
Unraveling the Potential of ALK-Targeted Therapies in Non-Small Cell Lung Cancer: Comprehensive Insights and Future Directions

 ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
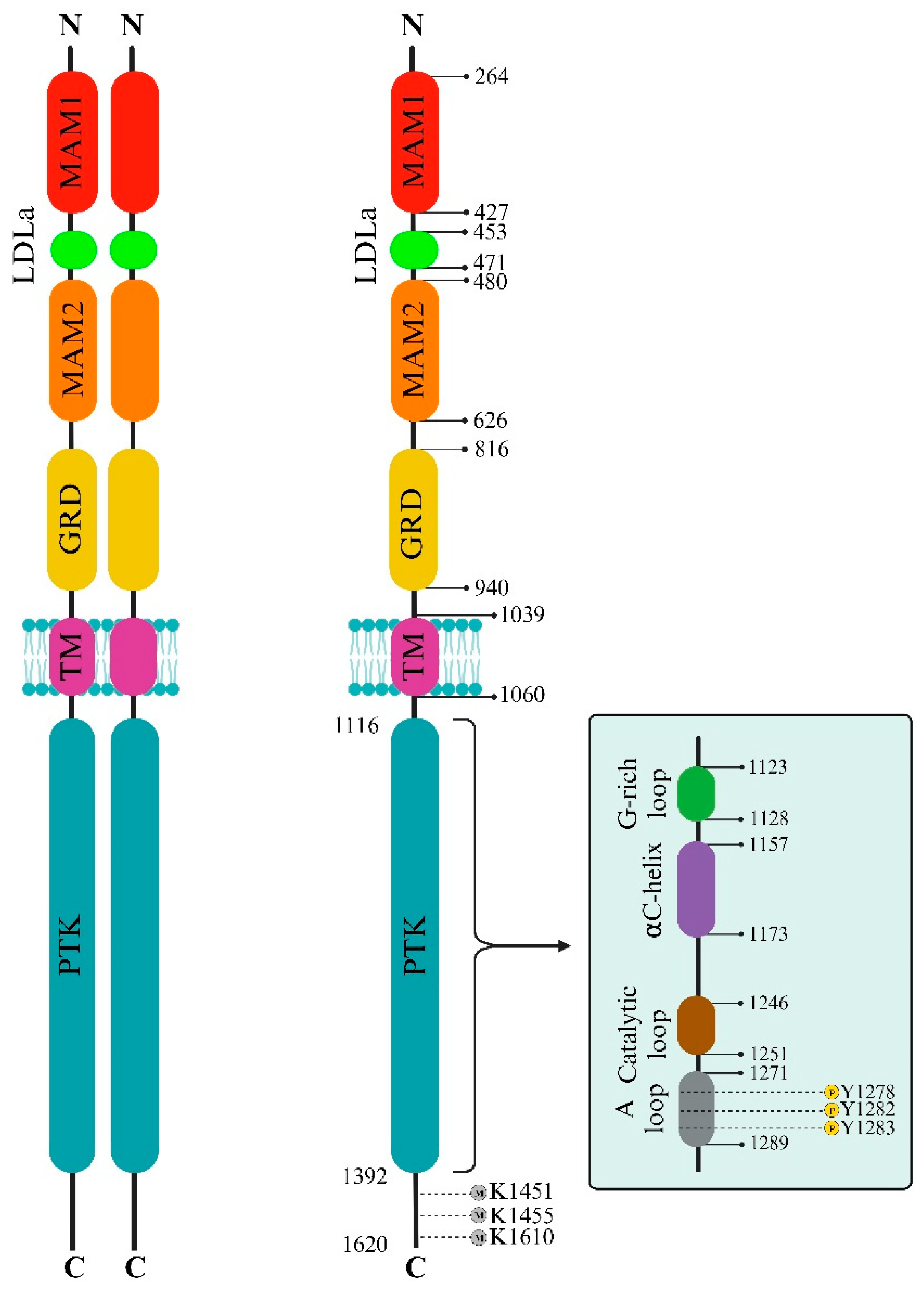
2. The ALK Structural Biology
2.1. ALK Extracellular Side
2.2. ALK Intracellular Side
2.3. Crizotinib and ALK Inhibition versus c-MET
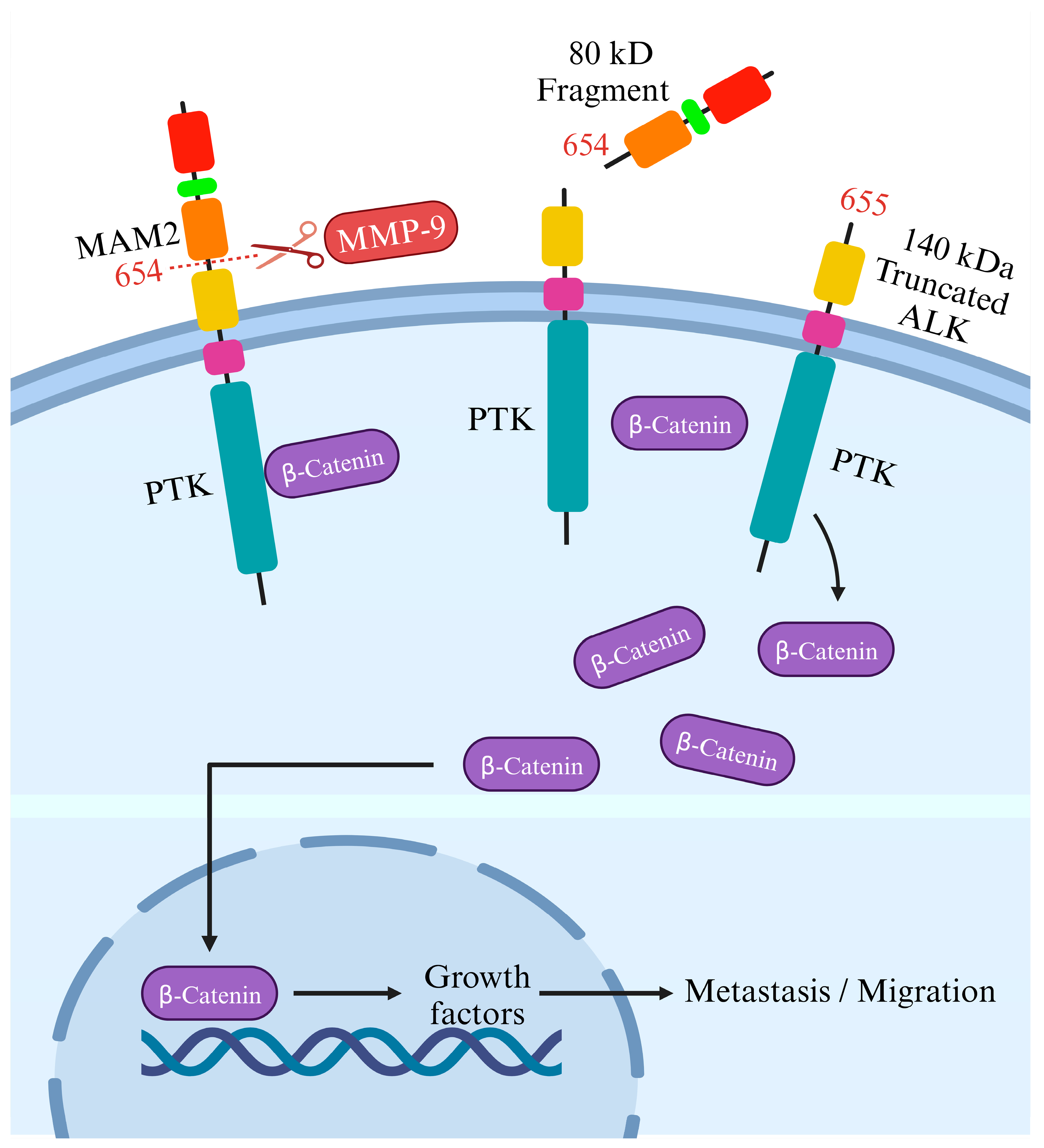
3. ALK Cleavage and Modifications
4. ALK Signaling and TKI Resistance
5. FDA-Approved ALK Inhibitors
5.1. Main Clinical Trials
5.2. Efficacy and Tolerability Profiles
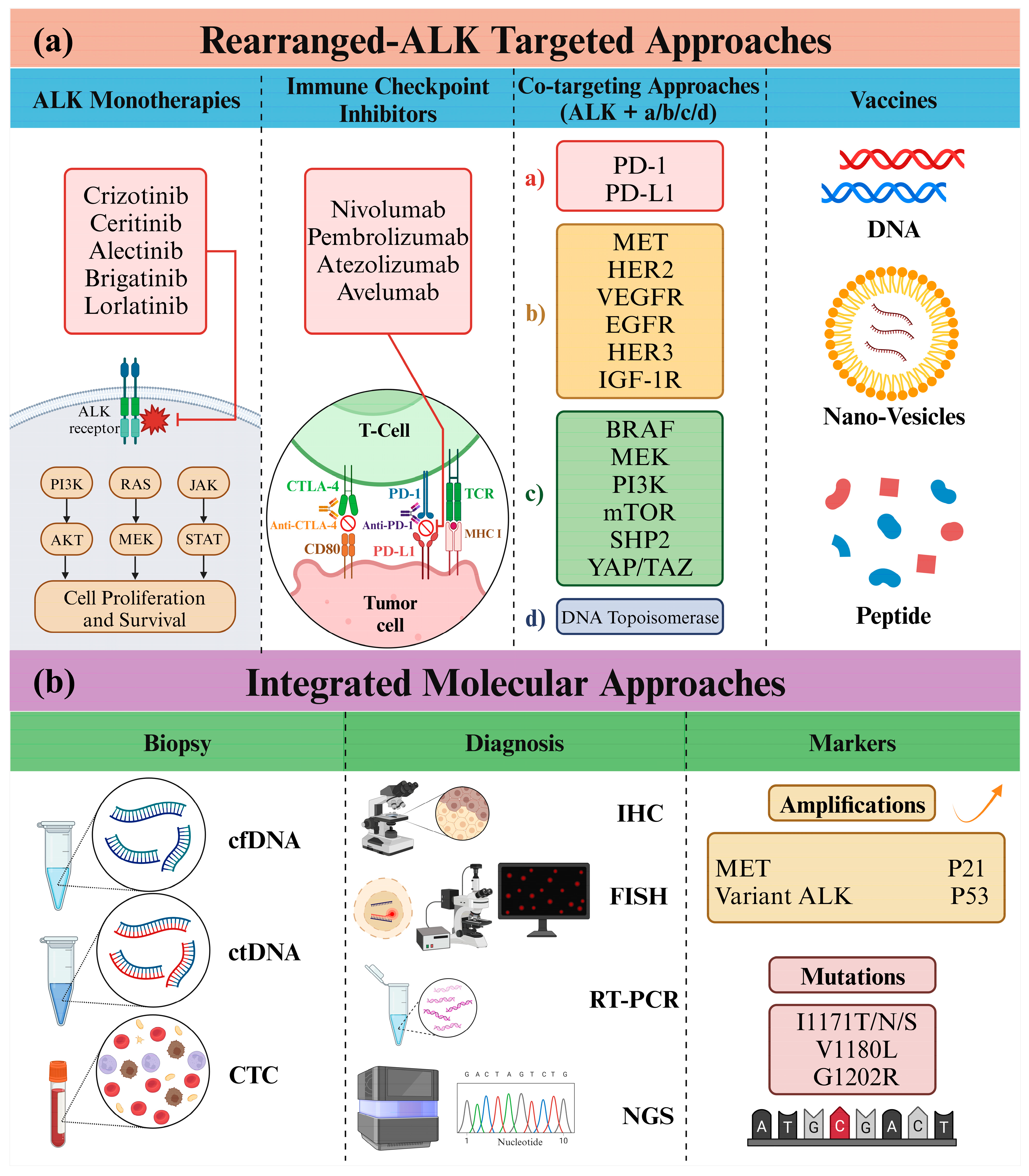
6. Advancements in ALK-Targeted Therapy and Future Horizons
6.1. ALK and Immunotherapy
6.2. Molecular Diagnosis of ALK: Insights from Next-Generation Sequencing
6.3. ALK and Co-Targeting Approaches
6.4. Other ALK-Innovative Approaches
7. Concluding Remarks
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Akt | Protein Kinase B |
| ALCL | Anaplastic large cell lymphoma |
| ALK | Anaplastic lymphoma kinase |
| CDK | Cyclin-dependent kinase |
| c-MET | Mesenchymal-epithelial transition factor |
| ECD | Extracellular domain |
| EGFR | Epidermal growth factor receptor |
| EML4 | Echinoderm microtubule-associated protein-like 4 |
| ERK | Extracellular Signal-Regulated Kinase |
| FDA | Food and Drug Administration |
| GRB2 | Growth factor receptor-bound protein 2 |
| GRD | Glycine-rich domain |
| ICI | Immune checkpoint inhibitor |
| LDLa | Low-density lipoprotein receptor class A |
| MAM | Meprin, A5 protein and receptor protein tyrosine phosphatase mu |
| MMP-9 | Matrix metallopeptidase 9 |
| NPM | Nucleolar phosphoprotein |
| NSCLC | Non-small cell lung cancer |
| ORR | Objective response rate |
| OS | Overall survival |
| PD-1 | Programmed cell death protein 1 |
| PD-L1 | Programmed cell death-1 ligand-1 |
| PFS | Progression-free survival |
| PI3K | Phosphatidylinositol 3 kinase |
| PTK | Protein tyrosine kinase |
| RAS | Rat sarcoma viral oncogene |
| STAT | Signal transducer and activator of transcription |
| TKI | Tyrosine kinase inhibitor |
| VEGF | Vascular endothelial growth factor |
References
- Panagiotidis, E. The Role of Positron Computed Tomography (PET/CT) in Lung Cancer Staging. Hell. J. Nucl. Med. 2023, 26, 22–29. [Google Scholar]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Hallberg, B.; Palmer, R.H. Mechanistic Insight into ALK Receptor Tyrosine Kinase in Human Cancer Biology. Nat. Rev. Cancer 2013, 13, 685–700. [Google Scholar] [CrossRef]
- Pearson, J.D.; Lee, J.K.H.; Bacani, J.T.C.; Lai, R.; Ingham, R.J. NPM-ALK: The Prototypic Member of a Family of Oncogenic Fusion Tyrosine Kinases. J. Signal Transduct. 2012, 2012, 123253. [Google Scholar] [CrossRef]
- Reshetnyak, A.V.; Rossi, P.; Myasnikov, A.G.; Sowaileh, M.; Mohanty, J.; Nourse, A.; Miller, D.J.; Lax, I.; Schlessinger, J.; Kalodimos, C.G. Mechanism for the Activation of the Anaplastic Lymphoma Kinase Receptor. Nature 2021, 600, 153–157. [Google Scholar] [CrossRef]
- Huang, H. Anaplastic Lymphoma Kinase (ALK) Receptor Tyrosine Kinase: A Catalytic Receptor with Many Faces. Int. J. Mol. Sci. 2018, 19, 3448. [Google Scholar] [CrossRef]
- Andraos, E.; Dignac, J.; Meggetto, F. NPM-ALK: A Driver of Lymphoma Pathogenesis and a Therapeutic Target. Cancers 2021, 13, 144. [Google Scholar] [CrossRef]
- Sankar, K.; Nagrath, S.; Ramnath, N. Immunotherapy for ALK-Rearranged Non-Small Cell Lung Cancer: Challenges Inform Promising Approaches. Cancers 2021, 13, 1476. [Google Scholar] [CrossRef]
- Cameron, L.B.; Hitchen, N.; Chandran, E.; Morris, T.; Manser, R.; Solomon, B.J.; Jordan, V. Targeted Therapy for Advanced Anaplastic Lymphoma Kinase (<I>ALK</I>)-Rearranged Non-Small Cell Lung Cancer. Cochrane Libr. Cochrane Rev. 2022, 1, CD013453. [Google Scholar] [CrossRef]
- Guo, Y.; Guo, H.; Zhang, Y.; Cui, J. Anaplastic Lymphoma Kinase-Special Immunity and Immunotherapy. Front. Immunol. 2022, 13, 908894. [Google Scholar] [CrossRef] [PubMed]
- Soda, M.; Choi, Y.L.; Enomoto, M.; Takada, S.; Yamashita, Y.; Ishikawa, S.; Fujiwara, S.-I.; Watanabe, H.; Kurashina, K.; Hatanaka, H.; et al. Identification of the Transforming EML4-ALK Fusion Gene in Non-Small-Cell Lung Cancer. Nature 2007, 448, 561–566. [Google Scholar] [CrossRef]
- Liu, S.; Huang, T.; Liu, M.; He, W.; Zhao, Y.; Yang, L.; Long, Y.; Zong, D.; Zeng, H.; Liu, Y.; et al. The Genomic Characteristics of ALK Fusion Positive Tumors in Chinese NSCLC Patients. Front. Oncol. 2020, 10, 726. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Yeap, B.Y.; Mino-Kenudson, M.; Digumarthy, S.R.; Costa, D.B.; Heist, R.S.; Solomon, B.; Stubbs, H.; Admane, S.; McDermott, U.; et al. Clinical Features and Outcome of Patients with Non–Small-Cell Lung Cancer Who Harbor EML4-ALK. J. Clin. Oncol. 2009, 27, 4247–4253. [Google Scholar] [CrossRef] [PubMed]
- Chia, P.L.; Mitchell, P.; Dobrovic, A.; John, T. Prevalence and Natural History of ALK Positive Non-Small-Cell Lung Cancer and the Clinical Impact of Targeted Therapy with ALK Inhibitors. Clin. Epidemiol. 2014, 6, 423–432. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.-H.I.; Lee, A.T.M.; Nagasaka, M. From Preclinical Efficacy to 2022 (36.7 Months Median Follow -up) Updated CROWN Trial, Lorlatinib Is the Preferred 1st-Line Treatment of Advanced ALK+ NSCLC. Crit. Rev. Oncol. 2023, 187, 104019. [Google Scholar] [CrossRef] [PubMed]
- Solomon, B.J.; Mok, T.; Kim, D.-W.; Wu, Y.-L.; Nakagawa, K.; Mekhail, T.; Felip, E.; Cappuzzo, F.; Paolini, J.; Usari, T.; et al. First-Line Crizotinib versus Chemotherapy in ALK-Positive Lung Cancer. N. Engl. J. Med. 2014, 371, 2167–2177. [Google Scholar] [CrossRef]
- Silva, A.P.S.; Coelho, P.V.; Anazetti, M.; Simioni, P.U. Targeted Therapies for the Treatment of Non-Small-Cell Lung Cancer: Monoclonal Antibodies and Biological Inhibitors. Hum. Vaccines Immunother. 2017, 13, 843–853. [Google Scholar] [CrossRef] [PubMed]
- Jahanzeb, M.; Lin, H.M.; Pan, X.; Yin, Y.; Baumann, P.; Langer, C.J. Immunotherapy Treatment Patterns and Outcomes Among ALK-Positive Patients with Non-Small-Cell Lung Cancer. Clin. Lung Cancer 2021, 22, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Mazieres, J.; Drilon, A.; Lusque, A.; Mhanna, L.; Cortot, A.B.; Mezquita, L.; Thai, A.A.; Mascaux, C.; Couraud, S.; Veillon, R.; et al. Immune Checkpoint Inhibitors for Patients with Advanced Lung Cancer and Oncogenic Driver Alterations: Results from the IMMUNOTARGET Registry. Ann. Oncol. 2019, 30, 1321–1328. [Google Scholar] [CrossRef]
- Tabbò, F.; Passiglia, F.; Novello, S. Upfront Management of ALK-Rearranged Metastatic Non-Small Cell Lung Cancer: One Inhibitor Fits All? Curr. Oncol. Rep. 2021, 23, 10. [Google Scholar] [CrossRef]
- Shaw, A.T.; Kim, T.M.; Crinò, L.; Gridelli, C.; Kiura, K.; Liu, G.; Novello, S.; Bearz, A.; Gautschi, O.; Mok, T.; et al. Ceritinib versus Chemotherapy in Patients with ALK-Rearranged Non-Small-Cell Lung Cancer Previously given Chemotherapy and Crizotinib (ASCEND-5): A Randomised, Controlled, Open-Label, Phase 3 Trial. Lancet. Oncol. 2017, 18, 874–886. [Google Scholar] [CrossRef]
