
Shear Stress and the AMP-Activated Protein Kinase Independently Protect the Vascular Endothelium from Palmitate Lipotoxicity
 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cloning the Anti-Human AMPK-α shRNAs
2.3. Cell Culture and Treatment
2.4. Lentivirus Assembly and HUVEC Transduction
2.4.1. Flow Cytometry
2.4.2. Cell Culture under Flow/Shear Stress
2.4.3. Western Blotting
2.4.4. Statistics
3. Results
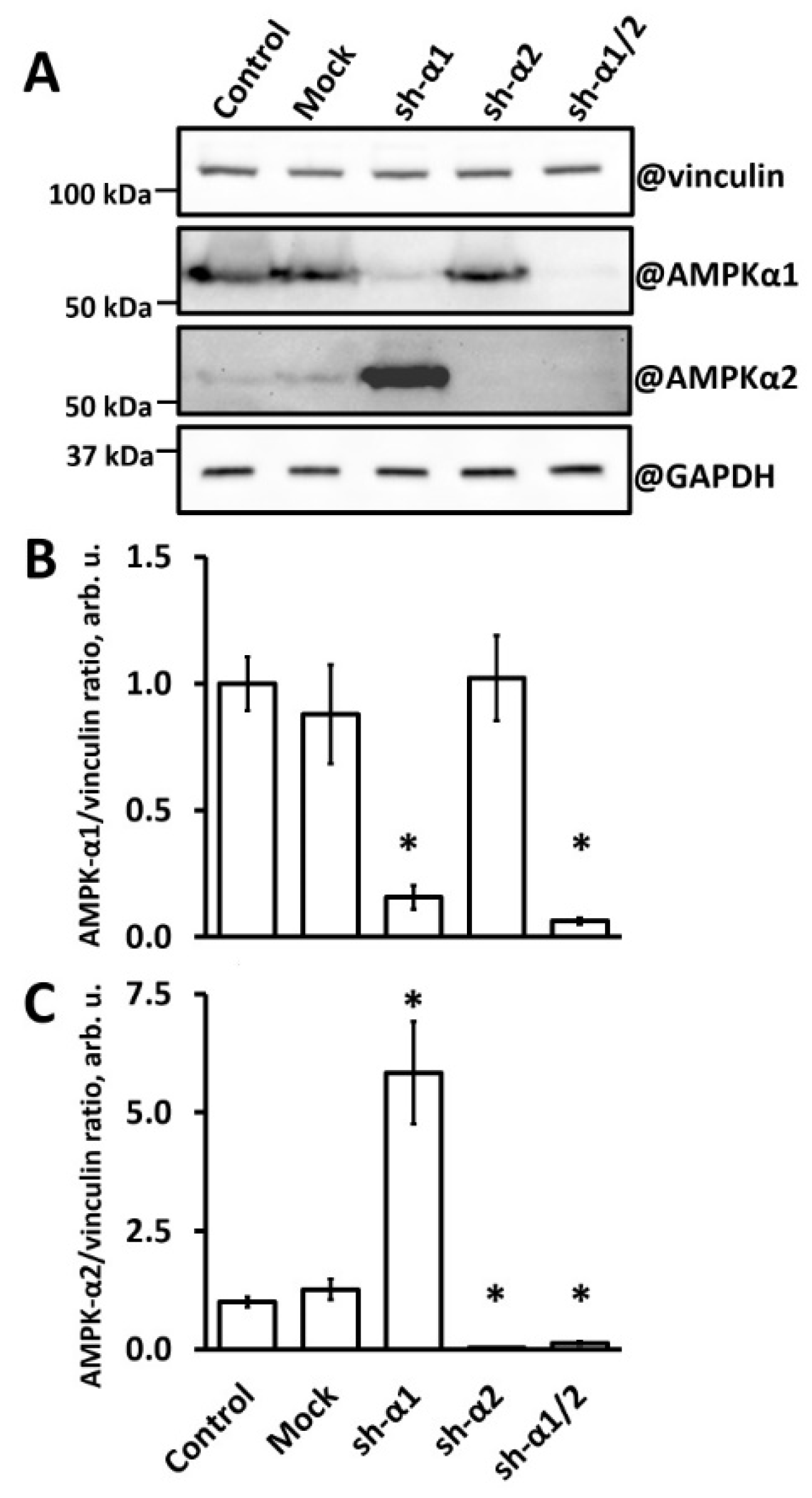
3.1. AMPK-α1 Knockdown in HUVEC Leads to AMPK-α2 Accumulation
3.2. AMPK-α1 Is Critical for HUVEC Protection from the Palmitate Lipotoxicity
3.3. Shear Stress Protects HUVEC from Palmitate Lipotoxicity Independently of AMPK
3.4. Shear Stress Protects Diabetic Microvascular Endothelial Cells from Palmitate Lipotoxicity
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Henderson, G.C. Plasma Free Fatty Acid Concentration as a Modifiable Risk Factor for Metabolic Disease. Nutrients 2021, 13, 2590. [Google Scholar] [CrossRef]
- Spiller, S.; Bluher, M.; Hoffmann, R. Plasma levels of free fatty acids correlate with type 2 diabetes mellitus. Diabetes Obes. Metab. 2018, 20, 2661–2669. [Google Scholar] [CrossRef]
- Alemany, M. Regulation of adipose tissue energy availability through blood flow control in the metabolic syndrome. Free Radic. Biol. Med. 2012, 52, 2108–2119. [Google Scholar] [CrossRef]
- Fuster, J.J.; Ouchi, N.; Gokce, N.; Walsh, K. Obesity-Induced Changes in Adipose Tissue Microenvironment and Their Impact on Cardiovascular Disease. Circ. Res. 2016, 118, 1786–1807. [Google Scholar] [CrossRef]
- Vorotnikov, A.V.; Khapchaev, A.Y.; Nickashin, A.V.; Shirinsky, V.P. In Vitro Modeling of Diabetes Impact on Vascular Endothelium: Are Essentials Engaged to Tune Metabolism? Biomedicines 2022, 10, 3181. [Google Scholar] [CrossRef]
- Arner, P.; Ryden, M. Fatty Acids, Obesity and Insulin Resistance. Obes. Facts 2015, 8, 147–155. [Google Scholar] [CrossRef]
- Roden, M.; Shulman, G.I. The integrative biology of type 2 diabetes. Nature 2019, 576, 51–60. [Google Scholar] [CrossRef]
- Yamagata, K. Fatty acids act on vascular endothelial cells and influence the development of cardiovascular disease. Prostaglandins Other Lipid Mediat. 2023, 165, 106704. [Google Scholar] [CrossRef]
- Ghosh, A.; Gao, L.; Thakur, A.; Siu, P.M.; Lai, C.W.K. Role of free fatty acids in endothelial dysfunction. J. Biomed. Sci. 2017, 24, 50. [Google Scholar] [CrossRef]
- Mallick, R.; Duttaroy, A.K. Modulation of endothelium function by fatty acids. Mol. Cell. Biochem. 2022, 477, 15–38. [Google Scholar] [CrossRef]
