
Hippocampal Leptin Resistance and Cognitive Decline: Mechanisms, Therapeutic Strategies and Clinical Implications


Abstract
:1. Introduction
2. Leptin: Biological Role and Actions
3. Leptin Receptors
3.1. Intracellular Signaling of Leptin Coupled to the Leptin Receptor
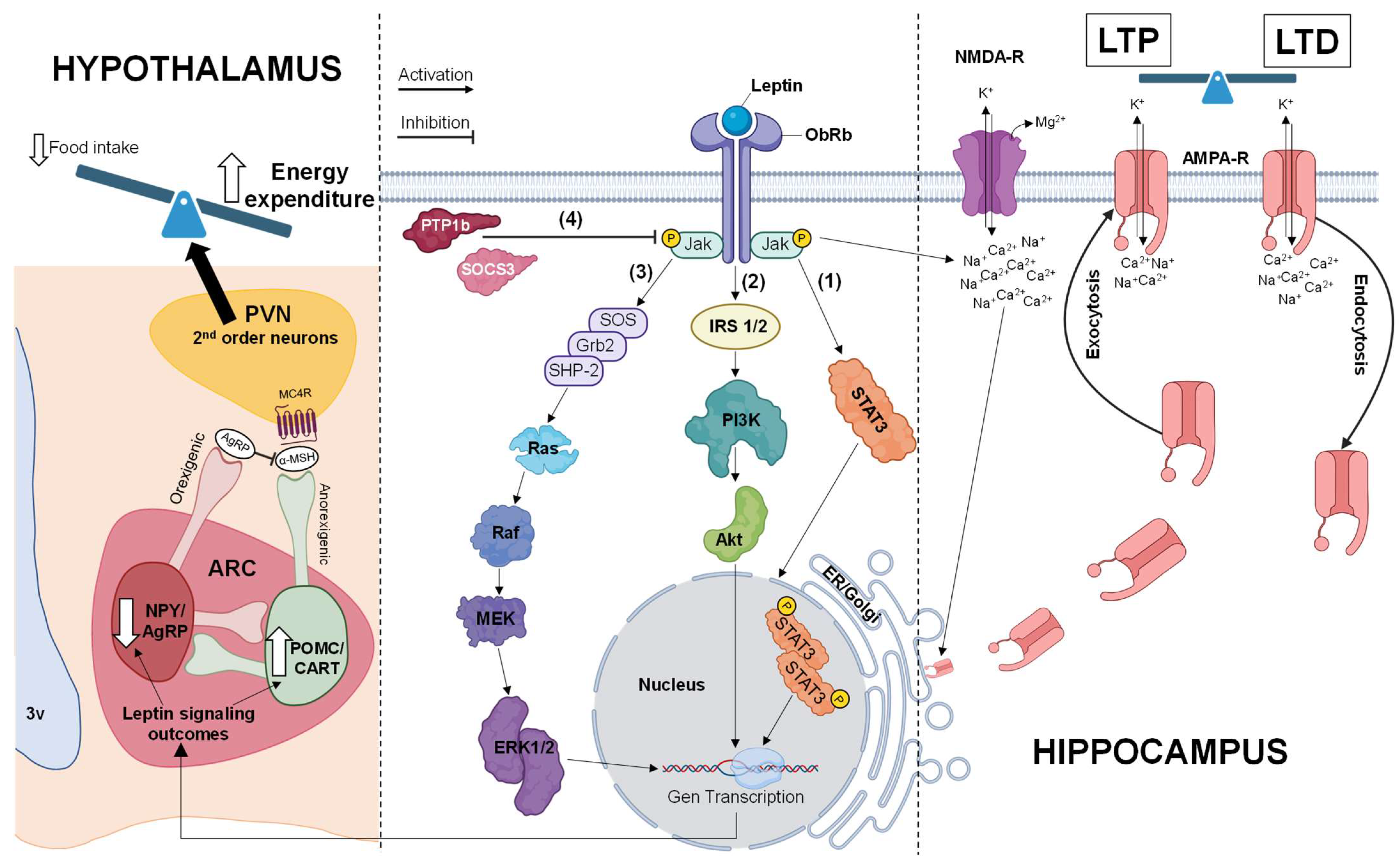
3.1.1. Jak/STAT Pathway
3.1.2. PI3K/Akt Pathway
3.1.3. MAPK Pathway
4. Physiological Effects of Leptin
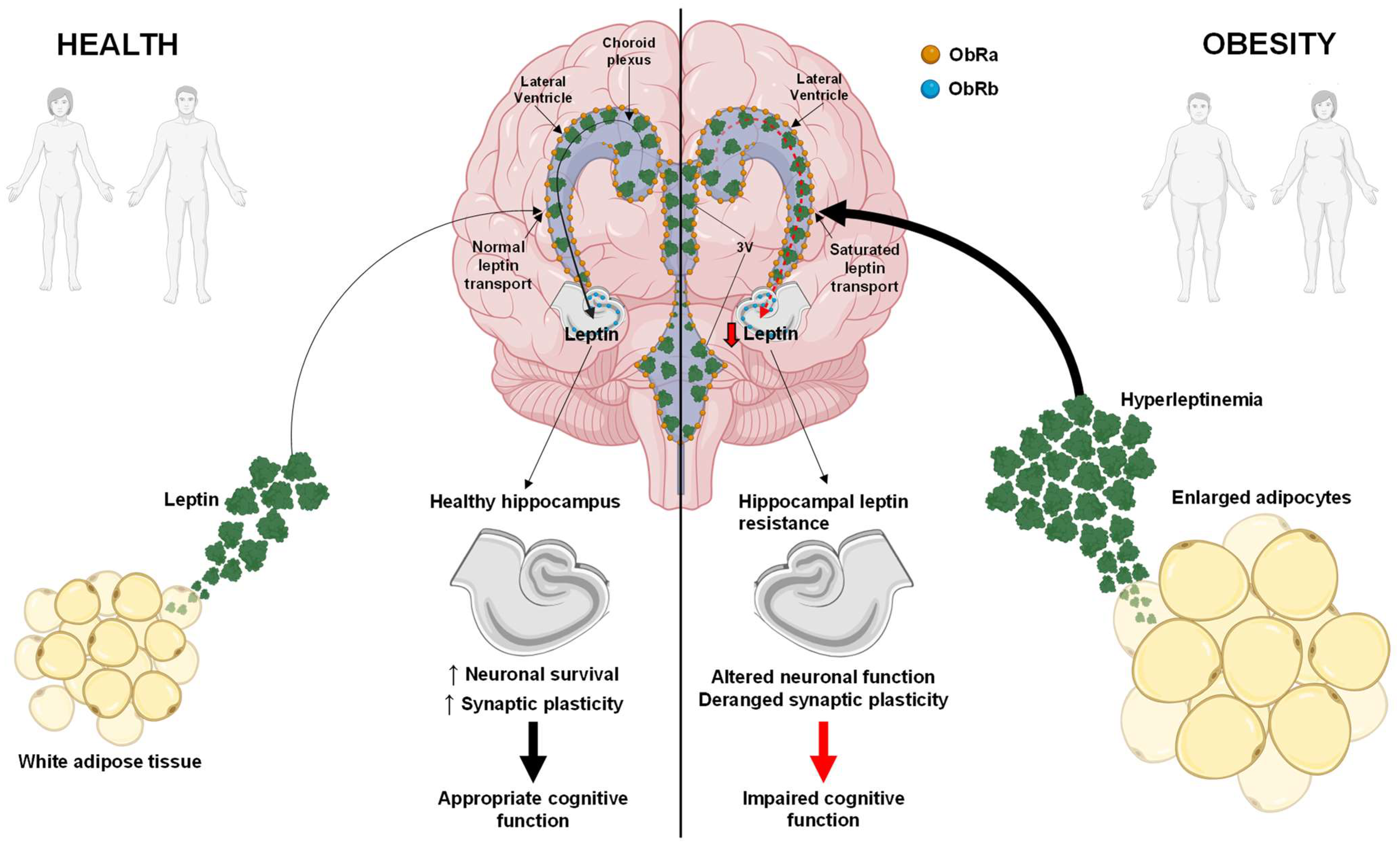
5. Hippocampal Leptin Resistance
6. Cognitive Consequences of Hippocampal Leptin Resistance
7. Implications of Hippocampal Leptin Resistance for Executive Function and Alzheimer’s Disease
8. Therapeutic Interventions and Future Directions
9. Clinical Implications and Outlook
10. Conclusions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, Y.; Proenca, R.; Maffei, M.; Barone, M.; Leopold, L.; Friedman, J.M. Positional cloning of the mouse obese gene and its human homologue. Nature 1994, 372, 425–432, Erratum in Nature 1995, 374, 479. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.M. Leptin and the endocrine control of energy balance. Nat. Metab. 2019, 1, 754–764. [Google Scholar] [CrossRef] [PubMed]
- Mercer, J.G.; Hoggard, N.; Williams, L.M.; Lawrence, C.B.; Hannah, L.T.; Trayhurn, P. Localization of leptin receptor mRNA and the long form splice variant (Ob-Rb) in mouse hypothalamus and adjacent brain regions by in situ hybridization. FEBS Lett. 1996, 387, 113–116. [Google Scholar] [CrossRef] [PubMed]
- Bjørbaek, C.; Uotani, S.; da Silva, B.; Flier, J.S. Divergent signaling capacities of the long and short isoforms of the leptin receptor. J. Biol. Chem. 1997, 272, 32686–32695. [Google Scholar] [CrossRef]
- Shioda, S.; Funahashi, H.; Nakajo, S.; Yada, T.; Maruta, O.; Nakai, Y. Immunohistochemical localization of leptin receptor in the rat brain. Neurosci. Lett. 1998, 243, 41–44. [Google Scholar] [CrossRef]
- Muoio, D.M.; Lynis Dohm, G. Peripheral metabolic actions of leptin. Best Pract. Res. Clin. Endocrinol. Metab. 2002, 16, 653–666. [Google Scholar] [CrossRef]
- Huo, L.; Grill, H.J.; Bjørbaek, C. Divergent regulation of proopiomelanocortin neurons by leptin in the nucleus of the solitary tract and in the arcuate hypothalamic nucleus. Diabetes 2006, 55, 567–573. [Google Scholar] [CrossRef]
- Fulton, S.; Pissios, P.; Manchon, R.P.; Stiles, L.; Frank, L.; Pothos, E.N.; Maratos-Flier, E.; Flier, J.S. Leptin regulation of the mesoaccumbens dopamine pathway. Neuron 2006, 51, 811–822. [Google Scholar] [CrossRef]
- Walker, C.D.; Long, H.; Williams, S.; Richard, D. Long-lasting effects of elevated neonatal leptin on rat hippocampal function, synaptic proteins and NMDA receptor subunits. J. Neurosci. Res. 2007, 85, 816–828. [Google Scholar] [CrossRef]
- Bjørbaek, C. Central leptin receptor action and resistance in obesity. J. Investig. Med. 2009, 57, 789–794. [Google Scholar] [CrossRef]
- Casado, M.E.; Collado-Pérez, R.; Frago, L.M.; Barrios, V. Recent Advances in the Knowledge of the Mechanisms of Leptin Physiology and Actions in Neurological and Metabolic Pathologies. Int. J. Mol. Sci. 2023, 24, 1422. [Google Scholar] [CrossRef] [PubMed]
- Tartaglia, L.A.; Dembski, M.; Weng, X.; Deng, N.; Culpepper, J.; Devos, R.; Richards, G.J.; Campfield, L.A.; Clark, F.T.; Deeds, J.; et al. Identification and expression cloning of a leptin receptor, OB-R. Cell 1995, 83, 1263–1271. [Google Scholar] [CrossRef] [PubMed]
- Gorska, E.; Popko, K.; Stelmaszczyk-Emmel, A.; Ciepiela, O.; Kucharska, A.; Wasik, M. Leptin receptors. Eur. J. Med. Res. 2010, 15 (Suppl. S2), 50–54. [Google Scholar] [CrossRef]
- Obradovic, M.; Sudar-Milovanovic, E.; Soskic, S.; Essack, M.; Arya, S.; Stewart, A.J.; Gojobori, T.; Isenovic, E.R. Leptin and Obesity: Role and Clinical Implication. Front. Endocrinol. 2021, 12, 585887. [Google Scholar] [CrossRef]
- Schwartz, M.W.; Seeley, R.J.; Campfield, L.A.; Burn, P.; Baskin, D.G. Identification of targets of leptin action in rat hypothalamus. J. Clin. Investig. 1996, 98, 1101–1106. [Google Scholar] [CrossRef] [PubMed]
- Couce, M.E.; Burguera, B.; Parisi, J.E.; Jensen, M.D.; Lloyd, R.V. Localization of leptin receptor in the human brain. Neuroendocrinology 1997, 66, 145–150. [Google Scholar] [CrossRef]
- Boyle, C.A.; Kola, P.K.; Oraegbuna, C.S.; Lei, S. Leptin excites basolateral amygdala principal neurons and reduces food intake by LepRb-JAK2-PI3K-dependent depression of GIRK channels. J. Cell. Physiol. 2024, 239, e31117. [Google Scholar] [CrossRef]
- Löllmann, B.; Grüninger, S.; Stricker-Krongrad, A.; Chiesi, M. Detection and quantification of the leptin receptor splice variants Ob-Ra, b, and, e in different mouse tissues. Biochem. Biophys. Res. Commun. 1997, 238, 648–652, Erratum in Biochem. Biophys. Res. Commun. 1997, 241, 803. [Google Scholar] [CrossRef]
- Lammert, A.; Kiess, W.; Bottner, A.; Glasow, A.; Kratzsch, J. Soluble leptin receptor represents the main leptin binding activity in human blood. Biochem. Biophys. Res. Commun. 2001, 283, 982–988. [Google Scholar] [CrossRef]
- Chan, J.L.; Blüher, S.; Yiannakouris, N.; Suchard, M.A.; Kratzsch, J.; Mantzoros, C.S. Regulation of circulating soluble leptin receptor levels by gender, adiposity, sex steroids, and leptin: Observational and interventional studies in humans. Diabetes 2002, 51, 2105–2112. [Google Scholar] [CrossRef]
- Weinland, C.; Tanovska, P.; Kornhuber, J.; Mühle, C.; Lenz, B. Serum lipids, leptin, and soluble leptin receptor in alcohol dependence: A cross-sectional and longitudinal study. Drug Alcohol Depend. 2020, 209, 107898. [Google Scholar] [CrossRef] [PubMed]
- Bouna-Pyrrou, P.; Muehle, C.; Kornhuber, J.; Weinland, C.; Lenz, B. Body mass index and serum levels of soluble leptin receptor are sex-specifically related to alcohol binge drinking behavior. Psychoneuroendocrinology 2021, 127, 105179. [Google Scholar] [CrossRef] [PubMed]
- Vaisse, C.; Halaas, J.L.; Horvath, C.M.; Darnell, J.E., Jr.; Stoffel, M.; Friedman, J.M. Leptin activation of Stat3 in the hypothalamus of wild-type and ob/ob mice but not db/db mice. Nat. Genet. 1996, 14, 95–97. [Google Scholar] [CrossRef]
- Uotani, S.; Bjørbaek, C.; Tornøe, J.; Flier, J.S. Functional properties of leptin receptor isoforms: Internalization and degradation of leptin and ligand-induced receptor downregulation. Diabetes 1999, 48, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Banks, A.S.; Davis, S.M.; Bates, S.H.; Myers, M.G., Jr. Activation of downstream signals by the long form of the leptin receptor. J. Biol. Chem. 2000, 275, 14563–14572. [Google Scholar] [CrossRef]