- Gadgeel, S.M.; Gandhi, L.; Riely, G.J.; Chiappori, A.A.; West, H.L.; Azada, M.C.; Morcos, P.N.; Lee, R.-M.; Garcia, L.; Yu, L.; et al. Safety and Activity of Alectinib against Systemic Disease and Brain Metastases in Patients with Crizotinib-Resistant ALK-Rearranged Non-Small-Cell Lung Cancer (AF-002JG): Results from the Dose-Finding Portion of a Phase 1/2 Study. Lancet. Oncol. 2014, 15, 1119–1128. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.-H.I.; Ahn, J.S.; De Petris, L.; Govindan, R.; Yang, J.C.-H.; Hughes, B.; Lena, H.; Moro-Sibilot, D.; Bearz, A.; Ramirez, S.V.; et al. Alectinib in Crizotinib-Refractory ALK-Rearranged Non-Small-Cell Lung Cancer: A Phase II Global Study. J. Clin. Oncol. 2016, 34, 661–668. [Google Scholar] [CrossRef]
- Novello, S.; Mazières, J.; Oh, I.-J.; deCastro, J.; Migliorino, M.R.; Helland, Å.; Dziadziuszko, R.; Griesinger, F.; Kotb, A.; Zeaiter, A.; et al. Alectinib versus Chemotherapy in Crizotinib-Pretreated Anaplastic Lymphoma Kinase (ALK)-Positive Non-Small-Cell Lung Cancer: Results from the Phase III ALUR Study. Ann. Oncol. 2018, 29, 1409–1416. [Google Scholar] [CrossRef]
- Kim, D.-W.; Tiseo, M.; Ahn, M.-J.; Reckamp, K.L.; Hansen, K.H.; Kim, S.-W.; Huber, R.M.; West, H.L.; Groen, H.J.M.; Hochmair, M.J.; et al. Brigatinib in Patients with Crizotinib-Refractory Anaplastic Lymphoma Kinase-Positive Non-Small-Cell Lung Cancer: A Randomized, Multicenter Phase II Trial. J. Clin. Oncol. 2017, 35, 2490–2498. [Google Scholar] [CrossRef] [PubMed]
- Cognigni, V.; Pecci, F.; Lupi, A.; Pinterpe, G.; DeFilippis, C.; Felicetti, C.; Cantini, L.; Berardi, R. The Landscape of ALK-Rearranged Non-Small Cell Lung Cancer: A Comprehensive Review of Clinicopathologic, Genomic Characteristics, and Therapeutic Perspectives. Cancers 2022, 14, 4765. [Google Scholar] [CrossRef]
- Pan, Y.; Deng, C.; Qiu, Z.; Cao, C.; Wu, F. The Resistance Mechanisms and Treatment Strategies for ALK-Rearranged Non-Small Cell Lung Cancer. Front. Oncol. 2021, 11, 713530. [Google Scholar] [CrossRef]
- Palmer, R.H.; Vernersson, E.; Grabbe, C.; Hallberg, B. Anaplastic Lymphoma Kinase: Signalling in Development and Disease. Biochem. J. 2009, 420, 345–361. [Google Scholar] [CrossRef]
- Li, T.; Stayrook, S.E.; Tsutsui, Y.; Zhang, J.; Wang, Y.; Li, H.; Proffitt, A.; Krimmer, S.G.; Ahmed, M.; Belliveau, O.; et al. Structural Basis for Ligand Reception by Anaplastic Lymphoma Kinase. Nature 2021, 600, 148–152. [Google Scholar] [CrossRef]
- Guan, J.; Umapathy, G.; Yamazaki, Y.; Wolfstetter, G.; Mendoza, P.; Pfeifer, K.; Mohammed, A.; Hugosson, F.; Zhang, H.; Hsu, A.W.; et al. FAM150A and FAM150B Are Activating Ligands for Anaplastic Lymphoma Kinase. Elife 2015, 4, e09811. [Google Scholar] [CrossRef]
- Reshetnyak, A.V.; Murray, P.B.; Shi, X.; Mo, E.S.; Mohanty, J.; Tome, F.; Bai, H.; Gunel, M.; Lax, I.; Schlessinger, J. Augmentor α and β (FAM150) Are Ligands of the Receptor Tyrosine Kinases ALK and LTK: Hierarchy and Specificity of Ligand-Receptor Interactions. Proc. Natl. Acad. Sci. USA 2015, 112, 15862–15867. [Google Scholar] [CrossRef]
- Bossi, R.T.; Saccardo, M.B.; Ardini, E.; Menichincheri, M.; Rusconi, L.; Magnaghi, P.; Orsini, P.; Avanzi, N.; Borgia, A.L.; Nesi, M.; et al. Crystal Structures of Anaplastic Lymphoma Kinase in Complex with ATP Competitive Inhibitors. Biochemistry 2010, 49, 6813–6825. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.J.; Tran-Dubé, M.; Shen, H.; Nambu, M.; Kung, P.-P.; Pairish, M.; Jia, L.; Meng, J.; Funk, L.; Botrous, I.; et al. Structure Based Drug Design of Crizotinib (PF-02341066), a Potent and Selective Dual Inhibitor of Mesenchymal-Epithelial Transition Factor (c-MET) Kinase and Anaplastic Lymphoma Kinase (ALK). J. Med. Chem. 2011, 54, 6342–6363. [Google Scholar] [CrossRef]
- Huang, Q.; Johnson, T.W.; Bailey, S.; Brooun, A.; Bunker, K.D.; Burke, B.J.; Collins, M.R.; Cook, A.S.; Cui, J.J.; Dack, K.N.; et al. Design of Potent and Selective Inhibitors to Overcome Clinical Anaplastic Lymphoma Kinase Mutations Resistant to Crizotinib. J. Med. Chem. 2014, 57, 1170–1187. [Google Scholar] [CrossRef] [PubMed]
- Kornev, A.P.; Taylor, S.S. Dynamics-Driven Allostery in Protein Kinases. Trends Biochem. Sci. 2015, 40, 628–647. [Google Scholar] [CrossRef] [PubMed]
- Donella-Deana, A.; Marin, O.; Cesaro, L.; Gunby, R.H.; Ferrarese, A.; Coluccia, A.M.L.; Tartari, C.J.; Mologni, L.; Scapozza, L.; Gambacorti-Passerini, C.; et al. Unique Substrate Specificity of Anaplastic Lymphoma Kinase (ALK): Development of Phosphoacceptor Peptides for the Assay of ALK Activity. Biochemistry 2005, 44, 8533–8542. [Google Scholar] [CrossRef] [PubMed]
- Tartari, C.J.; Gunby, R.H.; Coluccia, A.M.L.; Sottocornola, R.; Cimbro, B.; Scapozza, L.; Donella-Deana, A.; Pinna, L.A.; Gambacorti-Passerini, C. Characterization of Some Molecular Mechanisms Governing Autoactivation of the Catalytic Domain of the Anaplastic Lymphoma Kinase. J. Biol. Chem. 2008, 283, 3743–3750. [Google Scholar] [CrossRef]
- Wang, P.; Wu, F.; Zhang, J.; McMullen, T.; Young, L.C.; Ingham, R.J.; Li, L.; Lai, R. Serine Phosphorylation of NPM-ALK, Which Is Dependent on the Auto-Activation of the Kinase Activation Loop, Contributes to Its Oncogenic Potential. Carcinogenesis 2011, 32, 146–153. [Google Scholar] [CrossRef]
- Hallberg, B.; Palmer, R.H. The Role of the ALK Receptor in Cancer Biology. Ann. Oncol. 2016, 27 (Suppl. 3), iii4–iii15. [Google Scholar] [CrossRef]
- Shaw, A.T.; Friboulet, L.; Leshchiner, I.; Gainor, J.F.; Bergqvist, S.; Brooun, A.; Burke, B.J.; Deng, Y.-L.; Liu, W.; Dardaei, L.; et al. Resensitization to Crizotinib by the Lorlatinib ALK Resistance Mutation L1198F. N. Engl. J. Med. 2016, 374, 54–61. [Google Scholar] [CrossRef]
- Loong, H.H.; Mok, K.; Leung, L.K.S.; Mok, T.S.K. Crizotinib in the Management of Advanced-Stage Non-Small-Cell Lung Cancer. Future Oncol. 2015, 11, 735–745. [Google Scholar] [CrossRef]
- Kazandjian, D.; Blumenthal, G.M.; Chen, H.-Y.; He, K.; Patel, M.; Justice, R.; Keegan, P.; Pazdur, R. FDA Approval Summary: Crizotinib for the Treatment of Metastatic Non-Small Cell Lung Cancer with Anaplastic Lymphoma Kinase Rearrangements. Oncologist 2014, 19, e5–e11. [Google Scholar] [CrossRef]
- Huang, X. The Potential Role of HGF-MET Signaling and Autophagy in the War of Alectinib versus Crizotinib against ALK-Positive NSCLC. J. Exp. Clin. Cancer Res. 2018, 37, 33. [Google Scholar] [CrossRef]
- Morris, S.W.; Naeve, C.; Mathew, P.; James, P.L.; Kirstein, M.N.; Cui, X.; Witte, D.P. ALK, the Chromosome 2 Gene Locus Altered by the t(2;5) in Non-Hodgkin’s Lymphoma, Encodes a Novel Neural Receptor Tyrosine Kinase That Is Highly Related to Leukocyte Tyrosine Kinase (LTK). Oncogene 1997, 14, 2175–2188. [Google Scholar] [CrossRef]
- Degoutin, J.; Brunet-de Carvalho, N.; Cifuentes-Diaz, C.; Vigny, M. ALK (Anaplastic Lymphoma Kinase) Expression in DRG Neurons and Its Involvement in Neuron-Schwann Cells Interaction. Eur. J. Neurosci. 2009, 29, 275–286. [Google Scholar] [CrossRef]
