
Conjecturing about Small-Molecule Agonists and Antagonists of α4β1 Integrin: From Mechanistic Insight to Potential Therapeutic Applications

 , , and
, , and 
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
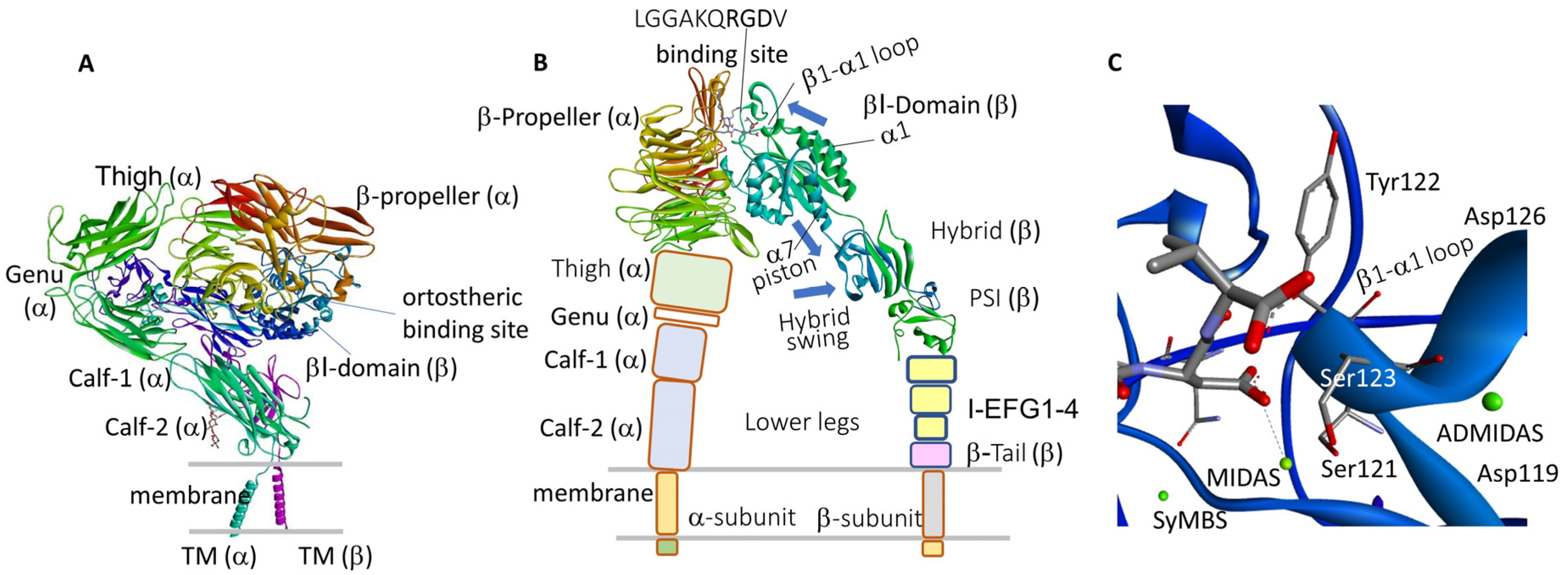
2. Biology and Functions of α4β1 Integrin
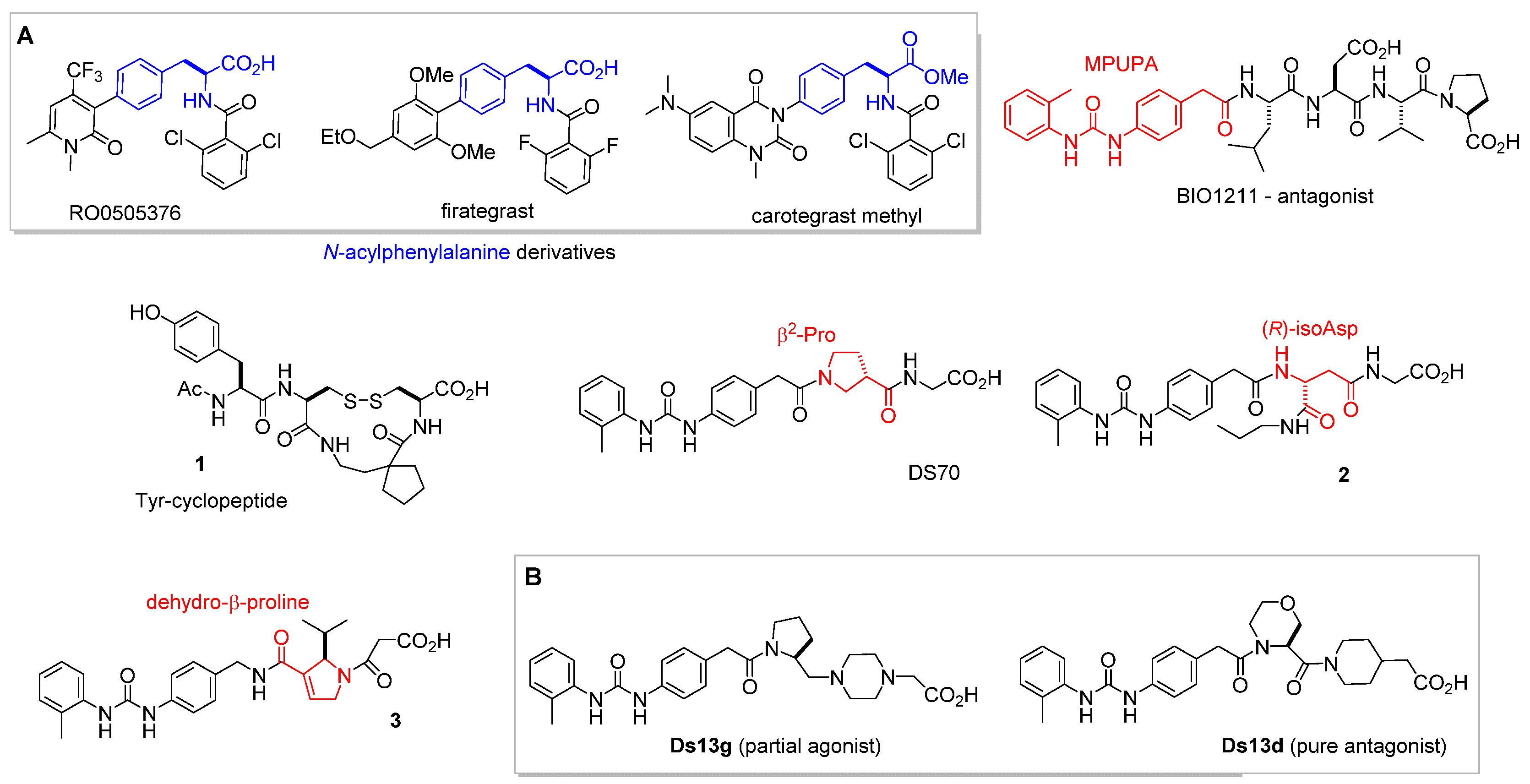
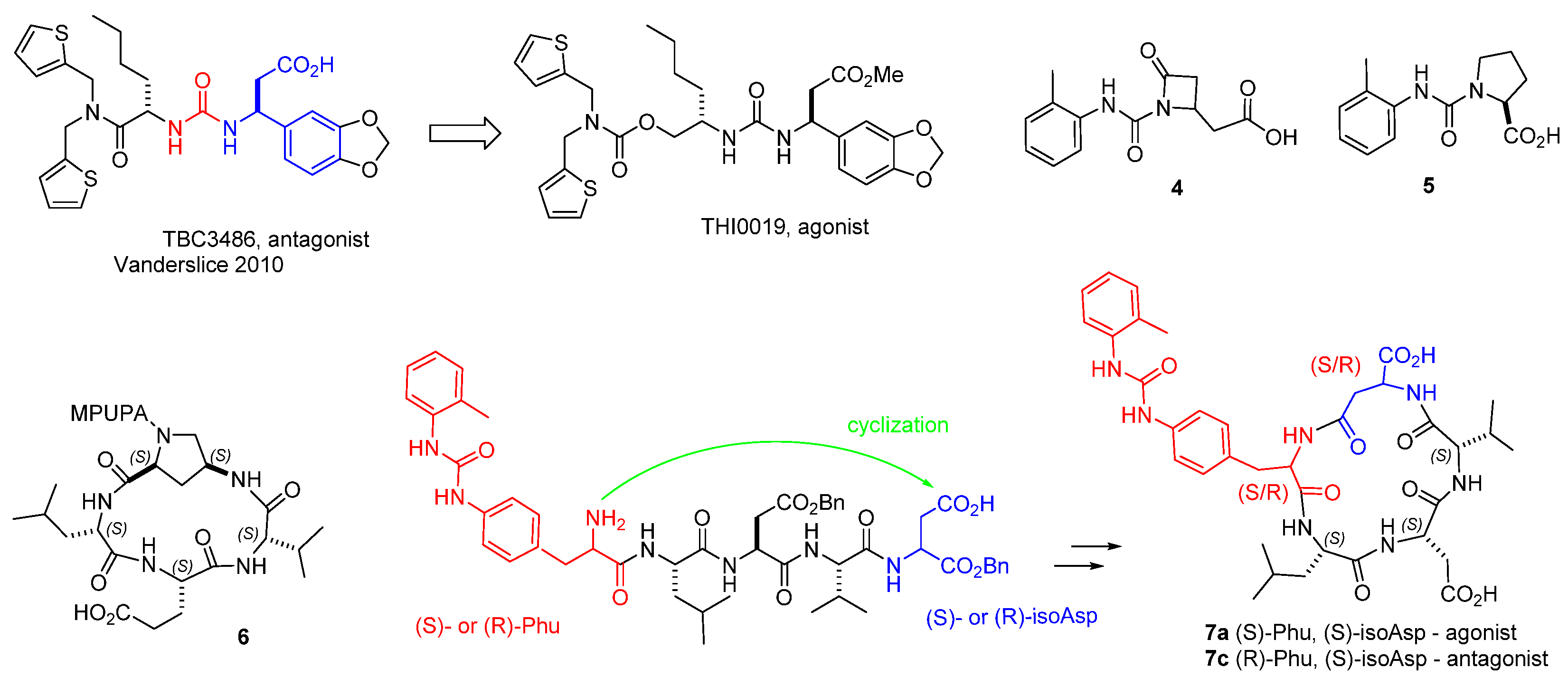
3. α4β1 Integrin Antagonists
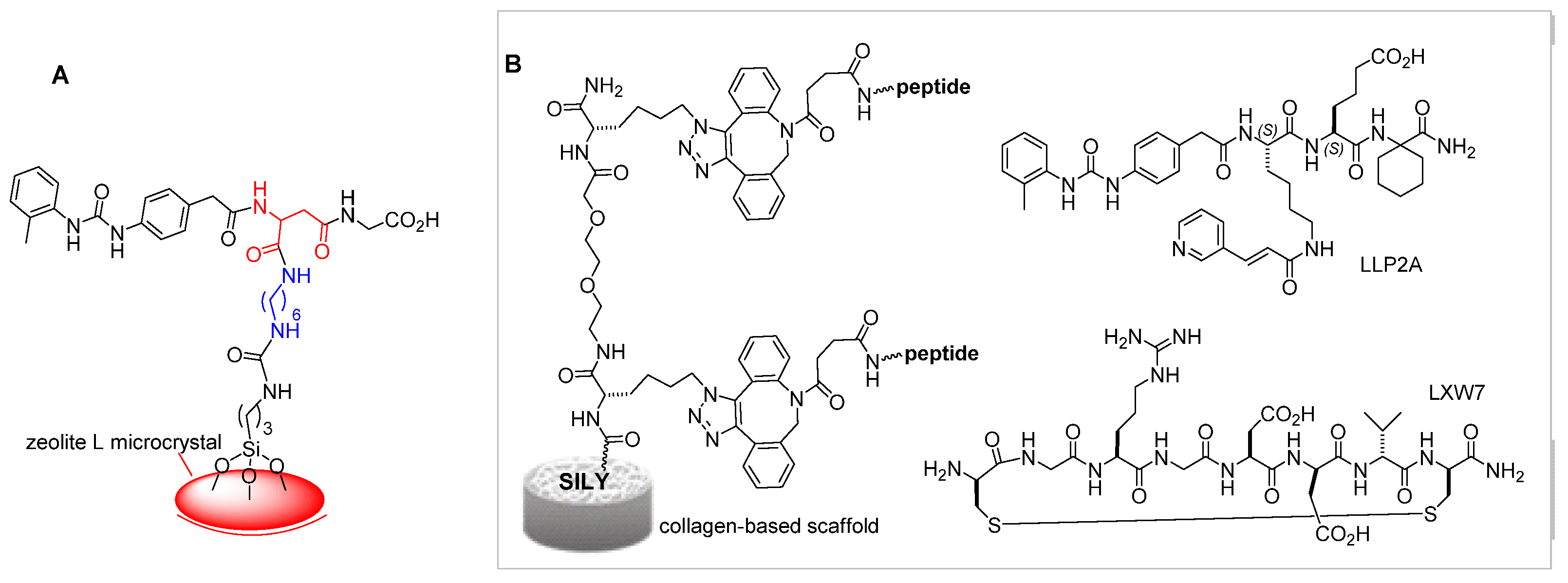
α4β1 Integrin Ligands in Diagnostics and Biomaterials
4. Small-Molecule Agonists of α4β1 Integrin
5. Therapeutic Opportunities of α4β1 Integrin Agonists
6. Ligand-Integrin Interactions and Conformational Implications
6.1. αIIbβ3 Integrin
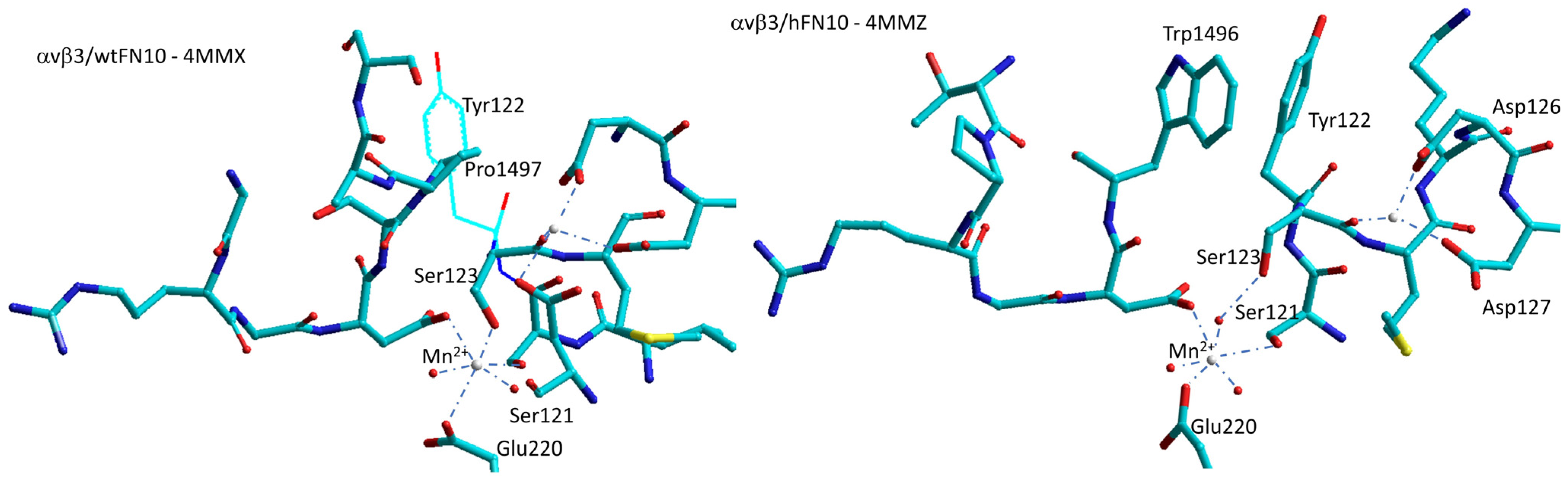
6.2. αvβ3 Integrin
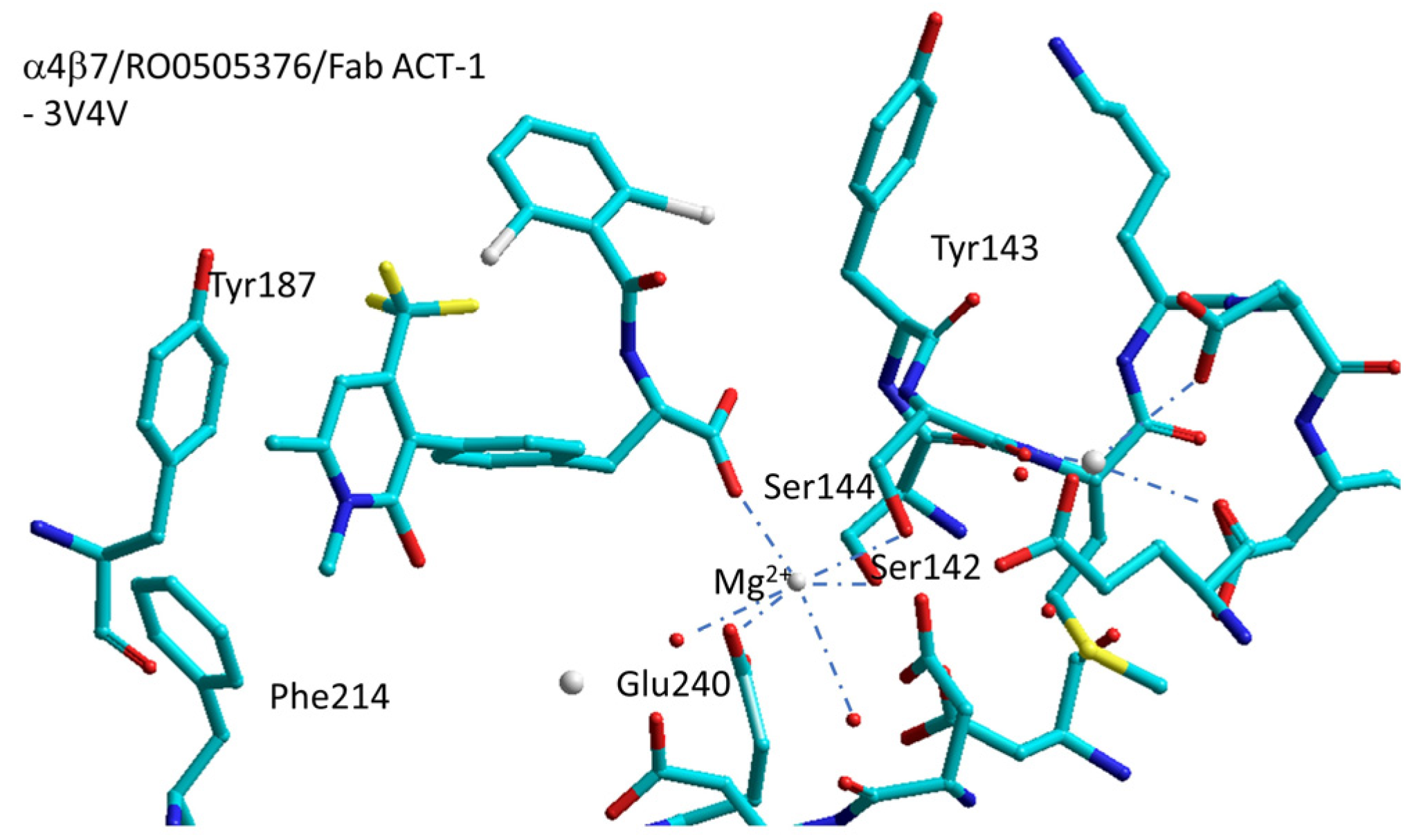
6.3. α4β7 Integrin
6.4. α5β1 Integrin
6.5. Agonism or Antagonism and Dynamics Nature of MIDAS
7. Simulations with Homology or Composite Models of α4β1 Integrin
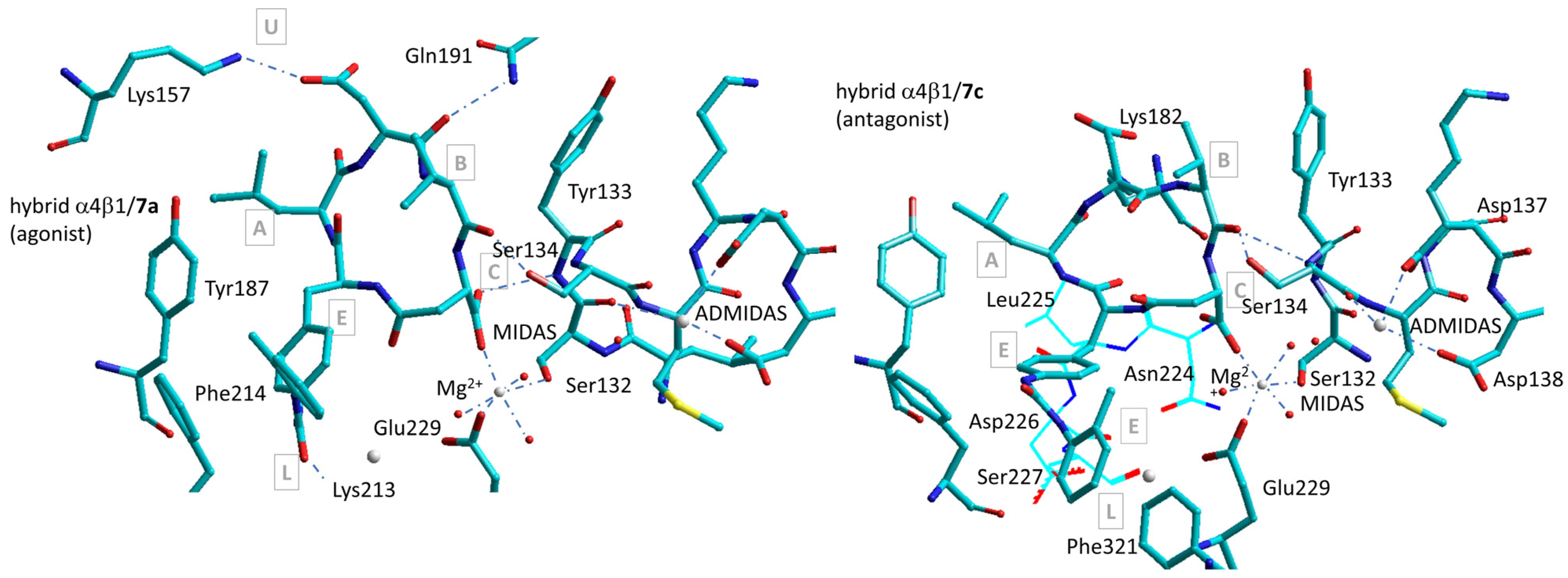
Simulations of α4β1 Integrin Agonists
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kadry, Y.A.; Calderwood, D.A. Chapter 22: Structural and signaling functions of integrins. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183206. [Google Scholar] [CrossRef]
- Cabodi, S.; Di Stefano, P.; del Pilar Camacho Leal, M.; Tinnirello, A.; Bisaro, B.; Morello, V.; Damiano, L.; Aramu, S.; Repetto, D.; Tornillo, G.; et al. Integrins and signal transduction. Adv. Exp. Med. Biol. 2010, 674, 43–54. [Google Scholar] [PubMed]
- Humphries, J.D.; Byron, A.; Humphries, M.J. Integrin ligands at a glance. J. Cell Sci. 2006, 119, 3901–3903. [Google Scholar] [CrossRef]
- Plow, E.F.; Haas, T.A.; Zhang, L.; Loftus, J.; Smith, J.W. Ligand binding to integrins. J. Biol. Chem. 2000, 275, 21785–21788. [Google Scholar] [CrossRef]
- Kolasangiani, R.; Bidone, T.C.; Schwartz, M.A. Integrin conformational dynamics and mechanotransduction. Cells 2022, 11, 3584. [Google Scholar] [CrossRef]
- Ley, K.; Rivera-Nieves, J.; Sandborn, W.J.; Shattil, S. Integrin-based therapeutics: Biological basis, clinical use and new drugs. Nat. Rev. Drug Discov. 2016, 15, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Slack, R.J.; Macdonald, S.J.F.; Roper, J.A.; Jenkins, R.G.; Hatley, R.J.D. Emerging therapeutic opportunities for integrin inhibitors. Nat. Rev. Drug Discov. 2022, 21, 60–78. [Google Scholar] [CrossRef]
- Adair, B.D.; Xiong, J.-P.; Yeager, M.; Arnaout, M.A. Cryo-EM structures of full-length integrin αIIbβ3 in native lipids. Nat. Commun. 2023, 14, 4168. [Google Scholar] [CrossRef] [PubMed]
- Springer, T.A.; Zhu, J.; Xiao, T. Structural basis for distinctive recognition of fibrinogen γC peptide by the platelet integrin αIIbβ3. J. Cell Biol. 2008, 182, 791–800. [Google Scholar] [CrossRef]
- Fagerholm, S.C. Integrins in health and disease. N. Engl. J. Med. 2022, 387, 1519–1521. [Google Scholar] [CrossRef]