- Bjørbaek, C.; Kahn, B.B. Leptin signaling in the central nervous system and the periphery. Recent Prog. Horm. Res. 2004, 59, 305–331. [Google Scholar] [CrossRef]
- Frühbeck, G. Intracellular signalling pathways activated by leptin. Biochem. J. 2006, 393 Pt 1, 7–20. [Google Scholar] [CrossRef]
- Saxton, R.A.; Caveney, N.A.; Moya-Garzon, M.D.; Householder, K.D.; Rodriguez, G.E.; Burdsall, K.A.; Long, J.Z.; Garcia, K.C. Structural insights into the mechanism of leptin receptor activation. Nat. Commun. 2023, 14, 1797. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lai, F.; Hou, Y.; Zheng, R. Leptin signaling and leptin resistance. Med. Rev. 2022, 2, 363–384. [Google Scholar] [CrossRef]
- Bjørbaek, C.; Elmquist, J.K.; Frantz, J.D.; Shoelson, S.E.; Flier, J.S. Identification of SOCS-3 as a potential mediator of central leptin resistance. Mol. Cell 1998, 1, 619–625. [Google Scholar] [CrossRef]
- Barsh, G.S.; Farooqi, I.S.; O’Rahilly, S. Genetics of body-weight regulation. Nature 2000, 404, 644–651. [Google Scholar] [CrossRef] [PubMed]
- Reed, A.S.; Unger, E.K.; Olofsson, L.E.; Piper, M.L.; Myers, M.G., Jr.; Xu, A.W. Functional role of suppressor of cytokine signaling 3 upregulation in hypothalamic leptin resistance and long-term energy homeostasis. Diabetes 2010, 59, 894–906. [Google Scholar] [CrossRef] [PubMed]
- Wunderlich, C.M.; Hövelmeyer, N.; Wunderlich, F.T. Mechanisms of chronic JAK-STAT3-SOCS3 signaling in obesity. JAKSTAT 2013, 2, e23878. [Google Scholar] [CrossRef] [PubMed]
- Abbott, K.N.; Arnott, C.K.; Westbrook, R.F.; Tran, D.M.D. The effect of high fat, high sugar, and combined high fat-high sugar diets on spatial learning and memory in rodents: A meta-analysis. Neurosci. Biobehav. Rev. 2019, 107, 399–421. [Google Scholar] [CrossRef] [PubMed]
- Bakoyiannis, I.; Ducourneau, E.G.; N’diaye, M.; Fermigier, A.; Ducroix-Crepy, C.; Bosch-Bouju, C.; Coutureau, E.; Trifilieff, P.; Ferreira, G. Obesogenic diet induces circuit-specific memory deficits in mice. eLife 2024, 13, e80388. [Google Scholar] [CrossRef]
- Madison, F.N.; Bingman, V.P.; Smulders, T.V.; Lattin, C.R. A bird’s eye view of the hippocampus beyond space: Behavioral, neuroanatomical, and neuroendocrine perspectives. Horm. Behav. 2024, 157, 105451. [Google Scholar] [CrossRef]
- Witter, M.P. The perforant path: Projections from the entorhinal cortex to the dentate gyrus. Prog. Brain Res. 2007, 163, 43–61. [Google Scholar]
- Kobayashi, K. Hippocampal mossy fiber synaptic transmission and its modulation. Vitam. Horm. 2010, 82, 65–85. [Google Scholar]
- Szirmai, I.; Buzsáki, G.; Kamondi, A. 120 years of hippocampal Schaffer collaterals. Hippocampus 2012, 22, 1508–1516. [Google Scholar] [CrossRef]
- Bin Ibrahim, M.Z.; Benoy, A.; Sajikumar, S. Long-term plasticity in the hippocampus: Maintaining within and ‘tagging’ between synapses. FEBS J. 2022, 289, 2176–2201. [Google Scholar] [CrossRef]
- Li, X.L.; Aou, S.; Oomura, Y.; Hori, N.; Fukunaga, K.; Hori, T. Impairment of long-term potentiation and spatial memory in leptin receptor-deficient rodents. Neuroscience 2002, 113, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Winocur, G.; Greenwood, C.E.; Piroli, G.G.; Grillo, C.A.; Reznikov, L.R.; Reagan, L.P.; McEwen, B.S. Memory impairment in obese Zucker rats: An investigation of cognitive function in an animal model of insulin resistance and obesity. Behav. Neurosci. 2005, 119, 1389–1395. [Google Scholar] [CrossRef] [PubMed]
- Stranahan, A.M.; Arumugam, T.V.; Cutler, R.G.; Lee, K.; Egan, J.M.; Mattson, M.P. Diabetes impairs hippocampal function through glucocorticoid-mediated effects on new and mature neurons. Nat. Neurosci. 2008, 11, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Valladolid-Acebes, I.; Stucchi, P.; Cano, V.; Fernández-Alfonso, M.S.; Merino, B.; Gil-Ortega, M.; Fole, A.; Morales, L.; Ruiz-Gayo, M.; Del Olmo, N. High-fat diets impair spatial learning in the radial-arm maze in mice. Neurobiol. Learn. Mem. 2011, 95, 80–85. [Google Scholar] [CrossRef]
- Kamal, A.; Ramakers, G.M.; Gispen, W.H.; Biessels, G.J. Hyperinsulinemia in rats causes impairment of spatial memory and learning with defects in hippocampal synaptic plasticity by involvement of postsynaptic mechanisms. Exp. Brain Res. 2013, 226, 45–51, Erratum in Exp. Brain Res. 2013, 227, 421. [Google Scholar] [CrossRef]
- Valladolid-Acebes, I.; Fole, A.; Martín, M.; Morales, L.; Cano, M.V.; Ruiz-Gayo, M.; Del Olmo, N. Spatial memory impairment and changes in hippocampal morphology are triggered by high-fat diets in adolescent mice. Is there a role of leptin? Neurobiol. Learn. Mem. 2013, 106, 18–25. [Google Scholar] [CrossRef]
- Roth, J.D.; Roland, B.L.; Cole, R.L.; Trevaskis, J.L.; Weyer, C.; Koda, J.E.; Anderson, C.M.; Parkes, D.G.; Baron, A.D. Leptin responsiveness restored by amylin agonism in diet-induced obesity: Evidence from nonclinical and clinical studies. Proc. Natl. Acad. Sci. USA 2008, 105, 7257–7262. [Google Scholar] [CrossRef]
- Liu, J.; Lee, J.; Salazar Hernandez, M.A.; Mazitschek, R.; Ozcan, U. Treatment of obesity with celastrol. Cell 2015, 161, 999–1011. [Google Scholar] [CrossRef]
- Boyle, C.N.; Zheng, Y.; Lutz, T.A. Mediators of Amylin Action in Metabolic Control. J. Clin. Med. 2022, 11, 2207. [Google Scholar] [CrossRef]
- Madej, T.; Boguski, M.S.; Bryant, S.H. Threading analysis suggests that the obese gene product may be a helical cytokine. FEBS Lett. 1995, 373, 13–18. [Google Scholar] [CrossRef]
- Caro, J.F.; Sinha, M.K.; Kolaczynski, J.W.; Zhang, P.L.; Considine, R.V. Leptin: The tale of an obesity gene. Diabetes 1996, 45, 1455–1462. [Google Scholar] [CrossRef] [PubMed]
- Considine, R.V.; Sinha, M.K.; Heiman, M.L.; Kriauciunas, A.; Stephens, T.W.; Nyce, M.R.; Ohannesian, J.P.; Marco, C.C.; McKee, L.J.; Bauer, T.L.; et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N. Engl. J. Med. 1996, 334, 292–295. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Qin, R.; Yuan, W.; Fan, J.S.; Huang, W.; Lin, Z. The solution structure of human leptin reveals a conformational plasticity important for receptor recognition. Structure 2024, 32, 18–23.e2. [Google Scholar] [CrossRef]
- Drougard, A.; Fournel, A.; Valet, P.; Knauf, C. Impact of hypothalamic reactive oxygen species in the regulation of energy metabolism and food intake. Front. Neurosci. 2015, 9, 56. [Google Scholar] [CrossRef]
- Coccurello, R.; Maccarrone, M. Hedonic Eating and the “Delicious Circle”: From Lipid-Derived Mediators to Brain Dopamine and Back. Front. Neurosci. 2018, 12, 271. [Google Scholar] [CrossRef]