- Mazot, P.; Cazes, A.; Boutterin, M.C.; Figueiredo, A.; Raynal, V.; Combaret, V.; Hallberg, B.; Palmer, R.H.; Delattre, O.; Janoueix-Lerosey, I.; et al. The Constitutive Activity of the ALK Mutated at Positions F1174 or R1275 Impairs Receptor Trafficking. Oncogene 2011, 30, 2017–2025. [Google Scholar] [CrossRef] [PubMed]
- Mazot, P.; Cazes, A.; Dingli, F.; Degoutin, J.; Irinopoulou, T.; Boutterin, M.-C.; Lombard, B.; Loew, D.; Hallberg, B.; Palmer, R.H.; et al. Internalization and Down-Regulation of the ALK Receptor in Neuroblastoma Cell Lines upon Monoclonal Antibodies Treatment. PLoS ONE 2012, 7, e33581. [Google Scholar] [CrossRef]
- Pischedda, F.; Ghirelli, A.; Tripathi, V.; Piccoli, G. Negr1-Derived Peptides Trigger ALK Degradation and Halt Neuroblastoma Progression In Vitro and In Vivo. Pharmaceutics 2023, 15, 2307. [Google Scholar] [CrossRef]
- Venkannagari, H.; Kasper, J.M.; Misra, A.; Rush, S.A.; Fan, S.; Lee, H.; Sun, H.; Seshadrinathan, S.; Machius, M.; Hommel, J.D.; et al. Highly Conserved Molecular Features in IgLONs Contrast Their Distinct Structural and Biological Outcomes. J. Mol. Biol. 2020, 432, 5287–5303. [Google Scholar] [CrossRef] [PubMed]
- Moog-Lutz, C.; Degoutin, J.; Gouzi, J.Y.; Frobert, Y.; Brunet-de Carvalho, N.; Bureau, J.; Créminon, C.; Vigny, M. Activation and Inhibition of Anaplastic Lymphoma Kinase Receptor Tyrosine Kinase by Monoclonal Antibodies and Absence of Agonist Activity of Pleiotrophin. J. Biol. Chem. 2005, 280, 26039–26048. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Gont, A.; Kee, L.; Dries, R.; Pfeifer, K.; Sharma, B.; Debruyne, D.N.; Harlow, M.; Sengupta, S.; Guan, J.; et al. Extracellular Domain Shedding of the ALK Receptor Mediates Neuroblastoma Cell Migration. Cell Rep. 2021, 36, 109363. [Google Scholar] [CrossRef]
- DelGrosso, F.; DeMariano, M.; Passoni, L.; Luksch, R.; Tonini, G.P.; Longo, L. Inhibition of N-Linked Glycosylation Impairs ALK Phosphorylation and Disrupts pro-Survival Signaling in Neuroblastoma Cell Lines. BMC Cancer 2011, 11, 525. [Google Scholar] [CrossRef]
- Contessa, J.N.; Bhojani, M.S.; Freeze, H.H.; Rehemtulla, A.; Lawrence, T.S. Inhibition of N-Linked Glycosylation Disrupts Receptor Tyrosine Kinase Signaling in Tumor Cells. Cancer Res. 2008, 68, 3803–3809. [Google Scholar] [CrossRef]
- Zhou, W.; Yan, L.-D.; Yu, Z.-Q.; Li, N.; Yang, Y.-H.; Wang, M.; Chen, Y.-Y.; Mao, M.-X.; Peng, X.-C.; Cai, J. Role of STK11 in ALK-Positive Non-Small Cell Lung Cancer. Oncol. Lett. 2022, 23, 181. [Google Scholar] [CrossRef]
- Spitaleri, G.; Trillo Aliaga, P.; Attili, I.; Del Signore, E.; Corvaja, C.; Corti, C.; Crimini, E.; Passaro, A.; de Marinis, F. Sustained Improvement in the Management of Patients with Non-Small-Cell Lung Cancer (NSCLC) Harboring ALK Translocation: Where Are We Running? Curr. Oncol. 2023, 30, 5072–5092. [Google Scholar] [CrossRef]
- Lei, Y.; Lei, Y.; Shi, X.; Wang, J. EML4-ALK Fusion Gene in Non-Small Cell Lung Cancer. Oncol. Lett. 2022, 24, 277. [Google Scholar] [CrossRef]
- Smolle, E.; Taucher, V.; Lindenmann, J.; Jost, P.J.; Pichler, M. Current Knowledge about Mechanisms of Drug Resistance against ALK Inhibitors in Non-Small Cell Lung Cancer. Cancers 2021, 13, 699. [Google Scholar] [CrossRef]
- Elshatlawy, M.; Sampson, J.; Clarke, K.; Bayliss, R. EML4-ALK Biology and Drug Resistance in Non-Small Cell Lung Cancer: A New Phase of Discoveries. Mol. Oncol. 2023, 17, 950–963. [Google Scholar] [CrossRef] [PubMed]
- Dedoni, S.; Scherma, M.; Camoglio, C.; Siddi, C.; Fratta, W.; Fadda, P. Anaplastic Lymphoma Kinase Receptor: Possible Involvement in Anorexia Nervosa. Nutrients 2023, 15, 2205. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.-J.; Pan, W.-W.; Liu, S.-B.; Shen, Z.-F.; Xu, Y.; Hu, L.-L. ERK/MAPK Signalling Pathway and Tumorigenesis. Exp. Ther. Med. 2020, 19, 1997–2007. [Google Scholar] [CrossRef] [PubMed]
- Ducray, S.P.; Natarajan, K.; Garland, G.D.; Turner, S.D.; Egger, G. The Transcriptional Roles of ALK Fusion Proteins in Tumorigenesis. Cancers 2019, 11, 1074. [Google Scholar] [CrossRef]
- Xin, S.; Fang, W.; Li, J.; Li, D.; Wang, C.; Huang, Q.; Huang, M.; Zhuang, W.; Wang, X.; Chen, L. Impact of STAT1 Polymorphisms on Crizotinib-Induced Hepatotoxicity in ALK-Positive Non-Small Cell Lung Cancer Patients. J. Cancer Res. Clin. Oncol. 2021, 147, 725–737. [Google Scholar] [CrossRef]
- Barreca, A.; Lasorsa, E.; Riera, L.; Machiorlatti, R.; Piva, R.; Ponzoni, M.; Kwee, I.; Bertoni, F.; Piccaluga, P.P.; Pileri, S.A.; et al. Anaplastic Lymphoma Kinase in Human Cancer. J. Mol. Endocrinol. 2011, 47, R11–R23. [Google Scholar] [CrossRef] [PubMed]
- Unno, K.; Chalmers, Z.R.; Pamarthy, S.; Vatapalli, R.; Rodriguez, Y.; Lysy, B.; Mok, H.; Sagar, V.; Han, H.; Yoo, Y.A.; et al. Activated ALK Cooperates with N-Myc via Wnt/β-Catenin Signaling to Induce Neuroendocrine Prostate Cancer. Cancer Res. 2021, 81, 2157–2170. [Google Scholar] [CrossRef] [PubMed]
- Pilling, A.B.; Kim, J.; Estrada-Bernal, A.; Zhou, Q.; Le, A.T.; Singleton, K.R.; Heasley, L.E.; Tan, A.C.; DeGregori, J.; Doebele, R.C. ALK Is a Critical Regulator of the MYC-Signaling Axis in ALK Positive Lung Cancer. Oncotarget 2018, 9, 8823–8835. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, R.; Sasaki, T.; Minami, Y.; Hibino, Y.; Okumura, S.; Sado, M.; Miyokawa, N.; Hayashi, S.; Kitada, M.; Ohsaki, Y. Activation of Src Signaling Mediates Acquired Resistance to ALK Inhibition in Lung Cancer. Int. J. Oncol. 2017, 51, 1533–1540. [Google Scholar] [CrossRef]
- Zhao, Y.; Yang, Y.; Xu, Y.; Lu, S.; Jian, H. AZD0530 Sensitizes Drug-Resistant ALK-Positive Lung Cancer Cells by Inhibiting SRC Signaling. FEBS Open Bio 2017, 7, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Tanimura, K.; Yamada, T.; Horinaka, M.; Katayama, Y.; Fukui, S.; Morimoto, K.; Nakano, T.; Tokuda, S.; Morimoto, Y.; Iwasaku, M.; et al. Inhibition of C-Jun N-Terminal Kinase Signaling Increased Apoptosis and Prevented the Emergence of ALK-TKI-Tolerant Cells in ALK-Rearranged Non-Small Cell Lung Cancer. Cancer Lett. 2021, 522, 119–128. [Google Scholar] [CrossRef]
- Tanizaki, J.; Okamoto, I.; Okabe, T.; Sakai, K.; Tanaka, K.; Hayashi, H.; Kaneda, H.; Takezawa, K.; Kuwata, K.; Yamaguchi, H.; et al. Activation of HER Family Signaling as a Mechanism of Acquired Resistance to ALK Inhibitors in EML4-ALK–Positive Non–Small Cell Lung Cancer. Clin. Cancer Res. 2012, 18, 6219–6226. [Google Scholar] [CrossRef]
- Shen, J.; Meng, Y.; Wang, K.; Gao, M.; Du, J.; Wang, J.; Li, Z.; Zuo, D.; Wu, Y. EML4-ALK G1202R Mutation Induces EMT and Confers Resistance to Ceritinib in NSCLC Cells via Activation of STAT3/Slug Signaling. Cell Signal. 2022, 92, 110264. [Google Scholar] [CrossRef]
- Tanizaki, J.; Okamoto, I.; Takezawa, K.; Sakai, K.; Azuma, K.; Kuwata, K.; Yamaguchi, H.; Hatashita, E.; Nishio, K.; Janne, P.A.; et al. Combined Effect of ALK and MEK Inhibitors in EML4-ALK-Positive Non-Small-Cell Lung Cancer Cells. Br. J. Cancer 2012, 106, 763–767. [Google Scholar] [CrossRef]