- Mas-Moruno, C.; Rechenmacher, F.; Kessler, H. Cilengitide: The first anti-angiogenic small molecule drug candidate design, synthesis and clinical evaluation. Anticancer Agents Med. Chem. 2010, 10, 753–768. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, A.; Hart, I.; Watson, A.; Welti, J.C.; Silva, R.G.; Robinson, S.D.; Da Violante, G.; Gourlaouen, M.; Salih, M.; Jones, M.C.; et al. Stimulation of tumor growth and angiogenesis by low concentrations of RGD-mimetic integrin inhibitors. Nat. Med. 2009, 15, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.H.; Weber, T.; Grove, R.; Wardell, C.; Horrigan, J.; Graff, O.; Atkinson, G.; Dua, P.; Yousry, T.; Macmanus, D.; et al. Firategrast for relapsing remitting multiple sclerosis: A phase 2, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2012, 11, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Chew, D.P.; Bhatt, D.L.; Sapp, S.; Topol, E.J. Increased mortality with oral platelet glycoprotein IIb/IIIa antagonists: A meta-analysis of phase III multicenter randomized trials. Circulation 2001, 103, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Topol, E.J.; Easton, D.; Harrington, R.A.; Amarenco, P.; Califf, R.M.; Graffagnino, C.; Davis, S.; Diener, H.C.; Ferguson, J.; Fitzgerald, D.; et al. Randomized, double-blind, placebo-controlled, international trial of the oral IIb/IIIa antagonist lotrafiban in coronary and cerebrovascular disease. Circulation 2003, 108, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Théroux, P.; Catella-Lawson, F.; Armstrong, P.; DeCani, J.; Hirsh, J.; Pepine, C.; Ryan, T.J.; Pelletier, G.; Davies, R.; Flather, M.; et al. Inhibition of the platelet glycoprotein IIb/IIIa receptor with tirofiban in unstable angina and non-Q-wave myocardial infarction. N. Engl. J. Med. 1998, 338, 1488–1497. [Google Scholar]
- Lin, F.-Y.; Li, J.; Xie, Y.; Zhu, J.; Huong Nguyen, T.-T.; Zhang, Y.; Zhu, J.; Springer, T.A. A general chemical principle for creating closure-stabilizing integrin inhibitors. Cell 2022, 185, 3533–3550.e27. [Google Scholar] [CrossRef]
- Muller, W. Leukocyte-endothelial cell interactions in the inflammatory response. Lab. Investig. 2002, 82, 521–534. [Google Scholar] [CrossRef]
- Ferreira, E.F.B.; Silva, L.B.; Cruz, J.V.; Araújo, P.H.F.; Kimani, N.M.; Leite, F.H.A.; Campos, J.M.; Santos, C.B.R. An overview of the α4β1 integrin and the potential therapeutic role of its antagonists. Curr. Med. Chem. 2021, 28, 5884–5895. [Google Scholar] [CrossRef]
- Ou, Z.; Dolmatova, E.; Lassègue, B.; Griendling, K.K. β1- and β2-integrins: Central players in regulating vascular permeability and leukocyte recruitment during acute inflammation. Am. J. Physiol. Heart Circ. Physiol. 2021, 320, H734–H739. [Google Scholar] [CrossRef]
- Hyun, Y.M.; Lefort, C.T.; Kim, M. Leukocyte integrins and their ligand interactions. Immunol. Res. 2009, 45, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Newham, P.; Craig, S.E.; Seddon, G.N.; Schofield, N.R.; Rees, A.; Edwards, R.M.; Jones, E.Y.; Humphries, M.J. α4 Integrin Binding Interfaces on VCAM-1 and MAdCAM-1. J. Biol. Chem. 1997, 272, 19429–19440. [Google Scholar] [CrossRef] [PubMed]
- Baiula, M.; Spampinato, S.; Gentilucci, L.; Tolomelli, A. Novel ligands targeting alpha4beta1 integrin: Therapeutic applications and perspectives. Front. Chem. 2019, 7, 489. [Google Scholar] [CrossRef] [PubMed]
- Lobb, R.R.; Hemler, M.E. The pathophysiologic role of α4 integrins in vivo. J. Clin. Investig. 1994, 94, 1722–1728. [Google Scholar] [CrossRef] [PubMed]
- Pyka-Fościak, G.; Litwin, J.A.; Lis, G.J. Osteopontin expression and the effect of anti-VLA-4 mAb treatment in experimental autoimmune encephalomyelitis, a mouse model of multiple sclerosis. Folia Neuropathol. 2023, 61, 129–137. [Google Scholar] [CrossRef]