- Wiesner, G.; Vaz, M.; Collier, G.; Seals, D.; Kaye, D.; Jennings, G.; Lambert, G.; Wilkinson, D.; Esler, M. Leptin is released from the human brain: Influence of adiposity and gender. J. Clin. Endocrinol. Metab. 1999, 84, 2270–2274. [Google Scholar] [CrossRef]
- Ahima, R.S.; Flier, J.S. Leptin. Annu. Rev. Physiol. 2000, 62, 413–437. [Google Scholar] [CrossRef] [PubMed]
- Cinti, S. The adipose organ: Morphological perspectives of adipose tissues. Proc. Nutr. Soc. 2001, 60, 319–328. [Google Scholar] [CrossRef]
- Considine, R.V. Regulation of leptin production. Rev. Endocr. Metab. Disord. 2001, 2, 357–363. [Google Scholar] [CrossRef]
- Park, H.K.; Ahima, R.S. Physiology of leptin: Energy homeostasis, neuroendocrine function and metabolism. Metabolism 2015, 64, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Lobo, A.M.; Donato, J., Jr. The role of leptin in health and disease. Temperature 2017, 4, 258–291. [Google Scholar] [CrossRef] [PubMed]
- Caron, A.; Lee, S.; Elmquist, J.K.; Gautron, L. Leptin and brain-adipose crosstalks. Nat. Rev. Neurosci. 2018, 19, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Lynes, M.D.; Tseng, Y.H. Deciphering adipose tissue heterogeneity. Ann. N. Y. Acad. Sci. 2018, 1411, 5–20. [Google Scholar] [CrossRef] [PubMed]
- Cinti, S. Adipose Organ Development and Remodeling. Compr. Physiol. 2018, 8, 1357–1431. [Google Scholar] [PubMed]
- Picó, C.; Palou, M.; Pomar, C.A.; Rodríguez, A.M.; Palou, A. Leptin as a key regulator of the adipose organ. Rev. Endocr. Metab. Disord. 2022, 23, 13–30. [Google Scholar] [CrossRef]
- Sasaki, T. Age-Associated Weight Gain, Leptin, and SIRT1: A Possible Role for Hypothalamic SIRT1 in the Prevention of Weight Gain and Aging through Modulation of Leptin Sensitivity. Front. Endocrinol. 2015, 6, 109. [Google Scholar] [CrossRef]
- Zhao, S.; Kusminski, C.M.; Elmquist, J.K.; Scherer, P.E. Leptin: Less Is More. Diabetes 2020, 69, 823–829. [Google Scholar] [CrossRef]
- Sinha, M.K.; Sturis, J.; Ohannesian, J.; Magosin, S.; Stephens, T.; Heiman, M.L.; Polonsky, K.S.; Caro, J.F. Ultradian oscillations of leptin secretion in humans. Biochem. Biophys. Res. Commun. 1996, 228, 733–738. [Google Scholar] [CrossRef]
- Sukumaran, S.; Xue, B.; Jusko, W.J.; Dubois, D.C.; Almon, R.R. Circadian variations in gene expression in rat abdominal adipose tissue and relationship to physiology. Physiol. Genom. 2010, 42, 141–152. [Google Scholar] [CrossRef]
- Ansarin, A.; Mahdavi, A.M.; Javadivala, Z.; Shanehbandi, D.; Zarredar, H.; Ansarin, K. The cross-talk between leptin and circadian rhythm signaling proteins in physiological processes: A systematic review. Mol. Biol. Rep. 2023, 50, 10427–10443. [Google Scholar] [CrossRef]
- Cumin, F.; Baum, H.P.; Levens, N. Leptin is cleared from the circulation primarily by the kidney. Int. J. Obes. Relat. Metab. Disord. 1996, 20, 1120–1126. [Google Scholar] [PubMed]
- Gao, Q.; Wolfgang, M.J.; Neschen, S.; Morino, K.; Horvath, T.L.; Shulman, G.I.; Fu, X.Y. Disruption of neural signal transducer and activator of transcription 3 causes obesity, diabetes, infertility, and thermal dysregulation. Proc. Natl. Acad. Sci. USA 2004, 101, 4661–4666. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.B.S. Phosphorylation of STAT3 in hypothalamic nuclei is stimulated by lower doses of leptin than are needed to inhibit food intake. Am. J. Physiol. Endocrinol. Metab. 2021, 321, E190–E201. [Google Scholar] [CrossRef] [PubMed]
- Flak, J.N.; Myers, M.G., Jr. Minireview: CNS Mechanisms of Leptin Action. Mol. Endocrinol. 2016, 30, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Jiang, H.; Xu, X.; Duan, W.; Mattson, M.P. Leptin-mediated cell survival signaling in hippocampal neurons mediated by JAK STAT3 and mitochondrial stabilization. J. Biol. Chem. 2008, 283, 1754–1763. [Google Scholar] [CrossRef]
- Shanley, L.J.; Irving, A.J.; Harvey, J. Leptin enhances NMDA receptor function and modulates hippocampal synaptic plasticity. J. Neurosci. 2001, 21, RC186. [Google Scholar] [CrossRef]
- McGregor, G.; Harvey, J. Leptin Regulation of Synaptic Function at Hippocampal TA-CA1 and SC-CA1 Synapses: Implications for Health and Disease. Neurochem. Res. 2019, 44, 650–660. [Google Scholar] [CrossRef]
- Farr, S.A.; Yamada, K.A.; Butterfield, D.A.; Abdul, H.M.; Xu, L.; Miller, N.E.; Banks, W.A.; Morley, J.E. Obesity and hypertriglyceridemia produce cognitive impairment. Endocrinology 2008, 149, 2628–2636. [Google Scholar] [CrossRef]
- Watanabe, T.; Sakamoto, K. Meta-analysis of cognitive and behavioral tests in leptin- and leptin receptor-deficient mice. Neurosci. Res. 2021, 170, 217–235. [Google Scholar] [CrossRef]
- Bonds, J.A.; Shetti, A.; Stephen, T.K.L.; Bonini, M.G.; Minshall, R.D.; Lazarov, O. Deficits in hippocampal neurogenesis in obesity-dependent and -independent type-2 diabetes mellitus mouse models. Sci. Rep. 2020, 10, 16368. [Google Scholar] [CrossRef]
- Hou, L.; Klann, E. Activation of the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin signaling pathway is required for metabotropic glutamate receptor-dependent long-term depression. J. Neurosci. 2004, 24, 6352–6361. [Google Scholar] [CrossRef] [PubMed]
- Durakoglugil, M.; Irving, A.J.; Harvey, J. Leptin induces a novel form of NMDA receptor-dependent long-term depression. J. Neurochem. 2005, 95, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Moult, P.R.; Harvey, J. Regulation of glutamate receptor trafficking by leptin. Biochem. Soc. Trans. 2009, 37 Pt 6, 1364–1368. [Google Scholar] [CrossRef]
- Zhao, F.; Siu, J.J.; Huang, W.; Askwith, C.; Cao, L. Insulin Modulates Excitatory Synaptic Transmission and Synaptic Plasticity in the Mouse Hippocampus. Neuroscience 2019, 411, 237–254. [Google Scholar] [CrossRef] [PubMed]
- Kommaddi, R.P.; Gowaikar, R.; Haseena, P.A.; Diwakar, L.; Singh, K.; Mondal, A. Akt activation ameliorates deficits in hippocampal-dependent memory and activity-dependent synaptic protein synthesis in an Alzheimer’s disease mouse model. J. Biol. Chem. 2024, 300, 105619. [Google Scholar] [CrossRef]
- Rosenkranz, J.A.; Frick, A.; Johnston, D. Kinase-dependent modification of dendritic excitability after long-term potentiation. J. Physiol. 2009, 587, 115–125. [Google Scholar] [CrossRef]
- Zhang, F.; Chen, J. Leptin protects hippocampal CA1 neurons against ischemic injury. J. Neurochem. 2008, 107, 578–587. [Google Scholar] [CrossRef]