- Kim, J.A.; Montagnani, M.; Chandrasekran, S.; Quon, M.J. Role of lipotoxicity in endothelial dysfunction. Heart Fail. Clin. 2012, 8, 589–607. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Song, S.E.; Kim, Y.W.; Kim, J.Y.; Park, S.C.; Park, Y.K.; Baek, S.H.; Lee, I.K.; Park, S.Y. Adiponectin inhibits palmitate-induced apoptosis through suppression of reactive oxygen species in endothelial cells: Involvement of cAMP/protein kinase A and AMP-activated protein kinase. J. Endocrinol. 2010, 207, 35–44. [Google Scholar] [CrossRef]
- Kim, J.E.; Kim, Y.W.; Lee, I.K.; Kim, J.Y.; Kang, Y.J.; Park, S.Y. AMP-activated protein kinase activation by 5-aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside (AICAR) inhibits palmitate-induced endothelial cell apoptosis through reactive oxygen species suppression. J. Pharmacol. Sci. 2008, 106, 394–403. [Google Scholar] [CrossRef]
- Kim, F.; Tysseling, K.A.; Rice, J.; Pham, M.; Haji, L.; Gallis, B.M.; Baas, A.S.; Paramsothy, P.; Giachelli, C.M.; Corson, M.A.; et al. Free fatty acid impairment of nitric oxide production in endothelial cells is mediated by IKKbeta. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 989–994. [Google Scholar] [CrossRef]
- Mugabo, Y.; Mukaneza, Y.; Renier, G. Palmitate induces C-reactive protein expression in human aortic endothelial cells. Relevance to fatty acid-induced endothelial dysfunction. Metabolism 2011, 60, 640–648. [Google Scholar] [CrossRef]
- Maloney, E.; Sweet, I.R.; Hockenbery, D.M.; Pham, M.; Rizzo, N.O.; Tateya, S.; Handa, P.; Schwartz, M.W.; Kim, F. Activation of NF-kappaB by palmitate in endothelial cells: A key role for NADPH oxidase-derived superoxide in response to TLR4 activation. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1370–1375. [Google Scholar] [CrossRef]
- Wang, L.; Chen, Y.; Li, X.; Zhang, Y.; Gulbins, E.; Zhang, Y. Enhancement of endothelial permeability by free fatty acid through lysosomal cathepsin B-mediated Nlrp3 inflammasome activation. Oncotarget 2016, 7, 73229–73241. [Google Scholar] [CrossRef]
- Chinen, I.; Shimabukuro, M.; Yamakawa, K.; Higa, N.; Matsuzaki, T.; Noguchi, K.; Ueda, S.; Sakanashi, M.; Takasu, N. Vascular lipotoxicity: Endothelial dysfunction via fatty-acid-induced reactive oxygen species overproduction in obese Zucker diabetic fatty rats. Endocrinology 2007, 148, 160–165. [Google Scholar] [CrossRef]
- Liu, Q.; Cheng, Z.; Huang, B.; Luo, S.; Guo, Y. Palmitic acid promotes endothelial-to-mesenchymal transition via activation of the cytosolic DNA-sensing cGAS-STING pathway. Arch. Biochem. Biophys. 2022, 727, 109321. [Google Scholar] [CrossRef]
