
Baseline Blood Levels of Mucin-1 Are Associated with Crucial On-Treatment Adverse Outcomes in Patients with Idiopathic Pulmonary Fibrosis Receiving Antifibrotic Pirfenidone

 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Methods
2.1. Design and Study Population
2.2. Measurement of Blood Mucin-1 Levels
2.3. Important Definitions
2.4. Statistical Analysis
3. Results
3.1. Study Population
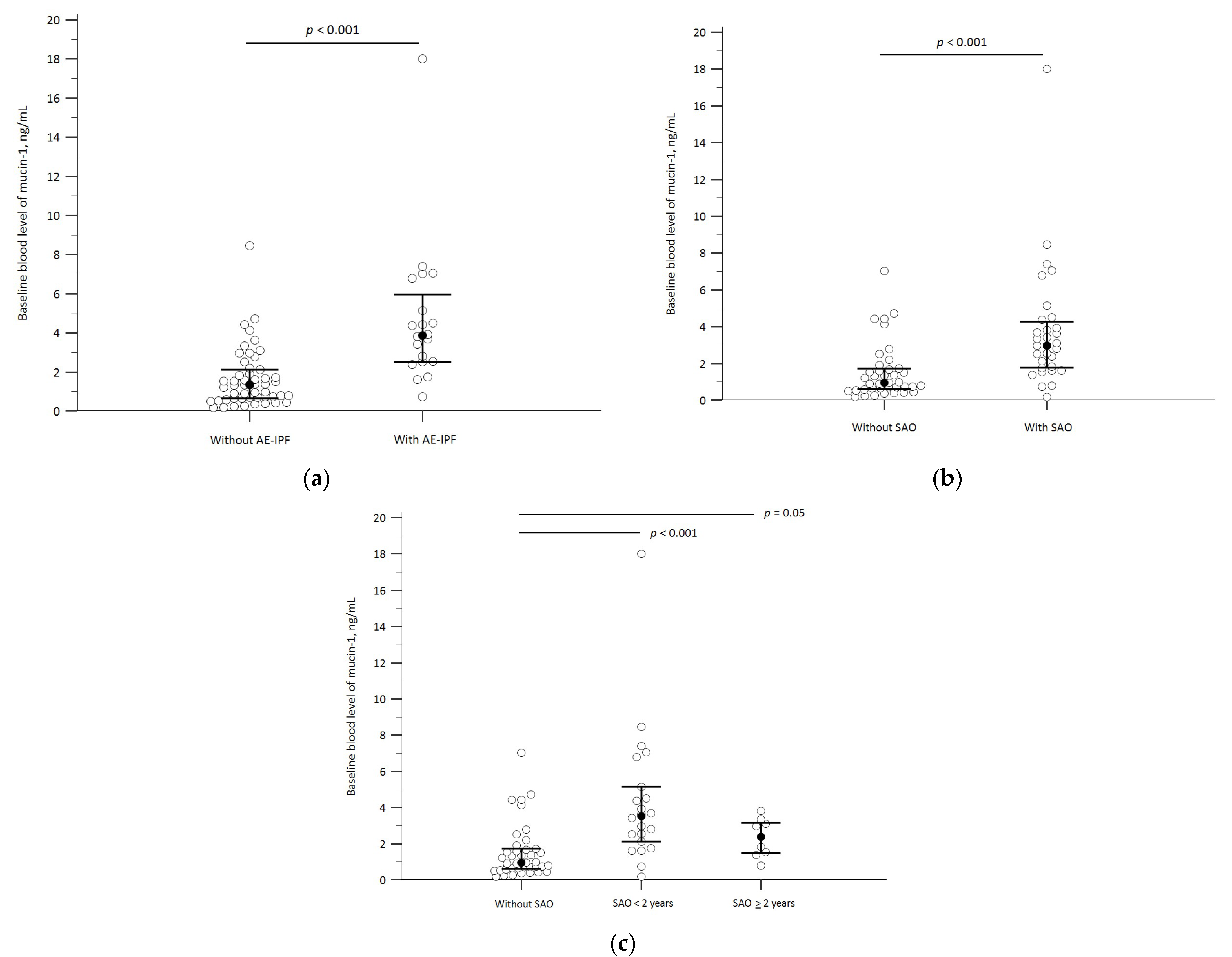