- Outteryck, O. Natalizumab in relapsing-remitting multiple sclerosis. Expert Rev. Neurother. 2016, 16, 471–481. [Google Scholar] [CrossRef]
- Shirani, A.; Stüve, O. Neuroimmunology in point for the impact of translational natalizumab for multiple sclerosis: A case. J. Immunol. 2017, 198, 1381–1386. [Google Scholar] [CrossRef] [PubMed]
- Nelson, S.M.; Nguyen, T.M.; McDonald, J.W.; MacDonald, J.K. Natalizumab for induction of remission in Crohn’s disease. Cochr. Database Syst. Rev. 2018, 8, CD006097. [Google Scholar] [CrossRef]
- Yang, G.X.; Hagmann, W.K. VLA-4 antagonists: Potent inhibitors of lymphocyte migration. Med. Res. Rev. 2003, 23, 369–392. [Google Scholar] [CrossRef]
- Sugiura, T.; Andou, A.; Hosoi, Y.; Koyama, T. Pharmacological and clinical data of oral alpha 4 integrin antagonist, Carotegrast methyl, CAROGRA. Nihon Yakurigaku Zasshi/Folia Pharmacol. Jpn. 2023, 158, 203–210. [Google Scholar] [CrossRef]
- Lin, K.C.; Ateeq, H.S.; Hsiung, S.H.; Chong, L.T.; Zimmerman, C.N.; Castro, A.; Lee, W.C.; Hammond, C.E.; Kalkunte, S.; Chen, L.L.; et al. Selective, tight-binding inhibitors of integrin α4β1 that inhibit allergic airway responses. J. Med. Chem. 1999, 42, 920–934. [Google Scholar] [CrossRef] [PubMed]
- Abraham, W.M.; Gill, A.; Ahmed, A.; Sielczak, M.W.; Lauredo, I.T.; Botinnikova, Y.; Lin, K.C.; Pepinsky, B.; Leone, D.R.; Lobb, R.R.; et al. A small-molecule, tight-binding inhibitor of the integrin alpha(4)beta(1) blocks antigen-induced airway responses and inflammation in experimental asthma in sheep. Am. J. Respir. Crit. Care Med. 2000, 162, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Karanam, B.V.; Jayra, A.; Rabe, M.; Wang, Z.; Keohane, C.; Strauss, J.; Vincent, S. Effect of enalapril on the in vitro and in vivo peptidyl cleavage of a potent VLA-4 antagonist. Xenobiotica 2007, 37, 487–502. [Google Scholar] [CrossRef]
- Singh, J.; Van Vlijmen, H.; Liao, Y.; Lee, W.C.; Cornebise, M.; Harris, M.; Shu, I.; Gill, A.; Cuervo, J.H.; Abraham, W.M.; et al. Identification of potent and novel alpha4beta1 antagonists using in silico screening. J. Med. Chem. 2002, 45, 2988–2993. [Google Scholar] [CrossRef] [PubMed]
- Vagner, J.; Qu, H.; Hruby, V.J. Peptidomimetics, a synthetic tool of drug discovery. Curr. Opin. Chem. Biol. 2008, 12, 292–296. [Google Scholar] [CrossRef]
- Jackson, D.Y. Alpha4 integrin antagonists. Curr. Pharm. Des. 2002, 8, 1229–1253. [Google Scholar] [CrossRef] [PubMed]
- De Marco, R.; Tolomelli, A.; Juaristi, E.; Gentilucci, L. Integrin ligands with alpha/beta-hybrid peptide structure: Design, bioactivity, and conformational aspects. Med. Res. Rev. 2016, 36, 389–424. [Google Scholar] [CrossRef]
- De Marco, R.; Mazzotti, G.; Dattoli, S.D.; Baiula, M.; Spampinato, S.; Greco, A.; Gentilucci, L. 5-Aminomethyloxazolidine-2,4-dione hybrid α/β-dipeptide scaffolds as inductors of constrained conformations: Applications to the synthesis of integrin antagonists. Pept. Sci. 2015, 104, 636–649. [Google Scholar] [CrossRef]
- Dattoli, S.D.; De Marco, R.; Baiula, M.; Spampinato, S.; Greco, A.; Tolomelli, A.; Gentilucci, L. Synthesis and assay of retro-α4β1 integrin-targeting motifs. Eur. J. Med. Chem. 2014, 73, 225–232. [Google Scholar] [CrossRef]