- Guimond, D.; Diabira, D.; Porcher, C.; Bader, F.; Ferrand, N.; Zhu, M.; Appleyard, S.M.; Wayman, G.A.; Gaiarsa, J.L. Leptin potentiates GABAergic synaptic transmission in the developing rodent hippocampus. Front. Cell. Neurosci. 2014, 8, 235. [Google Scholar] [CrossRef] [PubMed]
- Bland, T.; Sahin, G.S.; Zhu, M.; Dillon, C.; Impey, S.; Appleyard, S.M.; Wayman, G.A. USP8 Deubiquitinates the Leptin Receptor and Is Necessary for Leptin-Mediated Synapse Formation. Endocrinology 2019, 160, 1982–1998. [Google Scholar] [CrossRef]
- Benz, A.H.; Shajari, M.; Peruzki, N.; Dehghani, F.; Maronde, E. Early growth response-1 induction by fibroblast growth factor-1 via increase of mitogen-activated protein kinase and inhibition of protein kinase B in hippocampal neurons. Br. J. Pharmacol. 2010, 160, 1621–1630. [Google Scholar] [CrossRef]
- Mishra, P.; Narayanan, R. Stable continual learning through structured multiscale plasticity manifolds. Curr. Opin. Neurobiol. 2021, 70, 51–63. [Google Scholar] [CrossRef]
- Stephens, T.W.; Basinski, M.; Bristow, P.K.; Bue-Valleskey, J.M.; Burgett, S.G.; Craft, L.; Hale, J.; Hoffmann, J.; Hsiung, H.M.; Kriauciunas, A.; et al. The role of neuropeptide Y in the antiobesity action of the obese gene product. Nature 1995, 377, 530–532. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, T.M.; Mobbs, C.V. Hypothalamic agouti-related protein messenger ribonucleic acid is inhibited by leptin and stimulated by fasting. Endocrinology 1999, 140, 814–817. [Google Scholar] [CrossRef] [PubMed]
- Cowley, M.A.; Smart, J.L.; Rubinstein, M.; Cerdán, M.G.; Diano, S.; Horvath, T.L.; Cone, R.D.; Low, M.J. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature 2001, 411, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Gali Ramamoorthy, T.; Begum, G.; Harno, E.; White, A. Developmental programming of hypothalamic neuronal circuits: Impact on energy balance control. Front. Neurosci. 2015, 9, 126. [Google Scholar] [CrossRef]
- Timper, K.; Brüning, J.C. Hypothalamic circuits regulating appetite and energy homeostasis: Pathways to obesity. Dis. Model. Mech. 2017, 10, 679–689. [Google Scholar] [CrossRef]
- Ameroso, D.; Meng, A.; Chen, S.; Felsted, J.; Dulla, C.G.; Rios, M. Astrocytic BDNF signaling within the ventromedial hypothalamus regulates energy homeostasis. Nat. Metab. 2022, 4, 627–643. [Google Scholar] [CrossRef]
- Kanoski, S.E.; Meisel, R.L.; Mullins, A.J.; Davidson, T.L. The effects of energy-rich diets on discrimination reversal learning and on BDNF in the hippocampus and prefrontal cortex of the rat. Behav. Brain Res. 2007, 182, 57–66. [Google Scholar] [CrossRef]
- Park, H.R.; Park, M.; Choi, J.; Park, K.Y.; Chung, H.Y.; Lee, J. A high-fat diet impairs neurogenesis: Involvement of lipid peroxidation and brain-derived neurotrophic factor. Neurosci. Lett. 2010, 482, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Hoane, M.R.; Swan, A.A.; Heck, S.E. The effects of a high-fat sucrose diet on functional outcome following cortical contusion injury in the rat. Behav. Brain Res. 2011, 223, 119–124. [Google Scholar] [CrossRef]
- Scarpace, P.J.; Matheny, M.; Pollock, B.H.; Tümer, N. Leptin increases uncoupling protein expression and energy expenditure. Am. J. Physiol. 1997, 273 Pt 1, E226–E230. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.P.; Tall, A.R. Transcriptional profiling reveals global defects in energy metabolism, lipoprotein, and bile acid synthesis and transport with reversal by leptin treatment in ob/ob mouse liver. J. Biol. Chem. 2001, 276, 49066–49076. [Google Scholar] [CrossRef]
- Frühbeck, G.; Aguado, M.; Gómez-Ambrosi, J.; Martínez, J.A. Lipolytic effect of in vivo leptin administration on adipocytes of lean and ob/ob mice, but not db/db mice. Biochem. Biophys. Res. Commun. 1998, 250, 99–102. [Google Scholar] [CrossRef]
- Dube, J.J.; Bhatt, B.A.; Dedousis, N.; Bonen, A.; O’Doherty, R.M. Leptin, skeletal muscle lipids, and lipid-induced insulin resistance. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 293, R642–R650. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, L.L.; Fischer, M.A.; Lopaschuk, G.D. Leptin activates cardiac fatty acid oxidation independent of changes in the AMP-activated protein kinase-acetyl-CoA carboxylase-malonyl-CoA axis. J. Biol. Chem. 2002, 277, 29424–29430. [Google Scholar] [CrossRef] [PubMed]
- Rajapurohitam, V.; Gan, X.T.; Kirshenbaum, L.A.; Karmazyn, M. The obesity-associated peptide leptin induces hypertrophy in neonatal rat ventricular myocytes. Circ. Res. 2003, 93, 277–279. [Google Scholar] [CrossRef]
- Kanoski, S.E.; Hayes, M.R.; Greenwald, H.S.; Fortin, S.M.; Gianessi, C.A.; Gilbert, J.R.; Grill, H.J. Hippocampal leptin signaling reduces food intake and modulates food-related memory processing. Neuropsychopharmacology 2011, 36, 1859–1870. [Google Scholar] [CrossRef]
- Wayner, M.J.; Armstrong, D.L.; Phelix, C.F.; Oomura, Y. Orexin-A (Hypocretin-1) and leptin enhance LTP in the dentate gyrus of rats in vivo. Peptides 2004, 25, 991–996. [Google Scholar] [CrossRef]
- Farr, S.A.; Banks, W.A.; Morley, J.E. Effects of leptin on memory processing. Peptides 2006, 27, 1420–1425. [Google Scholar] [CrossRef]
- Oomura, Y.; Hori, N.; Shiraishi, T.; Fukunaga, K.; Takeda, H.; Tsuji, M.; Matsumiya, T.; Ishibashi, M.; Aou, S.; Li, X.L.; et al. Leptin facilitates learning and memory performance and enhances hippocampal CA1 long-term potentiation and CaMK II phosphorylation in rats. Peptides 2006, 27, 2738–2749. [Google Scholar] [CrossRef]
- O’Malley, D.; MacDonald, N.; Mizielinska, S.; Connolly, C.N.; Irving, A.J.; Harvey, J. Leptin promotes rapid dynamic changes in hippocampal dendritic morphology. Mol. Cell. Neurosci. 2007, 35, 559–572. [Google Scholar] [CrossRef] [PubMed]
- Porter, W.D.; Flatt, P.R.; Hölscher, C.; Gault, V.A. Liraglutide improves hippocampal synaptic plasticity associated with increased expression of Mash1 in ob/ob mice. Int. J. Obes. 2013, 37, 678–684. [Google Scholar] [CrossRef]
- Valladolid-Acebes, I.; Merino, B.; Principato, A.; Fole, A.; Barbas, C.; Lorenzo, M.P.; García, A.; Del Olmo, N.; Ruiz-Gayo, M.; Cano, V. High-fat diets induce changes in hippocampal glutamate metabolism and neurotransmission. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E396–E402. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, H.G.; Skau, S.; Nyberg, J. A lifetime perspective on risk factors for cognitive decline with a special focus on early events. Cereb. Circ. Cogn. Behav. 2024, 6, 100217. [Google Scholar] [CrossRef] [PubMed]