- Wang, R.; Deng, X.; Yoshioka, Y.; Vougiouklakis, T.; Park, J.-H.; Suzuki, T.; Dohmae, N.; Ueda, K.; Hamamoto, R.; Nakamura, Y. Effects of SMYD2-Mediated EML4-ALK Methylation on the Signaling Pathway and Growth in Non-Small-Cell Lung Cancer Cells. Cancer Sci. 2017, 108, 1203–1209. [Google Scholar] [CrossRef]
- Huang, M.-H.; Lee, J.-H.; Hung, P.-S.; Yang, J.C.-H. Potential Therapeutic Strategy for EGFR-Mutant Lung Cancer with Concomitant EML4-ALK Rearrangement-Combination of EGFR Tyrosine Kinase Inhibitors and ALK Inhibitors. JTO Clin. Res. Rep. 2022, 3, 100405. [Google Scholar] [CrossRef]
- Reyes, R.; Reguart, N. Neoadjuvant Treatment of Stage IIIA-N2 in EGFR-Mutant/ALK-Rearranged Non-Small Cell Lung Cancer. Transl. Lung Cancer Res. 2021, 10, 607–621. [Google Scholar] [CrossRef]
- Gou, W.; Li, Z.; Xu, X.; Shen, J.; Guo, M.; Zhou, X.; Zhang, X.; Wu, Y.; Zhai, X.; Zuo, D. ZX-29, a Novel ALK Inhibitor, Induces Apoptosis via ER Stress in ALK Rearrangement NSCLC Cells and Overcomes Cell Resistance Caused by an ALK Mutation. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2020, 1867, 118712. [Google Scholar] [CrossRef]
- Lu, Y.; Fan, Z.; Zhu, S.-J.; Huang, X.; Zhuang, Z.; Li, Y.; Deng, Z.; Gao, L.; Hong, X.; Zhang, T.; et al. A New ALK Inhibitor Overcomes Resistance to First- and Second-Generation Inhibitors in NSCLC. EMBO Mol. Med. 2022, 14, e14296. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-T.; Chiang, C.-L.; Hung, J.-Y.; Lee, M.-H.; Su, W.-C.; Wu, S.-Y.; Wei, Y.-F.; Lee, K.-Y.; Tseng, Y.-H.; Su, J.; et al. Resistance Profiles of Anaplastic Lymphoma Kinase Tyrosine Kinase Inhibitors in Advanced Non-Small-Cell Lung Cancer: A Multicenter Study Using Targeted next-Generation Sequencing. Eur. J. Cancer 2021, 156, 1–11. [Google Scholar] [CrossRef]
- Chu, Y.-Y.; Chen, M.-K.; Wei, Y.; Lee, H.-H.; Xia, W.; Wang, Y.-N.; Yam, C.; Hsu, J.L.; Wang, H.-L.; Chang, W.-C.; et al. Targeting the ALK-CDK9-Tyr19 Kinase Cascade Sensitizes Ovarian and Breast Tumors to PARP Inhibition via Destabilization of the P-TEFb Complex. Nat. Cancer 2022, 3, 1211–1227. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Yu, B.; Xu, Y.; Du, Z.; Zhang, Z.; Wang, Y.; Chen, H.; Zhang, L.A.; Chen, R.; Ma, F.; et al. Discovery of Selective and Potent Macrocyclic CDK9 Inhibitors for the Treatment of Osimertinib-Resistant Non-Small-Cell Lung Cancer. J. Med. Chem. 2023, 66, 15340–15361. [Google Scholar] [CrossRef]
- Chazan, G.; Solomon, B.J. Optimal First-Line Treatment for Metastatic ALK+ Non-Small Cell Lung Cancer—A Narrative Review. Transl. Lung Cancer Res. 2023, 12, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Knoll, S.; Bocharova, I.; Tang, W.; Signorovitch, J. Comparative Efficacy of First-Line Ceritinib and Crizotinib in Advanced or Metastatic Anaplastic Lymphoma Kinase-Positive Non-Small Cell Lung Cancer: An Adjusted Indirect Comparison with External Controls. Curr. Med. Res. Opin. 2019, 35, 105–111. [Google Scholar] [CrossRef]
- Wu, Y.-L.; Lu, S.; Lu, Y.; Zhou, J.; Shi, Y.-K.; Sriuranpong, V.; Ho, J.C.M.; Ong, C.K.; Tsai, C.-M.; Chung, C.-H.; et al. Results of PROFILE 1029, a Phase III Comparison of First-Line Crizotinib versus Chemotherapy in East Asian Patients with ALK-Positive Advanced Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2018, 13, 1539–1548. [Google Scholar] [CrossRef] [PubMed]
- Peters, S.; Camidge, D.R.; Shaw, A.T.; Gadgeel, S.; Ahn, J.S.; Kim, D.-W.; Ou, S.-H.I.; Pérol, M.; Dziadziuszko, R.; Rosell, R.; et al. Alectinib versus Crizotinib in Untreated ALK-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 377, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Gadgeel, S.; Peters, S.; Mok, T.; Shaw, A.T.; Kim, D.W.; Ou, S.I.; Pérol, M.; Wrona, A.; Novello, S.; Rosell, R.; et al. Alectinib versus Crizotinib in Treatment-Naive Anaplastic Lymphoma Kinase-Positive (ALK+) Non-Small-Cell Lung Cancer: CNS Efficacy Results from the ALEX Study. Ann. Oncol. 2018, 29, 2214–2222. [Google Scholar] [CrossRef] [PubMed]
- Dziadziuszko, R.; Peters, S.; Mok, T.; Camidge, D.R.; Gadgeel, S.M.; Ou, S.-H.I.; Konopa, K.; Noé, J.; Nowicka, M.; Bordogna, W.; et al. Circulating Cell-Free DNA as a Prognostic Biomarker in Patients with Advanced ALK+ Non-Small Cell Lung Cancer in the Global Phase III ALEX Trial. Clin. Cancer Res. 2022, 28, 1800–1808. [Google Scholar] [CrossRef] [PubMed]
- Mok, T.; Peters, S.; Camidge, D.R.; Noé, J.; Gadgeel, S.; Ou, S.-H.I.; Kim, D.-W.; Konopa, K.; Pozzi, E.; Liu, T.; et al. Outcomes According to ALK Status Determined by Central Immunohistochemistry or Fluorescence In Situ Hybridization in Patients With ALK-Positive NSCLC Enrolled in the Phase 3 ALEX Study. J. Thorac. Oncol. 2021, 16, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Dziadziuszko, R.; Peters, S.; Ruf, T.; Cardona, A.; Guerini, E.; Kurtsikidze, N.; Smoljanovic, V.; Planchard, D. Clinical Experience and Management of Adverse Events in Patients with Advanced ALK-Positive Non-Small-Cell Lung Cancer Receiving Alectinib. ESMO Open 2022, 7, 100612. [Google Scholar] [CrossRef] [PubMed]
- Camidge, D.R.; Kim, H.R.; Ahn, M.-J.; Yang, J.C.-H.; Han, J.-Y.; Lee, J.-S.; Hochmair, M.J.; Li, J.Y.-C.; Chang, G.-C.; Lee, K.H.; et al. Brigatinib versus Crizotinib in ALK-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 379, 2027–2039. [Google Scholar] [CrossRef]
- Camidge, D.R.; Kim, H.R.; Ahn, M.-J.; Yang, J.C.H.; Han, J.-Y.; Hochmair, M.J.; Lee, K.H.; Delmonte, A.; García Campelo, M.R.; Kim, D.-W.; et al. Brigatinib Versus Crizotinib in Advanced ALK Inhibitor-Naive ALK-Positive Non-Small Cell Lung Cancer: Second Interim Analysis of the Phase III ALTA-1L Trial. J. Clin. Oncol. 2020, 38, 3592–3603. [Google Scholar] [CrossRef]
- Camidge, D.R.; Kim, H.R.; Ahn, M.-J.; Yang, J.C.H.; Han, J.-Y.; Hochmair, M.J.; Lee, K.H.; Delmonte, A.; Garcia Campelo, M.R.; Kim, D.-W.; et al. Brigatinib Versus Crizotinib in ALK Inhibitor-Naive Advanced ALK-Positive NSCLC: Final Results of Phase 3 ALTA-1L Trial. J. Thorac. Oncol. 2021, 16, 2091–2108. [Google Scholar] [CrossRef]
- Ng, T.L.; Narasimhan, N.; Gupta, N.; Venkatakrishnan, K.; Kerstein, D.; Camidge, D.R. Early-Onset Pulmonary Events Associated With Brigatinib Use in Advanced NSCLC. J. Thorac. Oncol. 2020, 15, 1190–1199. [Google Scholar] [CrossRef]
- Popat, S.; Liu, G.; Lu, S.; Song, G.; Ma, X.; Yang, J.C.-H. Brigatinib vs Alectinib in Crizotinib-Resistant Advanced Anaplastic Lymphoma Kinase-Positive Non-Small-Cell Lung Cancer (ALTA-3). Future Oncol. 2021, 17, 4237–4247. [Google Scholar] [CrossRef]
- Soria, J.-C.; Tan, D.S.W.; Chiari, R.; Wu, Y.-L.; Paz-Ares, L.; Wolf, J.; Geater, S.L.; Orlov, S.; Cortinovis, D.; Yu, C.-J.; et al. First-Line Ceritinib versus Platinum-Based Chemotherapy in Advanced ALK-Rearranged Non-Small-Cell Lung Cancer (ASCEND-4): A Randomised, Open-Label, Phase 3 Study. Lancet 2017, 389, 917–929. [Google Scholar] [CrossRef] [PubMed]