- Dattoli, S.D.; Baiula, M.; De Marco, R.; Bedini, A.; Anselmi, M.; Gentilucci, L.; Spampinato, S. DS-70, a novel and potent α4 integrin antagonist, is an effective treatment for experimental allergic conjunctivitis in guinea pigs. Br. J. Pharmacol. 2018, 175, 3891–3910. [Google Scholar] [CrossRef]
- Baiula, M.; Anselmi, M.; Musiani, F.; Ghidini, A.; Carbone, J.; Caligiana, A.; Maurizio, A.; Spampinato, S.; Gentilucci, L. Design, pharmacological characterization, and molecular docking of minimalist peptidomimetic antagonists of α4β1 integrin. Int. J. Mol. Sci. 2023, 24, 9588. [Google Scholar] [CrossRef] [PubMed]
- Tolomelli, A.; Baiula, M.; Viola, A.; Ferrazzano, L.; Gentilucci, L.; Dattoli, S.D.; Spampinato, S.; Juaristi, E.; Escudero, M. Dehydro-β-proline containing α4β1 integrin antagonists: Stereochemical recognition in ligand−receptor interplay. ACS Med. Chem. Lett. 2015, 6, 70–706. [Google Scholar] [CrossRef] [PubMed]
- Chiba, J.; Machinaga, N.; Takashi, T.; Ejima, A.; Takayama, G.; Yokoyama, M.; Nakayama, A.; Baldwin, J.J.; McDonald, E.; Moriarty, K.J.; et al. Identified a morpholinyl-4-piperidinylacetic acid derivative as a potent oral active VLA-4 antagonist. Bioorg. Med. Chem. Lett. 2005, 15, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Krauss, A.H.; Corrales, R.M.; Pelegrino, F.S.A.; Tukler-Henriksson, J.; Pflugfelder, S.C.; de Paiva, C.S. Improvement of outcome measures of dry eye by a novel integrin antagonist in the murine desiccating stress model. Investig. Ophthalmol. Vis. Sci. 2015, 56, 5888–5895. [Google Scholar] [CrossRef]
- Baiula, M.; Caligiana, A.; Bedini, A.; Zhao, J.; Santino, F.; Cirillo, M.; Gentilucci, L.; Giacomini, D.; Spampinato, S. Leukocyte integrin antagonists as a novel option to treat dry age-related macular degeneration. Front. Pharmacol. 2021, 11, 617836. [Google Scholar] [CrossRef]
- Bellavia, M.C.; Nyiranshuti, L.; Latoche, J.D.; Ho, K.-V.; Fecek, R.J.; Taylor, J.L.; Day, K.E.; Nigam, S.; Pun, M.; Gallazzi, F.; et al. PET imaging of VLA-4 in a new BRAFV600E mouse model of melanoma. J. Mol. Biol. 2022, 24, 425–433. [Google Scholar] [CrossRef]
- Zhao, J.; Santino, F.; Giacomini, D.; Gentilucci, L. Integrin-Targeting Peptides for the Design of Functional Cell-Responsive Biomaterials. Biomedicines 2020, 8, 307. [Google Scholar] [CrossRef]
- Perkins, L.A.; Nyiranshuti, L.; Little-Ihrig, L.; Latoche, J.D.; Day, K.E.; Zhu, Q.; Tavakoli, S.; Sundd, P.; Novelli, E.M.; Anderson, C.J. Integrin VLA-4 as a PET imaging biomarker of hyper-adhesion in transgenic sickle mice. Blood Adv. 2020, 4, 4102–4112. [Google Scholar] [CrossRef]
- De Marco, R.; Greco, A.; Calonghi, N.; Dattoli, S.D.; Baiula, M.; Spampinato, S.; Picchetti, P.; De Cola, L.; Anselmi, M.; Cipriani, F.; et al. Selective detection of α4β1 integrin (VLA-4)-expressing cells using peptide-functionalized nanostructured materials mimicking endothelial surfaces adjacent to inflammatory sites. Pept. Sci. 2018, 110, e23081. [Google Scholar] [CrossRef]
- Anselmi, M.; Baiula, M.; Santino, F.; Zhao, J.; Spampinato, S.; Calonghi, N.; Gentilucci, L. Design of α/β-hybrid peptide ligands of α4β1 integrin equipped with a linkable side chain for chemoselective biofunctionalization of microstructured materials. Biomedicines 2021, 9, 1737. [Google Scholar] [CrossRef]
- Kakinoki, S.; Nishioka, S.; Arichi, Y.; Yamaoka, T. Stable and direct coating of fibronectin-derived Leu-Asp-Val peptide on ePTFE using one-pot tyrosine oxidation for endothelial cell adhesion. Colloids Surf. B 2022, 216, 112576. [Google Scholar] [CrossRef]