- Li, X.N.; Song, J.; Zhang, L.; LeMaire, S.A.; Hou, X.; Zhang, C.; Coselli, J.S.; Chen, L.; Wang, X.L.; Zhang, Y.; et al. Activation of the AMPK-FOXO3 pathway reduces fatty acid-induced increase in intracellular reactive oxygen species by upregulating thioredoxin. Diabetes 2009, 58, 2246–2257. [Google Scholar] [CrossRef]
- Zhou, J.; Li, Y.S.; Chien, S. Shear stress-initiated signaling and its regulation of endothelial function. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2191–2198. [Google Scholar] [CrossRef]
- Wragg, J.W.; Durant, S.; McGettrick, H.M.; Sample, K.M.; Egginton, S.; Bicknell, R. Shear stress regulated gene expression and angiogenesis in vascular endothelium. Microcirculation 2014, 21, 290–300. [Google Scholar] [CrossRef]
- Tamargo, I.A.; Baek, K.I.; Kim, Y.; Park, C.; Jo, H. Flow-induced reprogramming of endothelial cells in atherosclerosis. Nat. Rev. Cardiol. 2023, 20, 738–753. [Google Scholar] [CrossRef]
- Rennier, K.; Ji, J.Y. The role of death-associated protein kinase (DAPK) in endothelial apoptosis under fluid shear stress. Life Sci 2013, 93, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Doddaballapur, A.; Michalik, K.M.; Manavski, Y.; Lucas, T.; Houtkooper, R.H.; You, X.; Chen, W.; Zeiher, A.M.; Potente, M.; Dimmeler, S.; et al. Laminar shear stress inhibits endothelial cell metabolism via KLF2-mediated repression of PFKFB3. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 137–145. [Google Scholar] [CrossRef]
- Han, Y.; He, M.; Marin, T.; Shen, H.; Wang, W.T.; Lee, T.Y.; Hong, H.C.; Jiang, Z.L.; Garland, T., Jr.; Shyy, J.Y.; et al. Roles of KLF4 and AMPK in the inhibition of glycolysis by pulsatile shear stress in endothelial cells. Proc. Natl. Acad. Sci. USA 2021, 118, e2103982118. [Google Scholar] [CrossRef]
- Wang, Y.L.; Chen, C.T.; Tung, C.S.; Tsai, M.C. Laminar shear stress upregulates the expression of PPARs in vascular endothelial cells under high free fatty acid-induced stress. Exp. Ther. Med. 2021, 21, 438. [Google Scholar] [CrossRef]
- Maurya, M.R.; Gupta, S.; Li, J.Y.; Ajami, N.E.; Chen, Z.B.; Shyy, J.Y.; Chien, S.; Subramaniam, S. Longitudinal shear stress response in human endothelial cells to atheroprone and atheroprotective conditions. Proc. Natl. Acad. Sci. USA 2021, 118, e2023236118. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.; Ing, M.H.; Salazar, A.; Lassegue, B.; Griendling, K.; Navab, M.; Sevanian, A.; Hsiai, T.K. Pulsatile versus oscillatory shear stress regulates NADPH oxidase subunit expression: Implication for native LDL oxidation. Circ. Res. 2003, 93, 1225–1232. [Google Scholar] [CrossRef] [PubMed]
- Dimmeler, S.; Haendeler, J.; Rippmann, V.; Nehls, M.; Zeiher, A.M. Shear stress inhibits apoptosis of human endothelial cells. FEBS Lett. 1996, 399, 71–74. [Google Scholar] [CrossRef]