- Mou, Y.; Blok, E.; Barroso, M.; Jansen, P.W.; White, T.; Voortman, T. Dietary patterns, brain morphology and cognitive performance in children: Results from a prospective population-based study. Eur. J. Epidemiol. 2023, 38, 669–687. [Google Scholar] [CrossRef] [PubMed]
- Cao, G.Y.; Li, M.; Han, L.; Tayie, F.; Yao, S.S.; Huang, Z.; Ai, P.; Liu, Y.Z.; Hu, Y.H.; Xu, B. Dietary Fat Intake and Cognitive Function among Older Populations: A Systematic Review and Meta-Analysis. J. Prev. Alzheimer’s Dis. 2019, 6, 204–211. [Google Scholar] [CrossRef]
- Choi, S.H.; Hong, E.S.; Lim, S. Clinical implications of adipocytokines and newly emerging metabolic factors with relation to insulin resistance and cardiovascular health. Front. Endocrinol. 2013, 4, 97. [Google Scholar] [CrossRef]
- Bray, G.A.; Kim, K.K.; Wilding, J.P.H.; World Obesity Federation. Obesity: A chronic relapsing progressive disease process. A position statement of the World Obesity Federation. Obes. Rev. 2017, 18, 715–723. [Google Scholar] [CrossRef]
- Kawai, T.; Autieri, M.V.; Scalia, R. Adipose tissue inflammation and metabolic dysfunction in obesity. Am. J. Physiol. Cell. Physiol. 2021, 320, C375–C391. [Google Scholar] [CrossRef]
- Kojta, I.; Chacińska, M.; Błachnio-Zabielska, A. Obesity, Bioactive Lipids, and Adipose Tissue Inflammation in Insulin Resistance. Nutrients 2020, 12, 1305. [Google Scholar] [CrossRef]
- Knight, Z.A.; Hannan, K.S.; Greenberg, M.L.; Friedman, J.M. Hyperleptinemia is required for the development of leptin resistance. PLoS ONE 2010, 5, e11376. [Google Scholar] [CrossRef] [PubMed]
- Mori, H.; Hanada, R.; Hanada, T.; Aki, D.; Mashima, R.; Nishinakamura, H.; Torisu, T.; Chien, K.R.; Yasukawa, H.; Yoshimura, A. Socs3 deficiency in the brain elevates leptin sensitivity and confers resistance to diet-induced obesity. Nat. Med. 2004, 10, 739–743. [Google Scholar] [CrossRef] [PubMed]
- Bence, K.K.; Delibegovic, M.; Xue, B.; Gorgun, C.Z.; Hotamisligil, G.S.; Neel, B.G.; Kahn, B.B. Neuronal PTP1B regulates body weight, adiposity and leptin action. Nat. Med. 2006, 12, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Wauman, J.; Tavernier, J. Leptin receptor signaling: Pathways to leptin resistance. Front. Biosci. 2011, 16, 2771–2793. [Google Scholar] [CrossRef]
- Barnard, N.D.; Bunner, A.E.; Agarwal, U. Saturated and trans fats and dementia: A systematic review. Neurobiol. Aging 2014, 35 (Suppl. S2), S65–S73. [Google Scholar] [CrossRef]
- Corley, J.; Cox, S.R.; Taylor, A.M.; Hernandez, M.V.; Maniega, S.M.; Ballerini, L.; Wiseman, S.; Meijboom, R.; Backhouse, E.V.; Bastin, M.E.; et al. Dietary patterns, cognitive function, and structural neuroimaging measures of brain aging. Exp. Gerontol. 2020, 142, 111117. [Google Scholar] [CrossRef]
- Sharma, A.N.; Elased, K.M.; Garrett, T.L.; Lucot, J.B. Neurobehavioral deficits in db/db diabetic mice. Physiol. Behav. 2010, 101, 381–388. [Google Scholar] [CrossRef]
- Morales, L.; Del Olmo, N.; Valladolid-Acebes, I.; Fole, A.; Cano, V.; Merino, B.; Stucchi, P.; Ruggieri, D.; López, L.; Alguacil, L.F.; et al. Shift of circadian feeding pattern by high-fat diets is coincident with reward deficits in obese mice. PLoS ONE 2012, 7, e36139. [Google Scholar] [CrossRef]
- Davidson, T.L.; Chan, K.; Jarrard, L.E.; Kanoski, S.E.; Clegg, D.J.; Benoit, S.C. Contributions of the hippocampus and medial prefrontal cortex to energy and body weight regulation. Hippocampus 2009, 19, 235–252. [Google Scholar] [CrossRef]
- Stranahan, A.M.; Mattson, M.P. Impact of energy intake and expenditure on neuronal plasticity. Neuromol. Med. 2008, 10, 209–218. [Google Scholar] [CrossRef]
- Eichen, D.M.; Kang Sim, D.E.; Appleton-Knapp, S.L.; Strong, D.R.; Boutelle, K.N. Adults with overweight or obesity use less efficient memory strategies compared to adults with healthy weight on a verbal list learning task modified with food words. Appetite 2023, 181, 106402. [Google Scholar] [CrossRef] [PubMed]
- Hayes, A.M.R.; Lauer, L.T.; Kao, A.E.; Sun, S.; Klug, M.E.; Tsan, L.; Rea, J.J.; Subramanian, K.S.; Gu, C.; Tanios, N.; et al. Western diet consumption impairs memory function via dysregulated hippocampus acetylcholine signaling. Brain Behav. Immun. 2024, 118, 408–422. [Google Scholar] [CrossRef] [PubMed]
- Avena, N.M.; Bocarsly, M.E.; Hoebel, B.G. Animal models of sugar and fat bingeing: Relationship to food addiction and increased body weight. Methods Mol. Biol. 2012, 829, 351–365. [Google Scholar] [PubMed]
- Epstein, L.H.; Biondolillo, M.J.; Rizwan, A.; Ghanim, H.; Dandona, P.; Bickel, W.K.; Paluch, R.A. Insulin Resistance and Glycated Hemoglobin in Obesity Are Associated With Preference for Sugar-Sweetened Yogurt: A Pilot Study. Psychosom. Med. 2023, 85, 289–293. [Google Scholar] [CrossRef]
- Greenwood, C.E.; Winocur, G. High-fat diets, insulin resistance and declining cognitive function. Neurobiol. Aging 2005, 26 (Suppl. S1), 42–45. [Google Scholar] [CrossRef]
- Lindqvist, A.; Mohapel, P.; Bouter, B.; Frielingsdorf, H.; Pizzo, D.; Brundin, P.; Erlanson-Albertsson, C. High-fat diet impairs hippocampal neurogenesis in male rats. Eur. J. Neurol. 2006, 13, 1385–1388. [Google Scholar] [CrossRef]
- Erichsen, J.M.; Fadel, J.R.; Reagan, L.P. Peripheral versus central insulin and leptin resistance: Role in metabolic disorders, cognition, and neuropsychiatric diseases. Neuropharmacology 2022, 203, 108877. [Google Scholar] [CrossRef]
- Mota, B.; Brás, A.R.; Araújo-Andrade, L.; Silva, A.; Pereira, P.A.; Madeira, M.D.; Cardoso, A. High-Caloric Diets in Adolescence Impair Specific GABAergic Subpopulations, Neurogenesis, and Alter Astrocyte Morphology. Int. J. Mol. Sci. 2024, 25, 5524. [Google Scholar] [CrossRef]
- Del Olmo, N.; Ruiz-Gayo, M. Influence of High-Fat Diets Consumed During the Juvenile Period on Hippocampal Morphology and Function. Front. Cell. Neurosci. 2018, 12, 439. [Google Scholar] [CrossRef]
- Freeman, L.R.; Haley-Zitlin, V.; Rosenberger, D.S.; Granholm, A.C. Damaging effects of a high-fat diet to the brain and cognition: A review of proposed mechanisms. Nutr. Neurosci. 2014, 17, 241–251. [Google Scholar] [CrossRef]
- Pantiya, P.; Thonusin, C.; Chunchai, T.; Ongnok, B.; Nawara, W.; Arunsak, B.; Chattipakorn, N.; Chattipakorn, S.C. Higher untrained fitness exerts a neuroprotection in Independence to caloric restriction or exercise in high-fat diet-induced obesity. Exp. Neurol. 2023, 365, 114416. [Google Scholar] [CrossRef] [PubMed]