- Hao, D.; Liu, R.; Fernandez, T.G.; Pivetti, C.; Jackson, J.E.; Kulubya, E.S.; Jiang, H.-J.; Ju, H.-Y.; Liu, W.-L.; Panitch, A.; et al. A bioactive material with dual integrin-targeting ligands regulates specific endogenous cell adhesion and promotes vascularized bone regeneration in adult and fetal bone defects. Bioact. Mater. 2022, 20, 179–193. [Google Scholar] [CrossRef]
- Market, R.V.; Biediger, R.J.; Woodside, D.G. Piperazine-Based Agonists of LFA-1 and VLA-4. WO2023018471A1, 9 June 2022. [Google Scholar]
- Vanderslice, P.; Biediger, R.J.; Woodside, D.G.; Brown, W.S.; Khounlo, S.; Warier, N.D.; Gundlach IV, C.W.; Caivano, A.R.; Bornmann, W.G.; Maxwell, D.S.; et al. Small molecule agonist of very late antigen-4 (VLA- 4) integrin induces progenitor cell adhesion. J. Biol. Chem. 2013, 288, 19414–19428. [Google Scholar] [CrossRef]
- Galletti, P.; Soldati, R.; Pori, M.; Durso, M.; Tolomelli, A.; Gentilucci, L.; Dattoli, S.D.; Baiula, M.; Spampinato, S.; Giacomini, D. Targeting integrins αvβ3 and α5β1 with new β-lactam derivatives. Eur. J. Med. Chem. 2014, 83, 284–293. [Google Scholar] [CrossRef]
- Martelli, G.; Baiula, M.; Caligiana, A.; Galletti, P.; Gentilucci, L.; Artali, R.; Spampinato, S.; Giacomini, D. Could dissecting the molecular framework of β-lactam integrin ligands enhance selectivity? J. Med. Chem. 2019, 62, 10156–10166. [Google Scholar] [CrossRef]
- Sartori, A.; Bugatti, K.; Portioli, E.; Baiula, M.; Casamassima, I.; Bruno, A.; Bianchini, F.; Curti, C.; Zanardi, F.; Battistini, L. New 4-aminoproline-based small molecule cyclopeptidomimetics as potential modulators of a4b1 integrin. Molecules 2021, 26, 6066. [Google Scholar] [CrossRef] [PubMed]
- Anselmi, M.; Baiula, M.; Spampinato, S.; Artali, R.; He, T.; Gentilucci, L. Design and pharmacological characterization of α4β1 integrin cyclopeptide agonists: Computational investigation of ligand determinants for agonism versus antagonism. J. Med. Chem. 2023, 66, 5021–5040. [Google Scholar] [CrossRef] [PubMed]
- Faridi, M.H.; Maiguel, D.; Barth, C.J.; Stoub, D.; Day, R.; Schürer, S.; Gupta, V. Identification of novel agonists of the integrin CD11b/CD18. Bioorg. Med. Chem. Lett. 2009, 19, 6902–6906. [Google Scholar] [CrossRef]
- Maiguel, D.; Faridi, M.H.; Wei, C.; Kuwano, Y.; Balla, K.M.; Hernandez, D.; Barth, C.J.; Lugo, G.; Donnelly, M.; Nayer, A.; et al. Small molecule−mediated activation of the integrin CD11b/CD18 reduces inflammatory disease. Sci. Signal. 2011, 4, ra57. [Google Scholar] [CrossRef]
- Faridi, M.H.; Altintas, M.M.; Gomez, C.; Duque, J.C.; Vazquez-Padron, R.I.; Gupta, V. Small molecule agonists of integrin CD11b/CD18 do not induce global conformational changes and are significantly better than activating antibodies in reducing vascular injury. Biochim. Biophys. Acta 2013, 1830, 3696–3710. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Carman, C.V.; Kim, M.; Salas, A.; Shimaoka, M.; Springer, T.A. A small molecule agonist of an integrin, αLβ2. J. Biol. Chem. 2006, 281, 37904–37912. [Google Scholar] [CrossRef]
- Strozier, R.E. Integrin Agonists or Activating Compounds and Methods for Making and Using Same. US20220118086A1, 10 September 2021. [Google Scholar]