- Qin, X.; Tian, J.; Zhang, P.; Fan, Y.; Chen, L.; Guan, Y.; Fu, Y.; Zhu, Y.; Chien, S.; Wang, N. Laminar shear stress up-regulates the expression of stearoyl-CoA desaturase-1 in vascular endothelial cells. Cardiovasc. Res. 2007, 74, 506–514. [Google Scholar] [CrossRef]
- Dixit, M.; Bess, E.; Fisslthaler, B.; Hartel, F.V.; Noll, T.; Busse, R.; Fleming, I. Shear stress-induced activation of the AMP-activated protein kinase regulates FoxO1a and angiopoietin-2 in endothelial cells. Cardiovasc. Res. 2008, 77, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Young, A.; Wu, W.; Sun, W.; Benjamin Larman, H.; Wang, N.; Li, Y.S.; Shyy, J.Y.; Chien, S.; Garcia-Cardena, G. Flow activation of AMP-activated protein kinase in vascular endothelium leads to Kruppel-like factor 2 expression. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1902–1908. [Google Scholar] [CrossRef]
- Hirata, T.; Yamamoto, K.; Ikeda, K.; Arita, M. Functional lipidomics of vascular endothelial cells in response to laminar shear stress. FASEB J. 2021, 35, e21301. [Google Scholar] [CrossRef]
- Luo, J.Y.; Cheng, C.K.; He, L.; Pu, Y.; Zhang, Y.; Lin, X.; Xu, A.; Lau, C.W.; Tian, X.Y.; Ma, R.C.W.; et al. Endothelial UCP2 Is a Mechanosensitive Suppressor of Atherosclerosis. Circ. Res. 2022, 131, 424–441. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef]
- Steinberg, G.R.; Hardie, D.G. New insights into activation and function of the AMPK. Nat. Rev. Mol. Cell Biol. 2023, 24, 255–272. [Google Scholar] [CrossRef]
- Ruderman, N.B.; Carling, D.; Prentki, M.; Cacicedo, J.M. AMPK, insulin resistance, and the metabolic syndrome. J. Clin. Invest. 2013, 123, 2764–2772. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhao, J.; Ren, C.; Hu, B.; Ding, R.; He, Z.; Liang, C. Free fatty acids induce coronary microvascular dysfunction via inhibition of the AMPK/KLF2/eNOS signaling pathway. Int. J. Mol. Med. 2023, 51, 34. [Google Scholar] [CrossRef]
- Cacicedo, J.M.; Yagihashi, N.; Keaney, J.F., Jr.; Ruderman, N.B.; Ido, Y. AMPK inhibits fatty acid-induced increases in NF-kappaB transactivation in cultured human umbilical vein endothelial cells. Biochem. Biophys. Res. Commun. 2004, 324, 1204–1209. [Google Scholar] [CrossRef]
- Samsonov, M.V.; Podkuychenko, N.V.; Khapchaev, A.Y.; Efremov, E.E.; Yanushevskaya, E.V.; Vlasik, T.N.; Lankin, V.Z.; Stafeev, I.S.; Skulachev, M.V.; Shestakova, M.V.; et al. AICAR Protects Vascular Endothelial Cells from Oxidative Injury Induced by the Long-Term Palmitate Excess. Int. J. Mol. Sci. 2021, 23, 211. [Google Scholar] [CrossRef] [PubMed]
- Zou, M.H.; Wu, C. Adenosine Monophosphate-Activated Protein Kinase, Oxidative Stress, and Diabetic Endothelial Dysfunction. Cardiol. Discov. 2021, 1, 44–57. [Google Scholar] [CrossRef]