- Kiura, K.; Imamura, F.; Kagamu, H.; Matsumoto, S.; Hida, T.; Nakagawa, K.; Satouchi, M.; Okamoto, I.; Takenoyama, M.; Fujisaka, Y.; et al. Phase 3 Study of Ceritinib vs Chemotherapy in ALK-Rearranged NSCLC Patients Previously Treated with Chemotherapy and Crizotinib (ASCEND-5): Japanese Subset. Jpn. J. Clin. Oncol. 2018, 48, 367–375. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Woo, J.; Kim, S. A Systematic Review of Companion Diagnostic Tests by Immunohistochemistry for the Screening of Alectinib-Treated Patients in ALK-Positive Non-Small Cell Lung Cancer. Diagnostics 2022, 12, 1297. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Bauer, T.M.; de Marinis, F.; Felip, E.; Goto, Y.; Liu, G.; Mazieres, J.; Kim, D.-W.; Mok, T.; Polli, A.; et al. First-Line Lorlatinib or Crizotinib in Advanced ALK-Positive Lung Cancer. N. Engl. J. Med. 2020, 383, 2018–2029. [Google Scholar] [CrossRef] [PubMed]
- Mazieres, J.; Iadeluca, L.; Shaw, A.T.; Solomon, B.J.; Bauer, T.M.; deMarinis, F.; Felip, E.; Goto, Y.; Kim, D.-W.; Mok, T.; et al. Patient-Reported Outcomes from the Randomized Phase 3 CROWN Study of First-Line Lorlatinib versus Crizotinib in Advanced ALK-Positive Non-Small Cell Lung Cancer. Lung Cancer 2022, 174, 146–156. [Google Scholar] [CrossRef]
- Fabbri, L.; DiFederico, A.; Astore, M.; Marchiori, V.; Rejtano, A.; Seminerio, R.; Gelsomino, F.; De Giglio, A. From Development to Place in Therapy of Lorlatinib for the Treatment of ALK and ROS1 Rearranged Non-Small Cell Lung Cancer (NSCLC). Diagnostics 2023, 14, 48. [Google Scholar] [CrossRef]
- Cui, S.; Zhao, Y.; Gu, A.; Ge, X.; Song, Y.; Zhang, W.; Lou, Y.; Dong, L.; Han, B.; Jiang, L. Efficacy and Tolerability of Crizotinib in the Treatment of ALK-Positive, Advanced Non-Small Cell Lung Cancer in Chinese Patients. Med. Oncol. 2015, 32, 626. [Google Scholar] [CrossRef]
- Ou, S.-H.I.; Socinski, M.A.; Gadgeel, S.; Gandhi, L.; West, H.; Chiappori, A.A.; Cohen, V.; Riely, G.J.; Smoljanovic, V.; Bordogna, W.; et al. Patient-Reported Outcomes in a Phase II, North American Study of Alectinib in Patients with ALK-Positive, Crizotinib-Resistant, Non-Small Cell Lung Cancer. ESMO Open 2018, 3, e000364. [Google Scholar] [CrossRef]
- Hida, T.; Nokihara, H.; Kondo, M.; Kim, Y.H.; Azuma, K.; Seto, T.; Takiguchi, Y.; Nishio, M.; Yoshioka, H.; Imamura, F.; et al. Alectinib versus Crizotinib in Patients with ALK-Positive Non-Small-Cell Lung Cancer (J-ALEX): An Open-Label, Randomised Phase 3 Trial. Lancet 2017, 390, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Alexander, M.; Wei, J.; Parakh, S.; John, T.; Kao, S.; Nagrial, A.; Bowyer, S.; Warburton, L.; Moore, M.; Hughes, B.G.M.; et al. LOREALAUS: LOrlatinib REAL-World AUStralian Experience in Advanced ALK-Rearranged NSCLC. JTO Clin. Res. Rep. 2023, 4, 100490. [Google Scholar] [CrossRef] [PubMed]
- Drilon, A.; Siena, S.; Ou, S.-H.I.; Patel, M.; Ahn, M.J.; Lee, J.; Bauer, T.M.; Farago, A.F.; Wheler, J.J.; Liu, S.V.; et al. Safety and Antitumor Activity of the Multitargeted Pan-TRK, ROS1, and ALK Inhibitor Entrectinib: Combined Results from Two Phase I Trials (ALKA-372-001 and STARTRK-1). Cancer Discov. 2017, 7, 400–409. [Google Scholar] [CrossRef]
- Demetri, G.D.; DeBraud, F.; Drilon, A.; Siena, S.; Patel, M.R.; Cho, B.C.; Liu, S.V.; Ahn, M.-J.; Chiu, C.-H.; Lin, J.J.; et al. Updated Integrated Analysis of the Efficacy and Safety of Entrectinib in Patients with NTRK Fusion-Positive Solid Tumors. Clin. Cancer Res. 2022, 28, 1302–1312. [Google Scholar] [CrossRef]
- Zhang, B.; Zeng, J.; Zhang, H.; Zhu, S.; Wang, H.; He, J.; Yang, L.; Zhou, N.; Zu, L.; Xu, X.; et al. Characteristics of the Immune Microenvironment and Their Clinical Significance in Non-Small Cell Lung Cancer Patients with ALK-Rearranged Mutation. Front. Immunol. 2022, 13, 974581. [Google Scholar] [CrossRef]
- Liu, S.-Y.; Dong, Z.-Y.; Wu, S.-P.; Xie, Z.; Yan, L.-X.; Li, Y.-F.; Yan, H.-H.; Su, J.; Yang, J.-J.; Zhou, Q.; et al. Clinical Relevance of PD-L1 Expression and CD8+ T Cells Infiltration in Patients with EGFR-Mutated and ALK-Rearranged Lung Cancer. Lung Cancer 2018, 125, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Zheng, N.; Zhang, Y.; Zeng, Y.; Ma, Q.; Zhang, R.; Zhao, Q.; Lu, C.; Tian, J.; Wang, Z.; Tang, H.; et al. Pathological Response and Tumor Immune Microenvironment Remodeling Upon Neoadjuvant ALK-TKI Treatment in ALK-Rearranged Non-Small Cell Lung Cancer. Target. Oncol. 2023, 18, 625–636. [Google Scholar] [CrossRef]
- Mukherjee, S.; Patra, R.; Behzadi, P.; Masotti, A.; Paolini, A.; Sarshar, M. Toll-like Receptor-Guided Therapeutic Intervention of Human Cancers: Molecular and Immunological Perspectives. Front. Immunol. 2023, 14, 1244345. [Google Scholar] [CrossRef]
- Janik, J.E.; Morris, J.C.; Pittaluga, S.; McDonald, K.; Raffeld, M.; Jaffe, E.S.; Grant, N.; Gutierrez, M.; Waldmann, T.A.; Wilson, W.H. Elevated Serum-Soluble Interleukin-2 Receptor Levels in Patients with Anaplastic Large Cell Lymphoma. Blood 2004, 104, 3355–3357. [Google Scholar] [CrossRef]
- Matsuyama, H.; Suzuki, H.I.; Nishimori, H.; Noguchi, M.; Yao, T.; Komatsu, N.; Mano, H.; Sugimoto, K.; Miyazono, K. MiR-135b Mediates NPM-ALK-Driven Oncogenicity and Renders IL-17-Producing Immunophenotype to Anaplastic Large Cell Lymphoma. Blood 2011, 118, 6881–6892. [Google Scholar] [CrossRef]
- Knörr, F.; Damm-Welk, C.; Ruf, S.; Singh, V.K.; Zimmermann, M.; Reiter, A.; Woessmann, W. Blood Cytokine Concentrations in Pediatric Patients with Anaplastic Lymphoma Kinase-Positive Anaplastic Large Cell Lymphoma. Haematologica 2018, 103, 477–485. [Google Scholar] [CrossRef]
- Angeles, A.K.; Janke, F.; Daum, A.-K.; Reck, M.; Schneider, M.A.; Thomas, M.; Christopoulos, P.; Sültmann, H. Integrated Circulating Tumour DNA and Cytokine Analysis for Therapy Monitoring of ALK-Rearranged Lung Adenocarcinoma. Br. J. Cancer 2023, 129, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Li, Y.; Huang, Q.; Zeng, H.; Wei, Q.; Tian, P. High PD-L1 Expression Correlates with an Immunosuppressive Tumour Immune Microenvironment and Worse Prognosis in ALK-Rearranged Non-Small Cell Lung Cancer. Biomolecules 2023, 13, 991. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Chen, N.; Fang, W.; Zhan, J.; Liu, Q.; Kang, S.; He, X.; Liu, L.; Zhou, T.; Huang, J.; et al. Upregulation of PD-L1 by EML4-ALK Fusion Protein Mediates the Immune Escape in ALK Positive NSCLC: Implication for Optional Anti-PD-1/PD-L1 Immune Therapy for ALK-TKIs Sensitive and Resistant NSCLC Patients. Oncoimmunology 2016, 5, e1094598. [Google Scholar] [CrossRef] [PubMed]
- Jabbarzadeh Kaboli, P.; Shabani, S.; Sharma, S.; Partovi Nasr, M.; Yamaguchi, H.; Hung, M.-C. Shedding Light on Triple-Negative Breast Cancer with Trop2-Targeted Antibody-Drug Conjugates. Am. J. Cancer Res. 2022, 12, 1671–1685. [Google Scholar] [PubMed]
- Zhao, Q.; Guo, J.; Zhao, Y.; Shen, J.; Kaboli, P.J.; Xiang, S.; Du, F.; Wu, X.; Li, M.; Wan, L.; et al. Comprehensive Assessment of PD-L1 and PD-L2 Dysregulation in Gastrointestinal Cancers. Epigenomics 2020, 12, 2155–2171. [Google Scholar] [CrossRef] [PubMed]