- Yao, W.; Liu, W.; Lam, K.S.; Xiao, W.; Lane, N. Peptides for Activation of Cell Signaling in Osteoprogenitor Cells. EP3823630A4, 20 April 2022. [Google Scholar]
- Oh, J.; Magnuson, A.; Benoist, C.; Pittet, M.J.; Weissleder, R. Age related tumor growth in mice is related to integrin a 4 in CD8+ T cells. JCI Insight 2018, 3, e122961. [Google Scholar] [CrossRef] [PubMed]
- Hickman, A.; Koetsier, J.; Kurtanich, T.; Nielsen, M.C.; Winn, G.; Wang, Y.; Bentebibel, S.E.; Shi, L.; Punt, S.; Williams, L.; et al. LFA-1 activation enriches tumor-specific T cells in a cold tumor model and synergizes with CTLA-4 blockade. J. Clin. Investig. 2022, 132, e154152. [Google Scholar] [CrossRef] [PubMed]
- Lokugamage, N.; Chowdhury, I.H.; Biediger, R.J.; Market, R.V.; Khounlo, S.; Warier, N.D.; Hwang, S.A.; Actor, J.K.; Woodside, D.G.; Marathi, U.; et al. Use of a small molecule integrin activator as a systemically administered vaccine adjuvant in controlling Chagas disease. NPJ Vaccines 2021, 6, 114. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, V.; Li, X.; Jimenez, V.; Faridi, H.M.; Gupta, V. CD11b agonists offer a novel approach for treating lupus nephritis. Transl. Res. 2022, 245, 41–54. [Google Scholar] [CrossRef]
- Ehirchiou, D.; Bernabei, I.; Chobaz, V.; Castelblanco, M.; Hügle, T.; So, A.; Zhang, L.; Busso, N.; Nasi, S. CD11b Signaling prevents chondrocyte mineralization and attenuates the severity of osteoarthritis. Front. Cell Dev. Biol. 2020, 8, 611757. [Google Scholar] [CrossRef]
- Merlo, B.; Baldassarro, V.A.; Flagelli, A.; Marcoccia, R.; Giraldi, V.; Focarete, M.L.; Giacomini, D.; Iacono, E. Peptide mediated adhesion to beta-lactam ring of equine mesenchymal stem cells: A pilot study. Animals 2022, 12, 734. [Google Scholar] [CrossRef]
- Baldassarro, V.A.; Giraldi, V.; Giuliani, A.; Moretti, M.; Pagnotta, G.; Flagelli, A.; Clavenzani, P.; Lorenzini, L.; Giardino, L.; Focarete, M.L.; et al. Poly(l-lactic acid) scaffold releasing an α4β1 integrin agonist promotes nonfibrotic skin wound healing in diabetic mice. ACS Appl. Bio Mater. 2023, 6, 296–308. [Google Scholar] [CrossRef]
- Choi, W.-S.; Rice, W.J.; Stokes, D.L.; Coller, B.S. Three-dimensional reconstruction of intact human integrin αiibβ3: New implications for activation-dependent ligand binding. Blood 2013, 122, 4165–4171. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Huo, t.; Wu, h.; Moussa, Z.; Sen, M.; Dalton, V. Full-length αIIbβ3 CryoEM structure reveals intact integrin initiate-activation intrinsic architecture. Res. Sq. 2023, preprint. [Google Scholar]
- Li, J.; Springer, T.A. Energy landscape differences among integrins establish the framework for understanding activation. J. Cell Biol. 2018, 217, 397–412. [Google Scholar] [CrossRef]
- Zhu, J.; Zhu, J.; Springer, T.A. Complete integrin headpiece opening in eight steps. J. Cell Biol. 2013, 201, 1053–1068. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.P.; Kim, E.; Swift, M.; Smith, J.W.; Volkmann, N.; Hanein, D. Three-dimensional structures of full-length, membrane-embedded human α(iib)β(3) integrin complexes. Biophys. J. 2016, 110, 798–809. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.-Y.; Zhu, J.; Eng, E.T.; Hudson, N.E.; Springer, T.A. β-Subunit binding is sufficient for ligands to open the integrin αiibβ3 headpiece. J. Biol. Chem. 2016, 291, 4537–4546. [Google Scholar] [CrossRef]