- Paulo, S.L.; Miranda-Lourenço, C.; Belo, R.F.; Rodrigues, R.S.; Fonseca-Gomes, J.; Tanqueiro, S.R.; Geraldes, V.; Rocha, I.; Sebastião, A.M.; Xapelli, S.; et al. High Caloric Diet Induces Memory Impairment and Disrupts Synaptic Plasticity in Aged Rats. Curr. Issues Mol. Biol. 2021, 43, 2305–2319. [Google Scholar] [CrossRef] [PubMed]
- Akazawa, C.; Shigemoto, R.; Bessho, Y.; Nakanishi, S.; Mizuno, N. Differential expression of five N-methyl-D-aspartate receptor subunit mRNAs in the cerebellum of developing and adult rats. J. Comp. Neurol. 1994, 347, 150–160. [Google Scholar] [CrossRef] [PubMed]
- Monyer, H.; Burnashev, N.; Laurie, D.J.; Sakmann, B.; Seeburg, P.H. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron 1994, 12, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Law, A.J.; Weickert, C.S.; Webster, M.J.; Herman, M.M.; Kleinman, J.E.; Harrison, P.J. Changes in NMDA receptor subunit mRNAs and cyclophilin mRNA during development of the human hippocampus. Ann. N. Y. Acad. Sci. 2003, 1003, 426–430. [Google Scholar] [CrossRef]
- Huang, X.T.; Yue, S.J.; Li, C.; Guo, J.; Huang, Y.H.; Han, J.Z.; Feng, D.D.; Luo, Z.Q. Antenatal blockade of N-methyl-D-aspartate receptors by Memantine reduces the susceptibility to diabetes induced by a high-fat diet in rats with intrauterine growth restriction. Biol. Reprod. 2017, 96, 960–970. [Google Scholar] [CrossRef] [PubMed]
- Hermanussen, M.; Tresguerres, J.A. A new anti-obesity drug treatment: First clinical evidence that, antagonising glutamate-gated Ca2+ ion channels with memantine normalises binge-eating disorders. Econ. Hum. Biol. 2005, 3, 329–337. [Google Scholar] [CrossRef]
- Labban, R.S.M.; Alfawaz, H.; Almnaizel, A.T.; Hassan, W.M.; Bhat, R.S.; Moubayed, N.M.; Bjørklund, G.; El-Ansary, A. High-fat diet-induced obesity and impairment of brain neurotransmitter pool. Transl. Neurosci. 2020, 11, 147–160. [Google Scholar] [CrossRef]
- Boswell, R.G.; Potenza, M.N.; Grilo, C.M. The Neurobiology of Binge-eating Disorder Compared with Obesity: Implications for Differential Therapeutics. Clin. Ther. 2021, 43, 50–69. [Google Scholar] [CrossRef]
- Gómez-Apo, E.; Mondragón-Maya, A.; Ferrari-Díaz, M.; Silva-Pereyra, J. Structural Brain Changes Associated with Overweight and Obesity. J. Obes. 2021, 2021, 6613385. [Google Scholar] [CrossRef]
- McCabe, D.P.; Roediger, H.L.; McDaniel, M.A.; Balota, D.A.; Hambrick, D.Z. The relationship between working memory capacity and executive functioning: Evidence for a common executive attention construct. Neuropsychology 2010, 24, 222–243. [Google Scholar] [CrossRef] [PubMed]
- Higgs, S.; Spetter, M.S.; Thomas, J.M.; Rotshtein, P.; Lee, M.; Hallschmid, M.; Dourish, C.T. Interactions between metabolic, reward and cognitive processes in appetite control: Implications for novel weight management therapies. J. Psychopharmacol. 2017, 31, 1460–1474. [Google Scholar] [CrossRef] [PubMed]
- Davidson, T.L.; Jones, S.; Roy, M.; Stevenson, R.J. The Cognitive Control of Eating and Body Weight: It’s More Than What You “Think”. Front. Psychol. 2019, 10, 62. [Google Scholar] [CrossRef] [PubMed]
- Favieri, F.; Forte, G.; Casagrande, M. The Executive Functions in Overweight and Obesity: A Systematic Review of Neuropsychological Cross-Sectional and Longitudinal Studies. Front. Psychol. 2019, 10, 2126. [Google Scholar] [CrossRef] [PubMed]
- Yeomans, M.R.; Armitage, R.; Atkinson, R.; Francis, H.; Stevenson, R.J. Habitual intake of fat and sugar is associated with poorer memory and greater impulsivity in humans. PLoS ONE 2023, 18, e0290308. [Google Scholar] [CrossRef]
- Flores-Cordero, J.A.; Pérez-Pérez, A.; Jiménez-Cortegana, C.; Alba, G.; Flores-Barragán, A.; Sánchez-Margalet, V. Obesity as a Risk Factor for Dementia and Alzheimer’s Disease: The Role of Leptin. Int. J. Mol. Sci. 2022, 23, 5202. [Google Scholar] [CrossRef]
- Patel, V.; Edison, P. Cardiometabolic risk factors and neurodegeneration: A review of the mechanisms underlying diabetes, obesity and hypertension in Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 2024, 95, 581–589. [Google Scholar] [CrossRef]
- Maioli, S.; Lodeiro, M.; Merino-Serrais, P.; Falahati, F.; Khan, W.; Puerta, E.; Codita, A.; Rimondini, R.; Ramirez, M.J.; Simmons, A.; et al. Alterations in brain leptin signalling in spite of unchanged CSF leptin levels in Alzheimer’s disease. Aging Cell 2015, 14, 122–129. [Google Scholar] [CrossRef]
- Elinav, E.; Niv-Spector, L.; Katz, M.; Price, T.O.; Ali, M.; Yacobovitz, M.; Solomon, G.; Reicher, S.; Lynch, J.L.; Halpern, Z.; et al. Pegylated leptin antagonist is a potent orexigenic agent: Preparation and mechanism of activity. Endocrinology 2009, 150, 3083–3091. [Google Scholar] [CrossRef]
- Malekizadeh, Y.; Holiday, A.; Redfearn, D.; Ainge, J.A.; Doherty, G.; Harvey, J. A Leptin Fragment Mirrors the Cognitive Enhancing and Neuroprotective Actions of Leptin. Cereb. Cortex 2017, 27, 4769–4782. [Google Scholar] [CrossRef]
- Doherty, G.; Holiday, A.; Malekizadeh, Y.; Manolescu, C.; Duncan, S.; Flewitt, I.; Hamilton, K.; MacLeod, B.; Ainge, J.A.; Harvey, J. Leptin-based hexamers facilitate memory and prevent amyloid-driven AMPA receptor internalisation and neuronal degeneration. J. Neurochem. 2023, 165, 809–826. [Google Scholar] [CrossRef] [PubMed]
- Matochik, J.A.; London, E.D.; Yildiz, B.O.; Ozata, M.; Caglayan, S.; DePaoli, A.M.; Wong, M.L.; Licinio, J. Effect of leptin replacement on brain structure in genetically leptin-deficient adults. J. Clin. Endocrinol. Metab. 2005, 90, 2851–2854. [Google Scholar] [CrossRef]
- Dalamaga, M.; Chou, S.H.; Shields, K.; Papageorgiou, P.; Polyzos, S.A.; Mantzoros, C.S. Leptin at the intersection of neuroendocrinology and metabolism: Current evidence and therapeutic perspectives. Cell Metab. 2013, 18, 29–42. [Google Scholar] [CrossRef] [PubMed]
- Paz-Filho, G.J. The Effects of Leptin Replacement on Neural Plasticity. Neural Plast. 2016, 2016, 8528934. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.Y.; Kim, C.S.; Frazer, A.; Zhang, W. Leptin: A potential novel antidepressant. Proc. Natl. Acad. Sci. USA 2006, 103, 1593–1598. [Google Scholar] [CrossRef]
- London, E.D.; Berman, S.M.; Chakrapani, S.; Delibasi, T.; Monterosso, J.; Erol, H.K.; Paz-Filho, G.; Wong, M.L.; Licinio, J. Short-term plasticity of gray matter associated with leptin deficiency and replacement. J. Clin. Endocrinol. Metab. 2011, 96, E1212–E1220, Erratum in J. Clin. Endocrinol. Metab. 2011, 96, 3576. [Google Scholar] [CrossRef]
- Garza, J.C.; Guo, M.; Zhang, W.; Lu, X.Y. Leptin increases adult hippocampal neurogenesis in vivo and in vitro. J. Biol. Chem. 2008, 283, 18238–18247. [Google Scholar] [CrossRef]