- Riudavets, M.; Auclin, E.; Mosteiro, M.; Dempsey, N.; Majem, M.; Lobefaro, R.; López-Castro, R.; Bosch-Barrera, J.; Pilotto, S.; Escalera, E.; et al. Durvalumab Consolidation in Patients with Unresectable Stage III Non-Small Cell Lung Cancer with Driver Genomic Alterations. Eur. J. Cancer 2022, 167, 142–148. [Google Scholar] [CrossRef]
- Ota, K.; Azuma, K.; Kawahara, A.; Hattori, S.; Iwama, E.; Tanizaki, J.; Harada, T.; Matsumoto, K.; Takayama, K.; Takamori, S.; et al. Induction of PD-L1 Expression by the EML4-ALK Oncoprotein and Downstream Signaling Pathways in Non-Small Cell Lung Cancer. Clin. Cancer Res. 2015, 21, 4014–4021. [Google Scholar] [CrossRef]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef]
- Baldacci, S.; Grégoire, V.; Patrucco, E.; Chiarle, R.; Jamme, P.; Wasielewski, E.; Descarpentries, C.; Copin, M.-C.; Awad, M.M.; Cortot, A.B. Complete and Prolonged Response to Anti-PD1 Therapy in an ALK Rearranged Lung Adenocarcinoma. Lung Cancer 2020, 146, 366–369. [Google Scholar] [CrossRef]
- Hebart, H.; Lang, P.; Woessmann, W. Nivolumab for Refractory Anaplastic Large Cell Lymphoma: A Case Report. Ann. Intern. Med. 2016, 165, 607–608. [Google Scholar] [CrossRef] [PubMed]
- Rigaud, C.; Abbou, S.; Minard-Colin, V.; Geoerger, B.; Scoazec, J.Y.; Vassal, G.; Jaff, N.; Heuberger, L.; Valteau-Couanet, D.; Brugieres, L. Efficacy of Nivolumab in a Patient with Systemic Refractory ALK+ Anaplastic Large Cell Lymphoma. Pediatr. Blood Cancer 2018, 65. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.-W.; Gadgeel, S.; Gettinger, S.N.; Riely, G.J.; Oxnard, G.R.; Mekhail, T.; Schmid, P.; Dowlati, A.; Heist, R.S.; Wozniak, A.J.; et al. Brief Report: Safety and Antitumor Activity of Alectinib Plus Atezolizumab from a Phase 1b Study in Advanced ALK-Positive NSCLC. JTO Clin. Res. Rep. 2022, 3, 100367. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Liu, X.; Zhang, W.; Wang, W.; Wang, H.; Luo, L.; Jia, K.; Shao, C.; Mao, S.; Qiu, T.; et al. Association of PD-L1 Expression with Efficacy of Alectinib in Advanced NSCLC Patients with ALK Fusion. Lung Cancer 2023, 181, 107233. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.; Jabbour, S.K.; Malhotra, J. ALK Inhibitors and Checkpoint Blockade: A Cautionary Tale of Mixing Oil with Water? J. Thorac. Dis. 2018, 10, S2198–S2201. [Google Scholar] [CrossRef]
- Spigel, D.R.; Reynolds, C.; Waterhouse, D.; Garon, E.B.; Chandler, J.; Babu, S.; Thurmes, P.; Spira, A.; Jotte, R.; Zhu, J.; et al. Phase 1/2 Study of the Safety and Tolerability of Nivolumab Plus Crizotinib for the First-Line Treatment of Anaplastic Lymphoma Kinase Translocation—Positive Advanced Non-Small Cell Lung Cancer (CheckMate 370). J. Thorac. Oncol. 2018, 13, 682–688. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.; Reckamp, K.L.; Baas, P.; Crinò, L.; Eberhardt, W.E.E.; Poddubskaya, E.; Antonia, S.; Pluzanski, A.; Vokes, E.E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Socinski, M.A.; Jotte, R.M.; Cappuzzo, F.; Orlandi, F.; Stroyakovskiy, D.; Nogami, N.; Rodríguez-Abreu, D.; Moro-Sibilot, D.; Thomas, C.A.; Barlesi, F.; et al. Atezolizumab for First-Line Treatment of Metastatic Nonsquamous NSCLC. N. Engl. J. Med. 2018, 378, 2288–2301. [Google Scholar] [CrossRef]
- Tan, A.C.; Pavlakis, N. Anti-Angiogenic Therapy in ALK Rearranged Non-Small Cell Lung Cancer (NSCLC). Int. J. Mol. Sci. 2022, 23, 8863. [Google Scholar] [CrossRef]
- Hack, S.P.; Zhu, A.X.; Wang, Y. Augmenting Anticancer Immunity Through Combined Targeting of Angiogenic and PD-1/PD-L1 Pathways: Challenges and Opportunities. Front. Immunol. 2020, 11, 598877. [Google Scholar] [CrossRef]
- Pyo, K.-H.; Lim, S.M.; Park, C.-W.; Jo, H.-N.; Kim, J.H.; Yun, M.-R.; Kim, D.; Xin, C.-F.; Lee, W.; Gheorghiu, B.; et al. Comprehensive Analyses of Immunodynamics and Immunoreactivity in Response to Treatment in ALK-Positive Non-Small-Cell Lung Cancer. J. Immunother. Cancer 2020, 8, e000970. [Google Scholar] [CrossRef] [PubMed]
- McDermott, D.F.; Huseni, M.A.; Atkins, M.B.; Motzer, R.J.; Rini, B.I.; Escudier, B.; Fong, L.; Joseph, R.W.; Pal, S.K.; Reeves, J.A.; et al. Clinical Activity and Molecular Correlates of Response to Atezolizumab Alone or in Combination with Bevacizumab versus Sunitinib in Renal Cell Carcinoma. Nat. Med. 2018, 24, 749–757. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, N.J.; Young, R.J.; Sellitti, M.; Miller, A.; Drilon, A. Lorlatinib and Bevacizumab Activity in ALK-Rearranged Lung Cancers After Lorlatinib Progression. JCO Precis. Oncol. 2020, 4, 1333–1338. [Google Scholar] [CrossRef]
- Li, Y.; Lv, Y.; Zhang, C.; Fu, B.; Liu, Y.; Hu, J. Recent Advances in the Development of Dual ALK/ROS1 Inhibitors for Non-Small Cell Lung Cancer Therapy. Eur. J. Med. Chem. 2023, 257, 115477. [Google Scholar] [CrossRef]
- Marinelli, D.; Siringo, M.; Metro, G.; Ricciuti, B.; Gelibter, A.J. Non-Small-Cell Lung Cancer: How to Manage ALK-, ROS1- and NTRK-Rearranged Disease. Drugs Context 2022, 11. [Google Scholar] [CrossRef]
- Conde, E.; Rojo, F.; Gómez, J.; Enguita, A.B.; Abdulkader, I.; González, A.; Lozano, D.; Mancheño, N.; Salas, C.; Salido, M.; et al. Molecular Diagnosis in Non-Small-Cell Lung Cancer: Expert Opinion on ALK and ROS1 Testing. J. Clin. Pathol. 2022, 75, 145–153. [Google Scholar] [CrossRef]
- Xiang, Y.; Zhang, S.; Fang, X.; Jiang, Y.; Fang, T.; Liu, J.; Lu, K. Therapeutic Advances of Rare ALK Fusions in Non-Small Cell Lung Cancer. Curr. Oncol. 2022, 29, 7816–7831. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; An, Z.; Tang, Q.; Ma, Y.; Yan, J.; Chen, S.; Wang, Y. Mixed Responses to First-Line Alectinib in Non-Small Cell Lung Cancer Patients with Rare ALK Gene Fusions: A Case Series and Literature Review. J. Cell. Mol. Med. 2021, 25, 9476–9481. [Google Scholar] [CrossRef]
- Li, W.; Guo, L.; Liu, Y.; Dong, L.; Yang, L.; Chen, L.; Liu, K.; Shao, Y.; Ying, J. Potential Unreliability of Uncommon ALK, ROS1, and RET Genomic Breakpoints in Predicting the Efficacy of Targeted Therapy in NSCLC. J. Thorac. Oncol. 2021, 16, 404–418. [Google Scholar] [CrossRef]
- Wang, B.; Chen, R.; Wang, C.; Guo, J.; Yuan, M.; Chen, H.; Xia, X.; Zhong, D. Identification of Novel ALK Fusions Using DNA/RNA Sequencing in Immunohistochemistry/RT-PCR Discordant NSCLC Patients. Hum. Pathol. 2021, 114, 90–98. [Google Scholar] [CrossRef]
- Tabbò, F.; Muscarella, L.A.; Gobbini, E.; Trombetta, D.; Castellana, S.; Rigutto, A.; Galetta, D.; Maiello, E.; Martelli, O.; Tiseo, M.; et al. Detection of ALK Fusion Variants by RNA-Based NGS and Clinical Outcome Correlation in NSCLC Patients Treated with ALK-TKI Sequences. Eur. J. Cancer 2022, 174, 200–211. [Google Scholar] [CrossRef]
- Kuang, Y.; Xu, P.; Wang, J.; Zheng, Y.; Sun, X.; Li, Z.; Gan, R.; Li, H.; Guo, Y.; Yao, F.; et al. Detecting ALK Rearrangement with RT-PCR: A Reliable Approach Compared with Next-Generation Sequencing in Patients with NSCLC. Mol. Diagn. Ther. 2021, 25, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Hu, L.; Dong, D.; Guo, Z.; Wei, W.; Wang, C.; Shao, W.; Ma, T.; Chen, Y.; Li, Q.; et al. TP53 or CDKN2A/B Covariation in ALK/RET/ROS1-Rearranged NSCLC Is Associated with a High TMB, Tumor Immunosuppressive Microenvironment and Poor Prognosis. J. Cancer Res. Clin. Oncol. 2023, 149, 10041–10052. [Google Scholar] [CrossRef]