- Xiong, J.-P.; Stehle, T.; Zhang, R.; Joachimiak, A.; Frech, M.; Goodman, S.L.; Arnaout, M.A. Crystal structure of the extracellular segment of integrin αvβ3 in complex with an Arg-Gly-Asp ligand. Science 2002, 296, 151–155. [Google Scholar] [CrossRef]
- Van Agthoven, J.F.; Xiong, J.P.; Alonso, J.L.; Rui, X.; Adair, B.D.; Goodman, S.L.; Arnaout, M.A. Structural basis for pure antagonism of integrin αVβ3 by a high-affinity form of fibronectin. Nat. Struct. Mol. Biol. 2014, 21, 383–388. [Google Scholar] [CrossRef]
- Li, J.; Fukase, Y.; Shang, Y.; Zou, W.; Muñoz-Félix, J.M.; Buitrago, L.; van Agthoven, J.; Zhang, Y.; Hara, R.; Tanaka, Y.; et al. Novel pure αvβ3 integrin antagonists that do not induce receptor extension, prime the receptor, or enhance angiogenesis at low concentrations. ACS Pharmacol. Transl. Sci. 2019, 2, 387–401. [Google Scholar] [CrossRef]
- Yu, Y.; Schurpf, T.; Springer, T.A. How natalizumab binds and antagonizes alpha 4 integrins. J. Biol. Chem. 2013, 288, 32314–32325. [Google Scholar] [CrossRef]
- Yu, Y.; Zhu, J.; Mi, L.-Z.; Walz, T.; Sun, H.; Chen, J.-F.; Springer, T.A. Structural specializations of α4β7, an integrin that mediates rolling adhesion. J. Cell Biol. 2012, 196, 131–146. [Google Scholar] [CrossRef] [PubMed]
- Jo, M.H.; Li, J.; Jaumouillé, V.; Hao, Y.; Coppola, J.; Yan, J.; Waterman, C.M.; Springer, T.A.; Ha, T. Single-molecule characterization of subtype-specific β1 integrin mechanics. Nat. Commun. 2022, 13, 7471. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, N.; Iwasaki, K.; Takagi, J. A systematic survey of conformational states in β1 and β4 integrins using negative-stain electron microscopy. J. Cell Sci. 2018, 131, jcs21675. [Google Scholar]
- Su, Y.; Xia, W.; Li, J.; Walz, T.; Humphries, M.J.; Vestweber, D.; Cabañas, C.; Lu, C.; Springer, T.A. Relating conformation to function in integrin α5β1. Proc. Natl. Acad. Sci. USA 2016, 113, E3872–E3881. [Google Scholar] [CrossRef]
- Li, J.; Springer, T.A. Integrin extension enables ultrasensitive regulation by cytoskeletal force. Proc. Natl. Acad. Sci. USA 2017, 114, 4685–4690. [Google Scholar] [CrossRef]
- Nagae, M.; Re, S.; Mihara, E.; Nogi, T.; Sugita, Y.; Takagi, J. Crystal structure of α5β1 integrin ectodomain: Atomic details of the fibronectin receptor. J. Cell Biol. 2012, 197, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Xia, W.; Springer, T.A. Metal ion and ligand binding of integrin α5β1. Proc. Natl. Acad. Sci. USA 2014, 111, 17863–17868. [Google Scholar] [CrossRef]
- Anderson, J.M.; Li, J.; Springer, T.A. Regulation of integrin α5β1 conformational states and intrinsic affinities by metal ions and the ADMIDAS. MBoC 2022, 33, ar56. [Google Scholar] [CrossRef] [PubMed]
- You, T.J.; Maxwell, D.S.; Kogan, T.P.; Chen, Q.; Li, J.; Kassir, J.; Holland, G.W.; Dixon, R.A. A 3D structure model of integrin alpha 4 beta 1 complex: I. Construction of a homology model of beta 1 and ligand binding analysis. Biophys. J. 2002, 82, 447–457. [Google Scholar] [CrossRef]
- Macchiarulo, A.; Costantino, G.; Meniconi, M.; Pleban, K.; Ecker, G.; Bellocchi, D.; Pellicciari, R. Insights into phenylalanine derivatives recognition of VLA-4 integrin: From a pharmacophoric study to 3D-QSAR and molecular docking analyses. J. Chem. Inf. Comput. Sci. 2004, 44, 1829–1839. [Google Scholar] [CrossRef]