- Irving, A.J.; Harvey, J. Leptin regulation of hippocampal synaptic function in health and disease. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2013, 369, 20130155. [Google Scholar] [CrossRef]
- Forny-Germano, L.; De Felice, F.G.; Vieira, M.N.D.N. The Role of Leptin and Adiponectin in Obesity-Associated Cognitive Decline and Alzheimer’s Disease. Front. Neurosci. 2019, 12, 1027. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Ferrannini, E.; Groop, L.; Henry, R.R.; Herman, W.H.; Holst, J.J.; Hu, F.B.; Kahn, C.R.; Raz, I.; Shulman, G.I.; et al. Type 2 diabetes mellitus. Nat. Rev. Dis. Primers 2015, 1, 15019. [Google Scholar] [CrossRef]
- Müller, T.D.; Blüher, M.; Tschöp, M.H.; DiMarchi, R.D. Anti-obesity drug discovery: Advances and challenges. Nat. Rev. Drug Discov. 2022, 21, 201–223. [Google Scholar] [CrossRef] [PubMed]
- Moghazy, H.M.; Abdelhaliem, N.G.; Mohammed, S.A.; Hassan, A.; Abdelrahman, A. Liraglutide versus pramlintide in protecting against cognitive function impairment through affecting PI3K/AKT/GSK-3β/TTBK1 pathway and decreasing Tau hyperphosphorylation in high-fat diet-streptozocin rat model. Pflug. Arch. 2024, 476, 779–795. [Google Scholar] [CrossRef] [PubMed]
- Patrick, S.; Corrigan, R.; Grizzanti, J.; Mey, M.; Blair, J.; Pallas, M.; Camins, A.; Lee, H.G.; Casadesus, G. Neuroprotective Effects of the Amylin Analog, Pramlintide, on Alzheimer’s Disease Are Associated with Oxidative Stress Regulation Mechanisms. J. Alzheimer’s Dis. 2019, 69, 157–168. [Google Scholar] [CrossRef]
- Qiu, W.Q.; Zhu, H. Amylin and its analogs: A friend or foe for the treatment of Alzheimer’s disease? Front. Aging Neurosci. 2014, 6, 186. [Google Scholar] [CrossRef]
- D’Ascanio, A.M.; Mullally, J.A.; Frishman, W.H. Cagrilintide: A Long-Acting Amylin Analog for the Treatment of Obesity. Cardiol. Rev. 2024, 32, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Enebo, L.B.; Berthelsen, K.K.; Kankam, M.; Lund, M.T.; Rubino, D.M.; Satylganova, A.; Lau, D.C.W. Safety, tolerability, pharmacokinetics, and pharmacodynamics of concomitant administration of multiple doses of cagrilintide with semaglutide 2.4 mg for weight management: A randomised, controlled, phase 1b trial. Lancet 2021, 397, 1736–1748. [Google Scholar] [CrossRef]
- Gadde, K.M.; Allison, D.B. Long-acting amylin analogue for weight reduction. Lancet 2021, 398, 2132–2134. [Google Scholar] [CrossRef]
- Lau, D.C.W.; Erichsen, L.; Francisco, A.M.; Satylganova, A.; le Roux, C.W.; McGowan, B.; Pedersen, S.D.; Pietiläinen, K.H.; Rubino, D.; Batterham, R.L. Once-weekly cagrilintide for weight management in people with overweight and obesity: A multicentre, randomised, double-blind, placebo-controlled and active-controlled, dose-finding phase 2 trial. Lancet 2021, 398, 2160–2172. [Google Scholar] [CrossRef]
- Becerril, S.; Frühbeck, G. Cagrilintide plus semaglutide for obesity management. Lancet 2021, 397, 1687–1689. [Google Scholar] [CrossRef]
- Wu, T.; Gao, X.; Chen, M.; van Dam, R.M. Long-term effectiveness of diet-plus-exercise interventions vs. diet-only interventions for weight loss: A meta-analysis. Obes. Rev. 2009, 10, 313–323. [Google Scholar] [CrossRef]
- Miller, C.T.; Fraser, S.F.; Levinger, I.; Straznicky, N.E.; Dixon, J.B.; Reynolds, J.; Selig, S.E. The effects of exercise training in addition to energy restriction on functional capacities and body composition in obese adults during weight loss: A systematic review. PLoS ONE 2013, 8, e81692. [Google Scholar] [CrossRef]
- Khalafi, M.; Azali Alamdari, K.; Symonds, M.E.; Rohani, H.; Sakhaei, M.H. A comparison of the impact of exercise training with dietary intervention versus dietary intervention alone on insulin resistance and glucose regulation in individual with overweight or obesity: A systemic review and meta-analysis. Crit. Rev. Food Sci. Nutr. 2023, 63, 9349–9363. [Google Scholar] [CrossRef] [PubMed]
- Lecoultre, V.; Ravussin, E.; Redman, L.M. The fall in leptin concentration is a major determinant of the metabolic adaptation induced by caloric restriction independently of the changes in leptin circadian rhythms. J. Clin. Endocrinol. Metab. 2011, 96, E1512–E1516. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, K.C.D.C.; Pereira, R.M.; de Campos, T.D.P.; de Moura, R.F.; da Silva, A.S.R.; Cintra, D.E.; Ropelle, E.R.; Pauli, J.R.; de Araújo, M.B.; de Moura, L.P. The Role of Physical Exercise to Improve the Browning of White Adipose Tissue via POMC Neurons. Front. Cell. Neurosci. 2018, 12, 88. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Meng, F.; Lei, Y.; Liu, J.; Liu, J.; Zhang, J.; Liu, F.; Liu, C.; Guo, M.; Lu, X.Y. Leptin regulates exon-specific transcription of the Bdnf gene via epigenetic modifications mediated by an AKT/p300 HAT cascade. Mol. Psychiatry 2021, 26, 3701–3722. [Google Scholar] [CrossRef]
- Yu, W.H.; Kimura, M.; Walczewska, A.; Karanth, S.; McCann, S.M. Role of leptin in hypothalamic-pituitary function. Proc. Natl. Acad. Sci. USA 1997, 94, 1023–1028. [Google Scholar] [CrossRef] [PubMed]
- Ahima, R.S.; Prabakaran, D.; Mantzoros, C.; Qu, D.; Lowell, B.; Maratos-Flier, E.; Flier, J.S. Role of leptin in the neuroendocrine response to fasting. Nature 1996, 382, 250–252. [Google Scholar] [CrossRef]
- Harvey, J. Novel Leptin-Based Therapeutic Strategies to Limit Synaptic Dysfunction in Alzheimer’s Disease. Int. J. Mol. Sci. 2024, 25, 7352. [Google Scholar] [CrossRef]
- Horie, N.C.; Serrao, V.T.; Simon, S.S.; Gascon, M.R.; Dos Santos, A.X.; Zambone, M.A.; Del Bigio de Freitas, M.M.; Cunha-Neto, E.; Marques, E.L.; Halpern, A.; et al. Cognitive Effects of Intentional Weight Loss in Elderly Obese Individuals With Mild Cognitive Impairment. J. Clin. Endocrinol. Metab. 2016, 101, 1104–1112. [Google Scholar] [CrossRef]
- Wittekind, D.A.; Kratzsch, J.; Mergl, R.; Baber, R.; Wirkner, K.; Schroeter, M.L.; Witte, A.V.; Villringer, A.; Kluge, M. Leptin, but not ghrelin, is associated with food addiction scores in a population-based subject sample. Front. Psychiatry 2023, 14, 1200021. [Google Scholar] [CrossRef]
- Peters, T.; Antel, J.; Föcker, M.; Esber, S.; Hinney, A.; Schéle, E.; Dickson, S.L.; Albayrak, Ö.; Hebebrand, J. The association of serum leptin levels with food addiction is moderated by weight status in adolescent psychiatric inpatients. Eur. Eat. Disord. Rev. 2018, 26, 618–628. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). Available online: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight (accessed on 14 October 2024).