- Li, J.; Wang, Y.; Wang, Y.; Wen, X.; Ma, X.N.; Chen, W.; Huang, F.; Kou, J.; Qi, L.W.; Liu, B.; et al. Pharmacological activation of AMPK prevents Drp1-mediated mitochondrial fission and alleviates endoplasmic reticulum stress-associated endothelial dysfunction. J. Mol. Cell. Cardiol. 2015, 86, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Colombo, S.L.; Moncada, S. AMPKalpha1 regulates the antioxidant status of vascular endothelial cells. Biochem. J. 2009, 421, 163–169. [Google Scholar] [CrossRef]
- Stahmann, N.; Woods, A.; Spengler, K.; Heslegrave, A.; Bauer, R.; Krause, S.; Viollet, B.; Carling, D.; Heller, R. Activation of AMP-activated protein kinase by vascular endothelial growth factor mediates endothelial angiogenesis independently of nitric-oxide synthase. J. Biol. Chem. 2010, 285, 10638–10652. [Google Scholar] [CrossRef]
- Creighton, J.; Jian, M.; Sayner, S.; Alexeyev, M.; Insel, P.A. Adenosine monophosphate-activated kinase alpha1 promotes endothelial barrier repair. FASEB J. 2011, 25, 3356–3365. [Google Scholar] [CrossRef]
- Bess, E.; Fisslthaler, B.; Fromel, T.; Fleming, I. Nitric oxide-induced activation of the AMP-activated protein kinase alpha2 subunit attenuates IkappaB kinase activity and inflammatory responses in endothelial cells. PLoS ONE 2011, 6, e20848. [Google Scholar] [CrossRef]
- Yang, Q.; Xu, J.; Ma, Q.; Liu, Z.; Sudhahar, V.; Cao, Y.; Wang, L.; Zeng, X.; Zhou, Y.; Zhang, M.; et al. PRKAA1/AMPKalpha1-driven glycolysis in endothelial cells exposed to disturbed flow protects against atherosclerosis. Nat. Commun. 2018, 9, 4667. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, M.; Liang, B.; Xu, J.; Xie, Z.; Liu, C.; Viollet, B.; Yan, D.; Zou, M.H. AMPKalpha2 deletion causes aberrant expression and activation of NAD(P)H oxidase and consequent endothelial dysfunction in vivo: Role of 26S proteasomes. Circ. Res. 2010, 106, 1117–1128. [Google Scholar] [CrossRef]
- Zufferey, R.; Nagy, D.; Mandel, R.J.; Naldini, L.; Trono, D. Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nat. Biotechnol. 1997, 15, 871–875. [Google Scholar] [CrossRef] [PubMed]
- Khapchaev, A.Y.; Antonova, O.A.; Kazakova, O.A.; Samsonov, M.V.; Vorotnikov, A.V.; Shirinsky, V.P. Long-Term Experimental Hyperglycemia Does Not Impair Macrovascular Endothelial Barrier Integrity and Function in vitro. Biochemistry 2023, 88, 1126–1138. [Google Scholar] [CrossRef] [PubMed]