- Tvaroška, I.; Kozmon, S.; Kóňa, J. Molecular modeling insights into the structure and behavior of integrins: A review. Cells 2023, 12, 324. [Google Scholar] [CrossRef] [PubMed]
- Thangapandian, S.; John, S.; Sakkiah, S.; Lee, K.W. Discovery of potential integrin VLA-4 antagonists using pharmacophore modeling, virtual screening and molecular docking studies. Chem. Biol. Drug Des. 2011, 78, 289–300. [Google Scholar] [CrossRef]
- Vasconcelos, D.; Chaves, B.; Albuquerque, A.; Andrade, L.; Henriques, A.; Sartori, G.; Savino, W.; Caffarena, E.; Martins-Da- Silva, J.H. Development of New Potential Inhibitors of β1 Integrins through In Silico Methods- Screening and Computational Validation. Life 2022, 12, 932. [Google Scholar] [CrossRef]
- Hatley, R.J.D.; Barrett, T.N.; Slack, R.J.; Watson, M.E.; Baillache, D.J.; Gruszka, A.; Washio, Y.; Rowedder, J.E.; Pogány, P.; Pal, S.; et al. The design of potent, selective and drug-like RGD αvβ1 small-molecule inhibitors derived from non-RGD α4β1 antagonists. ChemMedChem 2019, 14, 1315–1320. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yan, H.; Tai, Y.; Xue, Y.; Wei, Y.; Wang, K.; Zhao, Q.; Wang, S.; Kong, D.; Midgley, A.C. Design and evaluation of a polypeptide that mimics the integrin binding site for EDA fibronectin to block profibrotic cell activity. Int. J. Mol. Sci. 2021, 22, 1575. [Google Scholar] [CrossRef] [PubMed]
- Carbone, J.; Ghidini, A.; Romano, A.; Gentilucci, L.; Musiani, F. PacDOCK: A Web server for positional distance-based and interaction-based analysis of docking results. Molecules 2022, 27, 6884. [Google Scholar] [CrossRef] [PubMed]
- Paladino, A.; Civera, M.; Belvisi, L.; Colombo, G. High Affinity vs. native fibronectin in the modulation of αvβ3 integrin conformational dynamics: Insights from computational analyses and implications for molecular design. PLoS Comput. Biol. 2017, 13, e1005334. [Google Scholar] [CrossRef]
- Schumacher, S.; Dedden, D.; Vazquez Nunez, R.; Matoba, K.; Takagi, J.I.; Biertümpfel, C.; Mizuno, N. Structural insights into integrin α5β1 opening by fibronectin ligand. Sci. Adv. 2021, 7, eabe9716. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, T.; Giacomini, D.; Tolomelli, A.; Baiula, M.; Gentilucci, L. Conjecturing about Small-Molecule Agonists and Antagonists of α4β1 Integrin: From Mechanistic Insight to Potential Therapeutic Applications. Biomedicines 2024, 12, 316. https://doi.org/10.3390/biomedicines12020316
He T, Giacomini D, Tolomelli A, Baiula M, Gentilucci L. Conjecturing about Small-Molecule Agonists and Antagonists of α4β1 Integrin: From Mechanistic Insight to Potential Therapeutic Applications. Biomedicines. 2024; 12(2):316. https://doi.org/10.3390/biomedicines12020316
Chicago/Turabian StyleHe, Tingting, Daria Giacomini, Alessandra Tolomelli, Monica Baiula, and Luca Gentilucci. 2024. "Conjecturing about Small-Molecule Agonists and Antagonists of α4β1 Integrin: From Mechanistic Insight to Potential Therapeutic Applications" Biomedicines 12, no. 2: 316. https://doi.org/10.3390/biomedicines12020316
APA StyleHe, T., Giacomini, D., Tolomelli, A., Baiula, M., & Gentilucci, L. (2024). Conjecturing about Small-Molecule Agonists and Antagonists of α4β1 Integrin: From Mechanistic Insight to Potential Therapeutic Applications. Biomedicines, 12(2), 316. https://doi.org/10.3390/biomedicines12020316