-severe asthma | - The combination therapy of itepekimab and dupilumab yielded similar improvement results to those achieved with dupilumab alone | [196] | |
| - | NCT03112577 | A Phase 1, Randomized, Placebo-Controlled, Parallel Panel Study to Assess the Effects of REGN3500, Dupilumab, and Combination of REGN3500 Plus Dupilumab | Mild allergic asthma | N/A | [197] | |
| Etokimab (ANB020) | - | NCT03469934 | A Phase 2, Placebo-Controlled Proof-of-Concept Study | Severe eosinophilic asthma | - Improved FEV1 - Reduced ACQ-5 score, sustained through Day 64 | [198] |
| Tozorakimab (MEDI3506) | FRONTIER-3 | NCT04570657 | A Phase 2, Randomized, Double-Blind, Placebo-Controlled Study to Assess the Efficacy and Safety | Uncontrolled moderate-to-severe asthma | N/A | [199] |
| ST2 Receptor | ||||||
| Melrilimab (GSK3772847) | - | NCT03207243 | A Phase 2, Randomized, Double-Blind, Parallel-Group, Multicenter, Stratified Study Evaluating the Efficacy and Safety of Repeat Doses of GSK3772847 | Moderate-to-severe asthma | - Lower chance of loss of asthma control | [200] |
| - | NCT03393806 | A Double-Blind (Sponsor Open) Placebo-Controlled, Stratified, Parallel-Group Study to Evaluate the Efficacy and Safety of Repeat Doses of GSK3772847 | Moderate-to-severe asthma with allergic fungal airway disease | - No significant difference between the placebo and drug arms | [201] | |
| Astegolimab | ZENYATTA | NCT02918019 | A Phase IIb, Randomized, Double-Blind, Placebo-Controlled, Multicenter, Dose-Ranging Study to Assess the Efficacy and Safety of MSTT1041A | Uncontrolled severe asthma | - Significant reduction in the AAER by 43% in patients with severe asthma | [202] |
| Target | Name | Mechanism of Action | Route and Dosing | Current Clinical Trial Phase | Current Clinical Trial ID | References |
|---|---|---|---|---|---|---|
| TSLP | ||||||
| TSLP Directly | CDX-622 | Humanized tetravalent bispecific antibody that depletes mast cells and neutralizes TSLP | 10 mg/kg intravenous (non-human primate) | Preclinical | N/A | [203] |
| HZ-1127 | Humanized monoclonal anti-TSLP antibody | - | Preclinical | N/A | [204] | |
| LQ043H | Bivalent TSLP neutralizing single-domain antibody | Inhaled | Preclinical | N/A | [205] | |
| MK-8226 | Monoclonal TSLP neutralizing antibody | Intravenous Dose range (0.3 mg/kg–10 mg/kg) | Phase 1b | NCT01732510 (terminated with no results) | [206] | |
| GB-0895 | Anti-TSLP neutralizing antibody | - | Phase 1 | - | [207] | |
| CM326 | Monoclonal TSLP neutralizing antibody | Subcutaneous 220 mg/2 mL of either low- or high-dose | Phase 2 | NCT05774340 (Ongoing) | [208] | |
| TQC2731 | Human IgG1 monoclonal anti-TSLP antibody | Subcutaneous Multiple doses (70 mg, 210 mg, and 420 mg) | Phase 2 | NCT05472324 (Ongoing) | [209] | |
| TSLP Receptor | BP79 | Small-molecule TSLP receptor inhibitor | Topical use targeting atopic dermatitis | Preclinical | N/A | [210] |
| IL-33 | ||||||
| IL-33 Directly | HpARI (Heligmosomoides polygrus Alarmin Release Inhibitor) | Product secreted by murine intestinal nematode H. polygrus that can interfere IL-33 pathway | - | Preclinical | N/A | [211] |
| IL-33trap | Neutralizing fusion protein that combines ST2 receptor with the IL1RAcP co-receptor to trap IL-33 | - | Preclinical | N/A | [212] | |
| 5H8 | Monoclonal IL-33 antibody that targets the FVLHN epitope on IL-33 | - | Preclinical | N/A | [213] | |
| ST2 Receptor | HpBARI (H. polygyrus Binds Alarmin Receptor and Inhibits) | Protein secreted by H. polygyrus, which consists of two complement control protein domains that have the ability to bind and block ST2 | - | Preclinical | N/A | [214] |
| IL1RAcP | Anti-IL1RAcP antibody | Monoclonal antibody to human IL-1R3 (also known as IL1RAcP) | - | Preclinical | N/A | [215] |
| IL-25 | ||||||
| IL-25 Directly | LNR125 | Anti-IL-25 monoclonal antibody | - | Preclinical | N/A | [216] |
| 22C7 | Human/mouse cross-reactive antibody against IL-25 | - | Preclinical | N/A | [217] | |
| XKH001 | Humanized monoclonal IgG1 IL-25 neutralizing antibody | Subcutaneous multiple doses (0.5, 1.67, 3.34, 5.0, or 10.0 mg/kg) | Phase 1 | NCT05128409 (Ongoing) | [218] | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hansi, R.K.; Ranjbar, M.; Whetstone, C.E.; Gauvreau, G.M. Regulation of Airway Epithelial-Derived Alarmins in Asthma: Perspectives for Therapeutic Targets. Biomedicines 2024, 12, 2312. https://doi.org/10.3390/biomedicines12102312
Hansi RK, Ranjbar M, Whetstone CE, Gauvreau GM. Regulation of Airway Epithelial-Derived Alarmins in Asthma: Perspectives for Therapeutic Targets. Biomedicines. 2024; 12(10):2312. https://doi.org/10.3390/biomedicines12102312
Chicago/Turabian StyleHansi, Ravneet K., Maral Ranjbar, Christiane E. Whetstone, and Gail M. Gauvreau. 2024. "Regulation of Airway Epithelial-Derived Alarmins in Asthma: Perspectives for Therapeutic Targets" Biomedicines 12, no. 10: 2312. https://doi.org/10.3390/biomedicines12102312