- Stewart, S.A.; Dykxhoorn, D.M.; Palliser, D.; Mizuno, H.; Yu, E.Y.; An, D.S.; Sabatini, D.M.; Chen, I.S.; Hahn, W.C.; Sharp, P.A.; et al. Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA 2003, 9, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Viollet, B.; Horman, S.; Leclerc, J.; Lantier, L.; Foretz, M.; Billaud, M.; Giri, S.; Andreelli, F. AMPK inhibition in health and disease. Crit. Rev. Biochem. Mol. Biol. 2010, 45, 276–295. [Google Scholar] [CrossRef]
- Phoenix, K.N.; Devarakonda, C.V.; Fox, M.M.; Stevens, L.E.; Claffey, K.P. AMPKalpha2 Suppresses Murine Embryonic Fibroblast Transformation and Tumorigenesis. Genes Cancer 2012, 3, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Zhang, M.; Liang, B.; Xie, Z.; Zhao, Z.; Asfa, S.; Choi, H.C.; Zou, M.H. Reduction of AMP-activated protein kinase alpha2 increases endoplasmic reticulum stress and atherosclerosis in vivo. Circulation 2010, 121, 792–803. [Google Scholar] [CrossRef] [PubMed]
- Kohlstedt, K.; Trouvain, C.; Boettger, T.; Shi, L.; Fisslthaler, B.; Fleming, I. AMP-activated protein kinase regulates endothelial cell angiotensin-converting enzyme expression via p53 and the post-transcriptional regulation of microRNA-143/145. Circ. Res. 2013, 112, 1150–1158. [Google Scholar] [CrossRef]
- Xing, J.; Wang, Q.; Coughlan, K.; Viollet, B.; Moriasi, C.; Zou, M.H. Inhibition of AMP-activated protein kinase accentuates lipopolysaccharide-induced lung endothelial barrier dysfunction and lung injury in vivo. Am. J. Pathol. 2013, 182, 1021–1030. [Google Scholar] [CrossRef]
- Aird, W.C. Phenotypic heterogeneity of the endothelium: I. Structure, function, and mechanisms. Circ. Res. 2007, 100, 158–173. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khapchaev, A.Y.; Vorotnikov, A.V.; Antonova, O.A.; Samsonov, M.V.; Shestakova, E.A.; Sklyanik, I.A.; Tomilova, A.O.; Shestakova, M.V.; Shirinsky, V.P. Shear Stress and the AMP-Activated Protein Kinase Independently Protect the Vascular Endothelium from Palmitate Lipotoxicity. Biomedicines 2024, 12, 339. https://doi.org/10.3390/biomedicines12020339
Khapchaev AY, Vorotnikov AV, Antonova OA, Samsonov MV, Shestakova EA, Sklyanik IA, Tomilova AO, Shestakova MV, Shirinsky VP. Shear Stress and the AMP-Activated Protein Kinase Independently Protect the Vascular Endothelium from Palmitate Lipotoxicity. Biomedicines. 2024; 12(2):339. https://doi.org/10.3390/biomedicines12020339
Chicago/Turabian StyleKhapchaev, Asker Y., Alexander V. Vorotnikov, Olga A. Antonova, Mikhail V. Samsonov, Ekaterina A. Shestakova, Igor A. Sklyanik, Alina O. Tomilova, Marina V. Shestakova, and Vladimir P. Shirinsky. 2024. "Shear Stress and the AMP-Activated Protein Kinase Independently Protect the Vascular Endothelium from Palmitate Lipotoxicity" Biomedicines 12, no. 2: 339. https://doi.org/10.3390/biomedicines12020339