- Rao, W.; Liu, Y.; Li, Y.; Guo, L.; Qiu, T.; Dong, L.; Ying, J.; Li, W. Potential Unreliability of ALK Variant Allele Frequency in the Efficacy Prediction of Targeted Therapy in NSCLC. Front. Med. 2023, 17, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Batra, U.; Nathany, S.; Sharma, M.; Pasricha, S.; Bansal, A.; Jain, P.; Mehta, A. IHC versus FISH versus NGS to Detect ALK Gene Rearrangement in NSCLC: All Questions Answered? J. Clin. Pathol. 2022, 75, 405–409. [Google Scholar] [CrossRef]
- Ma, X.; Zhang, K.; Xu, J.; Gao, H.; Yang, S.; Qin, H.; Wang, H.; Gao, F.; Liu, X. Dynamic Monitoring of Circulating Tumor DNA to Analyze Genetic Characteristics and Resistance Profile of Lorlatinib in ALK Positive Previously Treated NSCLC. Thorac. Cancer 2023, 14, 1980–1990. [Google Scholar] [CrossRef]
- Tan, D.S.-W.; Thomas, M.; Kim, D.-W.; Szpakowski, S.; Urban, P.; Mehra, R.; Chow, L.Q.M.; Sharma, S.; Solomon, B.J.; Felip, E.; et al. Genetic Landscape of Patients with ALK-Rearranged Non-Small-Cell Lung Cancer (NSCLC) and Response to Ceritinib in ASCEND-1 Study. Lung Cancer 2022, 163, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Dziadziuszko, R.; Mok, T.; Peters, S.; Han, J.-Y.; Alatorre-Alexander, J.; Leighl, N.; Sriuranpong, V.; Pérol, M.; de Castro Junior, G.; Nadal, E.; et al. Blood First Assay Screening Trial (BFAST) in Treatment-Naive Advanced or Metastatic NSCLC: Initial Results of the Phase 2 ALK-Positive Cohort. J. Thorac. Oncol. 2021, 16, 2040–2050. [Google Scholar] [CrossRef]
- Kwon, M.; Ku, B.M.; Olsen, S.; Park, S.; Lefterova, M.; Odegaard, J.; Jung, H.-A.; Sun, J.-M.; Lee, S.-H.; Ahn, J.S.; et al. Longitudinal Monitoring by Next-Generation Sequencing of Plasma Cell-Free DNA in ALK Rearranged NSCLC Patients Treated with ALK Tyrosine Kinase Inhibitors. Cancer Med. 2022, 11, 2944–2956. [Google Scholar] [CrossRef]
- Sánchez-Herrero, E.; Serna-Blasco, R.; Ivanchuk, V.; García-Campelo, R.; Dómine Gómez, M.; Sánchez, J.M.; Massutí, B.; Reguart, N.; Camps, C.; Sanz-Moreno, S.; et al. NGS-Based Liquid Biopsy Profiling Identifies Mechanisms of Resistance to ALK Inhibitors: A Step toward Personalized NSCLC Treatment. Mol. Oncol. 2021, 15, 2363–2376. [Google Scholar] [CrossRef]
- Volckmar, A.-L.; Leichsenring, J.; Kirchner, M.; Christopoulos, P.; Neumann, O.; Budczies, J.; Morais de Oliveira, C.M.; Rempel, E.; Buchhalter, I.; Brandt, R.; et al. Combined Targeted DNA and RNA Sequencing of Advanced NSCLC in Routine Molecular Diagnostics: Analysis of the First 3,000 Heidelberg Cases. Int. J. Cancer 2019, 145, 649–661. [Google Scholar] [CrossRef]
- Yang, S.-R.; Schultheis, A.M.; Yu, H.; Mandelker, D.; Ladanyi, M.; Büttner, R. Precision Medicine in Non-Small Cell Lung Cancer: Current Applications and Future Directions. Semin. Cancer Biol. 2022, 84, 184–198. [Google Scholar] [CrossRef]
- Moes-Sosnowska, J.; Szpechcinski, A.; Chorostowska-Wynimko, J. Clinical Significance of TP53 Alterations in Advanced NSCLC Patients Treated with EGFR, ALK and ROS1 Tyrosine Kinase Inhibitors: An Update. Tumour Biol. 2023. [Google Scholar] [CrossRef]
- Koole, S.N.; Vessies, D.C.L.; Schuurbiers, M.M.F.; Kramer, A.; Schouten, R.D.; Degeling, K.; Bosch, L.J.W.; van denHeuvel, M.M.; vanHarten, W.H.; van denBroek, D.; et al. Cell-Free DNA at Diagnosis for Stage IV Non-Small Cell Lung Cancer: Costs, Time to Diagnosis and Clinical Relevance. Cancers 2022, 14, 1783. [Google Scholar] [CrossRef]
- Rossi, E.; Aieta, M.; Tartarone, A.; Pezzuto, A.; Facchinetti, A.; Santini, D.; Ulivi, P.; Ludovini, V.; Possidente, L.; Fiduccia, P.; et al. A Fully Automated Assay to Detect the Expression of Pan-Cytokeratins and of EML4-ALK Fusion Protein in Circulating Tumour Cells (CTCs) Predicts Outcome of Non-Small Cell Lung Cancer (NSCLC) Patients. Transl. Lung Cancer Res. 2021, 10, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Zia, V.; Lengyel, C.G.; Tajima, C.C.; de Mello, R.A. Advancements of ALK Inhibition of Non-Small Cell Lung Cancer: A Literature Review. Transl. Lung Cancer Res. 2023, 12, 1563–1574. [Google Scholar] [CrossRef] [PubMed]
- Ceddia, S.; Codacci-Pisanelli, G. Treatment of Brain Metastases in ALK-Positive Non-Small Cell Lung Cancer. Crit. Rev. Oncol. 2021, 165, 103400. [Google Scholar] [CrossRef] [PubMed]
- Wrona, A.; Dziadziuszko, R.; Jassem, J. Combining Radiotherapy with Targeted Therapies in Non-Small Cell Lung Cancer: Focus on Anti-EGFR, Anti-ALK and Anti-Angiogenic Agents. Transl. Lung Cancer Res. 2021, 10, 2032–2047. [Google Scholar] [CrossRef] [PubMed]
- Nensi, S.; Ashton, J. ALK-Positive Non-Small Cell Lung Cancer; Potential Combination Drug Treatments. Curr. Cancer Drug Targets 2021, 21, 737–748. [Google Scholar] [CrossRef] [PubMed]
- Meador, C.B.; Piotrowska, Z. Biology and Impact of Lineage Plasticity in ALK-Positive NSCLC: A Narrative Review. Transl. Lung Cancer Res. 2023, 12, 837–856. [Google Scholar] [CrossRef]
- Fukuda, A.; Yoshida, T. Treatment of Advanced ALK-Rearranged NSCLC Following Second-Generation ALK-TKI Failure. Expert Rev. Anticancer Ther. 2023, 23, 1157–1167. [Google Scholar] [CrossRef]
- Desai, A.; Lovly, C.M. Strategies to Overcome Resistance to ALK Inhibitors in Non-Small Cell Lung Cancer: A Narrative Review. Transl. Lung Cancer Res. 2023, 12, 615–628. [Google Scholar] [CrossRef]
- Lovly, C.M.; McDonald, N.T.; Chen, H.; Ortiz-Cuaran, S.; Heukamp, L.C.; Yan, Y.; Florin, A.; Ozretić, L.; Lim, D.; Wang, L.; et al. Rationale for Co-Targeting IGF-1R and ALK in ALK Fusion-Positive Lung Cancer. Nat. Med. 2014, 20, 1027–1034. [Google Scholar] [CrossRef]
- Hutchinson, L. Lung Cancer: Combating Resistance through IGF-1R and ALK Co-Targeting. Nat. Rev. Clin. Oncol. 2014, 11, 622. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Borenäs, M.; Xiong, J.; Lai, W.-Y.; Palmer, R.H.; Hallberg, B. IGF1R Contributes to Cell Proliferation in ALK-Mutated Neuroblastoma with Preference for Activating the PI3K-AKT Signaling Pathway. Cancers 2023, 15, 4252. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Zhu, F.; Xu, F.; Chen, Z.; Jiang, Y.Y.; Chen, Y.Z. Co-Targeting Cancer Drug Escape Pathways Confers Clinical Advantage for Multi-Target Anticancer Drugs. Pharmacol. Res. 2015, 102, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Hrustanovic, G.; Bivona, T.G. RAS-MAPK in ALK Targeted Therapy Resistance. Cell Cycle 2015, 14, 3661–3662. [Google Scholar] [CrossRef] [PubMed]
- Berry, M.A.; Bland, A.R.; Ashton, J.C. Mechanisms of Synergistic Suppression of ALK-Positive Lung Cancer Cell Growth by the Combination of ALK and SHP2 Inhibitors. Sci. Rep. 2023, 13, 10041. [Google Scholar] [CrossRef] [PubMed]