{kind=link}
{kind=link}
| NCT Number Phase/Design | Drug Name(s) | Condition(s) | Primary Outcome(s) | Sponsor | Status | Last Updated Posted | |
|---|---|---|---|---|---|---|---|
| Pramlintide | |||||||
| 1 | NCT03560960 Early Phase I/Diagnostic | Pramlintide (Symlin) (monotherapy) | Alzheimer´s Disease; Mild Cognitive Impairment; | Plasma Aβ and t-tau changes. Plasma inflammatory changes. Metabolic changes in blood. All parameters determined 5, 30, 60, and 180 min after a Pramlintide challenge test. | Boston University | Recruiting | 31 July 2024 |
| 2 | NCT00691158 N/A/RTBPC | Pramlintide (monotherapy or combined with Metreleptin) or placebo | Obesity | Hypothalamic, brainstem and whole brain fMRI response measurements to different treatments. | Oregon Health and Science University | Active, not recruiting | 16 December 2022 |
| 3 | NCT00690235 IV RDBPC | Pramlintide (monotherapy) or placebo | Schizophrenia; Schizoaffective Disorder; Diabetes; Weight Gain | Weight loss from Pramlintide in Olanzapine- or Clozapine-induced weight gain patients with schizophrenia | University of Texas Southwestern Medical Center | Completed | 16 November 2018 |
| 4 | NCT00189514 II/RDBPC | Pramlintide acetate (monotherapy) or placebo | Obesity | Long-term effects of Pramlintide on body weight in obese subjects. Long-term safety and tolerability of the drug in obese patients. | AstraZeneca | Completed | 11 June 2015 |
| 5 | NCT00444561 II/RDBPC | Pramlintide acetate (AC137, monotherapy) or placebo | Overweight; Obesity | Pharmacodynamics of Pramlintide in obese subjects. | AstraZeneca | Completed | 11 June 2015 |
| 6 | NCT00673387 II/RDBPC | Pramlintide acetate (Smylin) or placebo versus Metreleptin or placebo | Overweight; Obesity | Measurement of the mean percentage of change in body weight from baseline to week 28 of the study. | AstraZeneca | Completed | 15 April 2015 |
| 7 | NCT00819234 II/RTBPC | Pramlintide (combined with Metreleptin) or placebo | Obesity | Measurement of the mean percentage of change in body weight from the baseline of the original study (NCT00673387) at week 52 in the extension study. | AstraZeneca | Completed | 15 April 2015 |
| 8 | NCT01235741 II/RDBPC | Pramlintide (combined with Metreleptin) or placebo | Obesity | Effect of 16 weeks of treatment on body weight in subjects with obesity. Studies of safety and tolerability of the drug combination. | AstraZeneca | Terminated | 15 April 2015 |
| 9 | NCT0039292 IIa/RDBPC | Pramlintide acetate (Smylin) or placebo; Metreleptin or placebo | Overweight; Obesity | Effect of 16 weeks of treatment on body weight in participants with overweight and/or obesity. | AstraZeneca | Completed | 14 April 2015 |
| 10 | NCT00112021 IIb/RDBPC | Pramlintide acetate (monotherapy) or placebo | Obesity | Effect of 16 weeks of treatment on body weight in subjects with obesity. Studies of safety and tolerability of Pramlintide in participants with obesity. | AstraZeneca | Completed | 10 April 2015 |
| 11 | NCT00402077 II/RSBPC | Pramlintide acetate (Symlin) compared to Sibutramine, Phentermine or placebo | Overweight; Obesity | Determination of treatment-related adverse events during 24-week treatment period. Change in body weight from baseline to week 12 of treatment. | AstraZeneca | Completed | 6 March 2015 |
| Cagrilintide | |||||||
| 12 | NCT06131372 II/RQBPC | CagriSema or placebo versus active comparators [Cagrilintide or Semaglutide, monotherapies] or placebo | Chronic Kidney Disease; Type 2 Diabetes; Obesity | Change in urinary albumin-to-creatinine ratio (UACR) from baseline (week 0) to end of treatment (week 26). | Novo Nordisk A/S | Recruiting | 26 August 2024 |
| 13 | NCT06388187 III/RQBPC | CagriSema (two different doses) or placebo (two diferent doses) | Obesity | Body weight changes from baseline (week 0) to end of treatment (week 68). Achievement of ≥5% weight reduction from baseline to the end of the treatmen. | Novo Nordisk A/S | Recruiting | 20 August 2024 |
| 14 | NCT06131437 III/ROL | CagriSema versus active comparator (Tirzepatide) | Obesity | Change in body weight from baseline (week 0) to end of treatment (week 72). | Novo Nordisk A/S | Active, not recruiting | 20 August 2024 |
| 15 | NCT05567796 (REDEFINE 1) III/RQBPC | CagriSema or placebo versus active comparators [Cagrilintide or Semaglutide, monotherapies] | Obesity | Body weight changes from baseline (week 0) to end of treatment (week 68). Achievement of ≥5% weight reduction from baseline to the end of the treatment. | Novo Nordisk A/S | Active, not recruiting | 20 August 2024 |
| 16 | NCT05813925 III/RQBPC | CagriSema versus active comparator (Semaglutide) | Overweight; Obesity | Body weight changes from baseline (week 0) to end of treatment (week 68). | Novo Nordisk A/S | Recruiting | 6 August 2024 |
| 17 | NCT05394519 (REDEFINE 2) III/RQBPC | CagriSema or placebo | Overweight; Obesity; Type 2 Diabetes Mellitus | Body weight changes from baseline (week 0) to end of treatment (week 68). Achievement of ≥5% weight reduction from baseline to the end of the treatment. | Novo Nordisk A/S | Active, not recruiting | 17 July 2024 |
| 18 | NCT05996848 (REDEFINE 6) III/RDBPC | CagriSema versus active comparator [Semaglutide, monotherapy] or placebo | Overweight; Obesity | Body weight changes from baseline (week 0) to end of treatment (week 68). Achievement of ≥5% weight reduction from baseline to the end of the treatment. | Novo Nordisk A/S | Recruiting | 1 July 2024 |
| 19 | NCT05804162 I/RQBPC | Cagrilintide or placebo versus Moxifloxacin and placebo | Obesity | Change from baseline in Fredericia’s heart rate-corrected QT interval (QTcF). | Novo Nordisk A/S | Completed | 30 October 2023 |
| 20 | NCT04940078 I/ROL | Cagrilintide (monotherapy) versus CagriSema (Cagrilintide+Semaglutide) | Overweight; Obesity | Pharmacokinetic characterization of treatments in the subject population. | Novo Nordisk A/S | Completed | 20 February 2024 |
| 21 | NCT06289504 I/ROL | CagriSema +Atorvastatin + Warfarin versus Semaglutide +Atorvastatin + Warfarin | Obesity | Pharmacokinetic characterization of treatments in the subject population. | Novo Nordisk A/S | Recruiting | 14 May 2024 |
| 22 | NCT06207877 I/RQBPC | CagriSema or placebo | Obesity | Changes in energy intake during ad libitum lunch, evening meal and snackbox from baseline to day 156 of the study. | Novo Nordisk A/S | Recruiting | 15 March 2024 |
| 23 | NCT06267092 I/RQBPC | CagriSema or placebo | Overweight; Obesity | Change in mean postprandial appetite score based on visual analog scale (VAS) from baseline to week 24 of the study. | Novo Nordisk A/S | Recruiting | 28 February 2024 |
| Davalintide (AC2307) | |||||||
| 24 | NCT00785408 II/RDBPC | AC2307 or placebo | Overweight; Obesity | Changes in body weight from baseline to week 24 of the study. Studies of safety and tolerability. | AstraZeneca | Completed | 19 January 2015 |
| GUBamy (GUB014295) | |||||||
| 25 | NCT06144684 I/RDBPC | GUB014295 (GUBamy) or placebo | Healthy Volunteers; Overweight | Adverse events (AE) incidence. Changes in vital signs. Safety laboratory parameters. All safety parameters determined from baseline (day 0) to the end of the trial’s part 1 (day 29), part 2A (day 64) and part 2B (day 106). | Gubra A/S | Recruiting | 21 August 2024 |
| Celastrol | |||||||
| 26 | NCT05494112 N/A/Safety OL | Celastrol (dietary supplement) | Healthy Volunteers | Effect of Celastrol in on liver function. | Legend Labz, Inc. | Unknown status | 9 September 2022 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valladolid-Acebes, I. Hippocampal Leptin Resistance and Cognitive Decline: Mechanisms, Therapeutic Strategies and Clinical Implications. Biomedicines 2024, 12, 2422. https://doi.org/10.3390/biomedicines12112422
Valladolid-Acebes I. Hippocampal Leptin Resistance and Cognitive Decline: Mechanisms, Therapeutic Strategies and Clinical Implications. Biomedicines. 2024; 12(11):2422. https://doi.org/10.3390/biomedicines12112422
Chicago/Turabian StyleValladolid-Acebes, Ismael. 2024. "Hippocampal Leptin Resistance and Cognitive Decline: Mechanisms, Therapeutic Strategies and Clinical Implications" Biomedicines 12, no. 11: 2422. https://doi.org/10.3390/biomedicines12112422
APA StyleValladolid-Acebes, I. (2024). Hippocampal Leptin Resistance and Cognitive Decline: Mechanisms, Therapeutic Strategies and Clinical Implications. Biomedicines, 12(11), 2422. https://doi.org/10.3390/biomedicines12112422