- Umapathy, G.; Guan, J.; Gustafsson, D.E.; Javanmardi, N.; Cervantes-Madrid, D.; Djos, A.; Martinsson, T.; Palmer, R.H.; Hallberg, B. MEK Inhibitor Trametinib Does Not Prevent the Growth of Anaplastic Lymphoma Kinase (ALK)-Addicted Neuroblastomas. Sci. Signal. 2017, 10, eaam7550. [Google Scholar] [CrossRef] [PubMed]
- Jokinen, E.; Laurila, N.; Koivunen, P.; Koivunen, J.P. Combining Targeted Drugs to Overcome and Prevent Resistance of Solid Cancers with Some Stem-like Cell Features. Oncotarget 2014, 5, 9295–9307. [Google Scholar] [CrossRef]
- Ku, B.M.; Bae, Y.H.; Lee, K.Y.; Sun, J.-M.; Lee, S.-H.; Ahn, J.S.; Park, K.; Ahn, M.-J. Entrectinib Resistance Mechanisms in ROS1-Rearranged Non-Small Cell Lung Cancer. Investig. New Drugs 2020, 38, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Honkanen, T.J.; Luukkainen, M.E.K.; Koivunen, J.P. Role of Human Epidermal Growth Factor Receptor 3 in Treatment Resistance of Anaplastic Lymphoma Kinase Translocated Non-Small Cell Lung Cancer. Cancer Biol. Ther. 2023, 24, 2256906. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Liao, Y.; Cao, J.; Fang, B.; Yun, S.Y.; Kinose, F.; Haura, E.B.; Lawrence, H.R.; Doebele, R.C.; Koomen, J.M.; et al. Differential Chemoproteomics Reveals MARK2/3 as Cell Migration-Relevant Targets of the ALK Inhibitor Brigatinib. Chembiochem 2023, 24, e202200766. [Google Scholar] [CrossRef]
- Chiarle, R.; Martinengo, C.; Mastini, C.; Ambrogio, C.; D’Escamard, V.; Forni, G.; Inghirami, G. The Anaplastic Lymphoma Kinase Is an Effective Oncoantigen for Lymphoma Vaccination. Nat. Med. 2008, 14, 676–680. [Google Scholar] [CrossRef] [PubMed]
- Voena, C.; Menotti, M.; Mastini, C.; Di Giacomo, F.; Longo, D.L.; Castella, B.; Merlo, M.E.B.; Ambrogio, C.; Wang, Q.; Minero, V.G.; et al. Efficacy of a Cancer Vaccine against ALK-Rearranged Lung Tumors. Cancer Immunol. Res. 2015, 3, 1333–1343. [Google Scholar] [CrossRef] [PubMed]
- Mota, I.; Patrucco, E.; Mastini, C.; Mahadevan, N.R.; Thai, T.C.; Bergaggio, E.; Cheong, T.-C.; Leonardi, G.; Karaca-Atabay, E.; Campisi, M.; et al. ALK Peptide Vaccination Restores the Immunogenicity of ALK-Rearranged Non-Small Cell Lung Cancer. Nat. Cancer 2023, 4, 1016–1035. [Google Scholar] [CrossRef]
- Codony-Servat, J.; García-Roman, S.; Molina-Vila, M.Á.; Bertran-Alamillo, J.; Viteri, S.; d’Hondt, E.; Rosell, R. Anti-Epidermal Growth Factor Vaccine Antibodies Increase the Antitumor Activity of Kinase Inhibitors in ALK and RET Rearranged Lung Cancer Cells. Transl. Oncol. 2021, 14, 100887. [Google Scholar] [CrossRef]
- Nouri, K.; Azad, T.; Lightbody, E.; Khanal, P.; Nicol, C.J.; Yang, X. A Kinome-Wide Screen Using a NanoLuc LATS Luminescent Biosensor Identifies ALK as a Novel Regulator of the Hippo Pathway in Tumorigenesis and Immune Evasion. FASEB J. 2019, 33, 12487–12499. [Google Scholar] [CrossRef]
- Kreutmair, S.; Erlacher, M.; Andrieux, G.; Istvanffy, R.; Mueller-Rudorf, A.; Zwick, M.; Rückert, T.; Pantic, M.; Poggio, T.; Shoumariyeh, K.; et al. Loss of the Fanconi Anemia-Associated Protein NIPA Causes Bone Marrow Failure. J. Clin. Investig. 2020, 130, 2827–2844. [Google Scholar] [CrossRef]
- Kreutmair, S.; Lippert, L.J.; Klingeberg, C.; Albers-Leischner, C.; Yacob, S.; Shlyakhto, V.; Mueller, T.; Mueller-Rudorf, A.; Yu, C.; Gorantla, S.P.; et al. NIPA (Nuclear Interaction Partner of ALK) Is Crucial for Effective NPM-ALK Mediated Lymphomagenesis. Front. Oncol. 2022, 12, 875117. [Google Scholar] [CrossRef]
- Xiao, J.; Zhang, L.; Yi, H.; Zou, L.; Mo, J.; Xue, F.; Zheng, J.; Huang, Y.; Lu, H.; Wu, H.; et al. Inhibiting ALK-TOPK Signaling Pathway Promotes Cell Apoptosis of ALK-Positive NSCLC. Cell Death Dis. 2022, 13, 828. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yang, X.; Wang, R.; Zhang, X. Prognostic Value of PDZ-Binding Kinase/T-LAK Cell-Originated Protein Kinase (PBK/TOPK) in Patients with Cancer. J. Cancer 2019, 10, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, Y.; Park, J.-H.; Miyamoto, T.; Takamatsu, N.; Kato, T.; Iwasa, A.; Okabe, S.; Imai, Y.; Fujiwara, K.; Nakamura, Y.; et al. T-LAK Cell-Originated Protein Kinase (TOPK) as a Prognostic Factor and a Potential Therapeutic Target in Ovarian Cancer. Clin. Cancer Res. 2016, 22, 6110–6117. [Google Scholar] [CrossRef] [PubMed]
- Zlobec, I.; Molinari, F.; Kovac, M.; Bihl, M.P.; Altermatt, H.J.; Diebold, J.; Frick, H.; Germer, M.; Horcic, M.; Montani, M.; et al. Prognostic and Predictive Value of TOPK Stratified by KRAS and BRAF Gene Alterations in Sporadic, Hereditary and Metastatic Colorectal Cancer Patients. Br. J. Cancer 2010, 102, 151–161. [Google Scholar] [CrossRef]
- Ohashi, T.; Komatsu, S.; Ichikawa, D.; Miyamae, M.; Okajima, W.; Imamura, T.; Kiuchi, J.; Kosuga, T.; Konishi, H.; Shiozaki, A.; et al. Overexpression of PBK/TOPK Relates to Tumour Malignant Potential and Poor Outcome of Gastric Carcinoma. Br. J. Cancer 2017, 116, 218–226. [Google Scholar] [CrossRef]
- Cicin, I.; Martin, C.; Haddad, C.K.; Kim, S.-W.; Smolin, A.; Abdillah, A.; Yang, X. ALK TKI Therapy in Patients with ALK-Positive Non-Small Cell Lung Cancer and Brain Metastases: A Review of the Literature and Local Experiences. Crit. Rev. Oncol. 2022, 180, 103847. [Google Scholar] [CrossRef]
- Lorlatinib (Lorbrena): CADTH Reimbursement Recommendation: Indication: As Monotherapy for the First-Line Treatment of Adult Patients with Anaplastic Lymphoma Kinase (ALK)-Positive Locally Advanced (Not Amenable to Curative Therapy) or Metastatic Non–Small. Available online: https://www.ncbi.nlm.nih.gov/books/NBK599021 (accessed on 18 January 2024).
- Ando, K.; Manabe, R.; Kishino, Y.; Kusumoto, S.; Yamaoka, T.; Tanaka, A.; Ohmori, T.; Sagara, H. Comparative Efficacy of ALK Inhibitors for Treatment-Naïve ALK-Positive Advanced Non-Small Cell Lung Cancer with Central Nervous System Metastasis: A Network Meta-Analysis. Int. J. Mol. Sci. 2023, 24, 2242. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parvaresh, H.; Roozitalab, G.; Golandam, F.; Behzadi, P.; Jabbarzadeh Kaboli, P. Unraveling the Potential of ALK-Targeted Therapies in Non-Small Cell Lung Cancer: Comprehensive Insights and Future Directions. Biomedicines 2024, 12, 297. https://doi.org/10.3390/biomedicines12020297
Parvaresh H, Roozitalab G, Golandam F, Behzadi P, Jabbarzadeh Kaboli P. Unraveling the Potential of ALK-Targeted Therapies in Non-Small Cell Lung Cancer: Comprehensive Insights and Future Directions. Biomedicines. 2024; 12(2):297. https://doi.org/10.3390/biomedicines12020297
Chicago/Turabian StyleParvaresh, Hannaneh, Ghazaal Roozitalab, Fatemeh Golandam, Payam Behzadi, and Parham Jabbarzadeh Kaboli. 2024. "Unraveling the Potential of ALK-Targeted Therapies in Non-Small Cell Lung Cancer: Comprehensive Insights and Future Directions" Biomedicines 12, no. 2: 297. https://doi.org/10.3390/biomedicines12020297
APA StyleParvaresh, H., Roozitalab, G., Golandam, F., Behzadi, P., & Jabbarzadeh Kaboli, P. (2024). Unraveling the Potential of ALK-Targeted Therapies in Non-Small Cell Lung Cancer: Comprehensive Insights and Future Directions. Biomedicines, 12(2), 297. https://doi.org/10.3390/biomedicines12020297