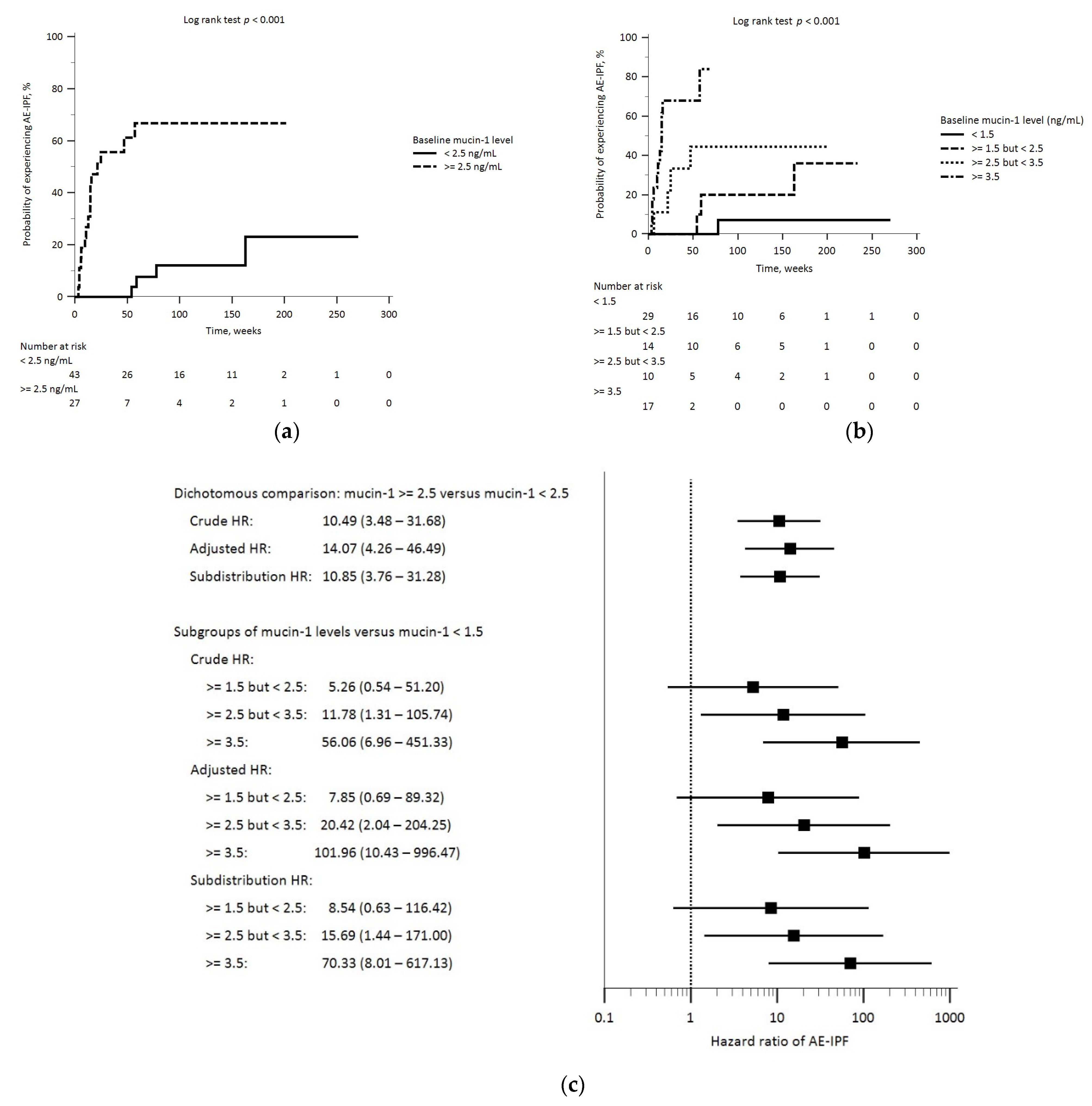
3.2. Baseline Mucin-1 Levels and AE-IPF
3.3. Baseline Mucin-1 and SAO
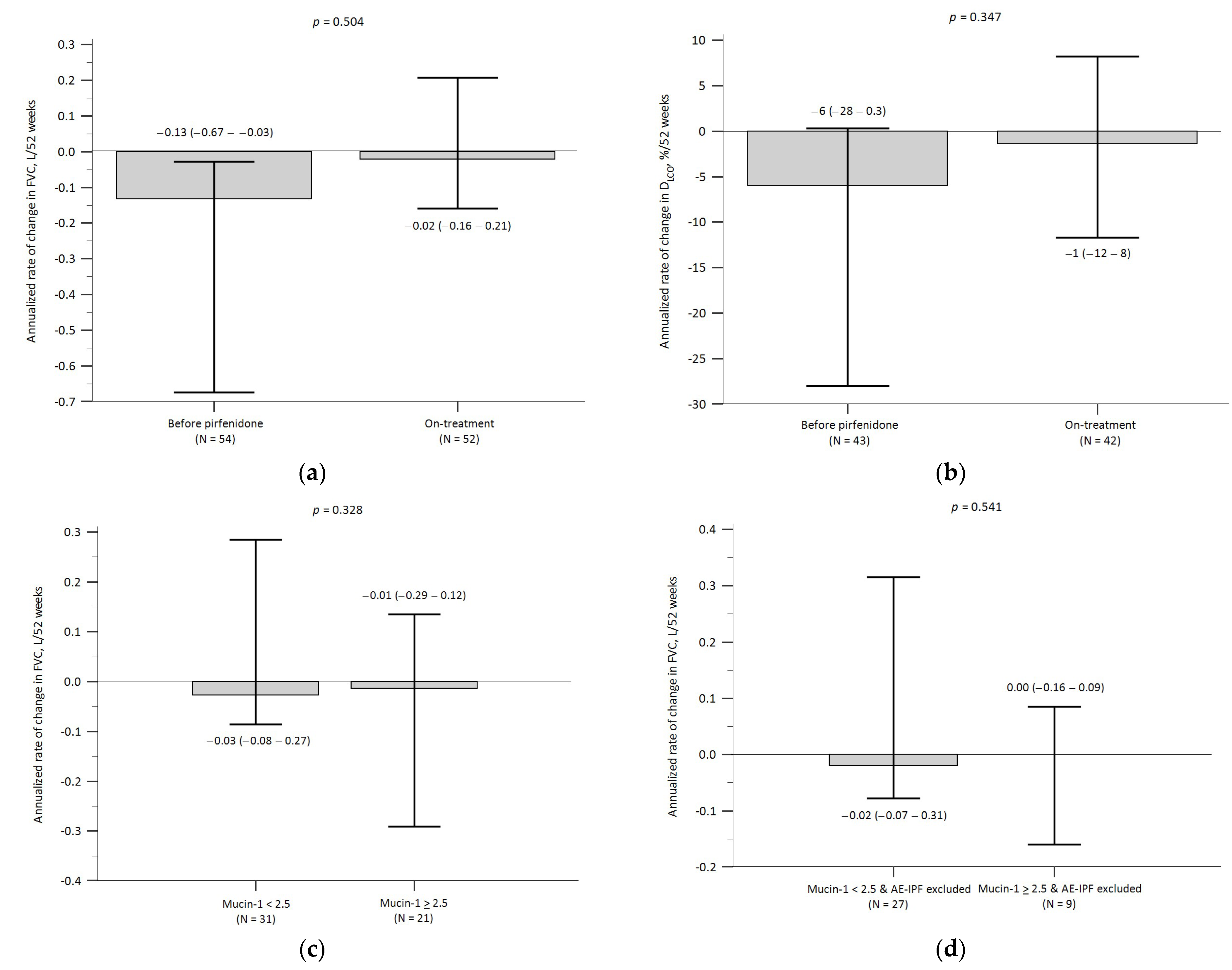
3.4. Sensitivity Analyses
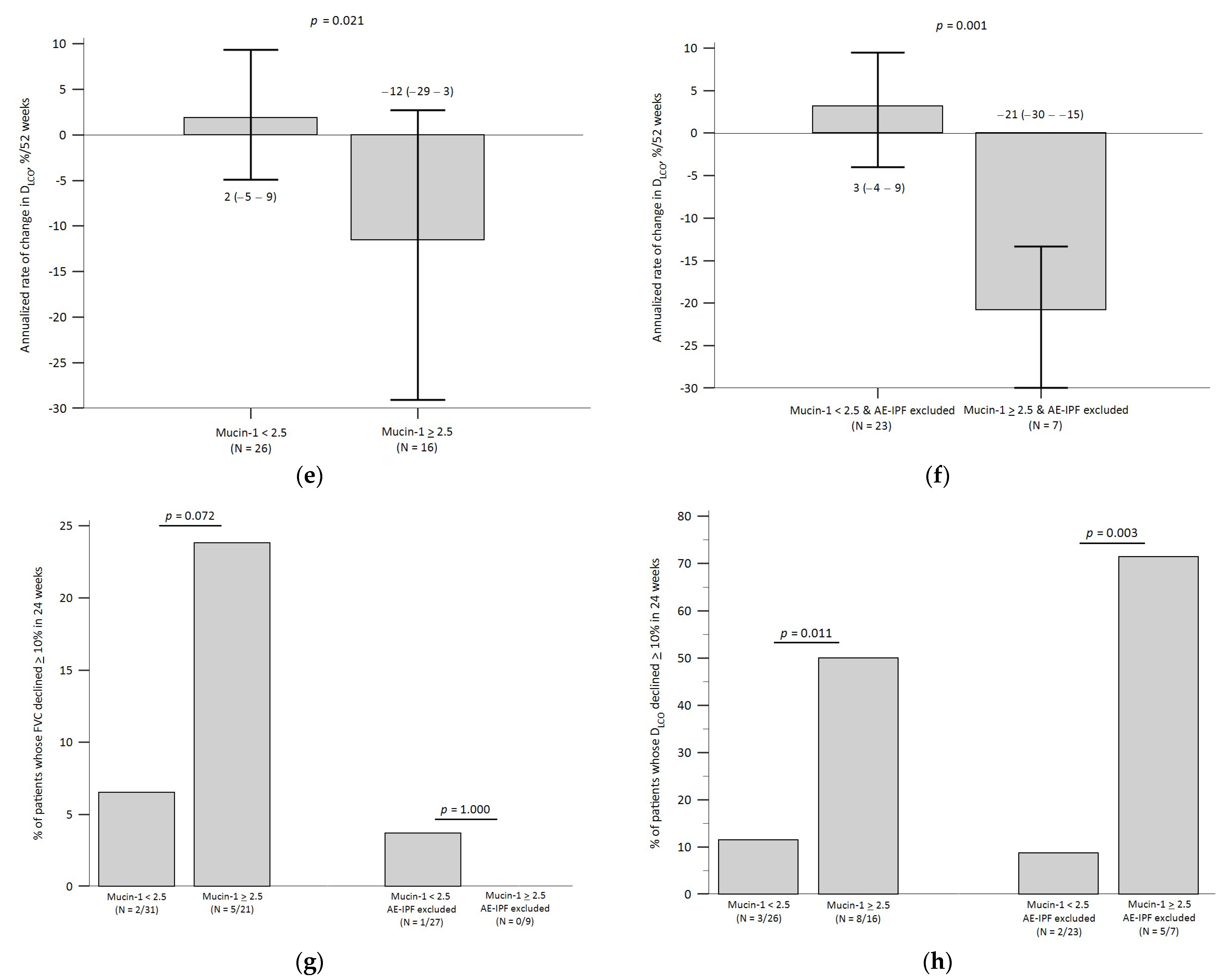
3.5. Baseline Mucin-1 and Pulmonary Function Decline
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AE-IPF | acute exacerbation of idiopathic pulmonary fibrosis |
| aHR | adjusted hazard ratio |
| CCI | Charlson comorbidity index |
| CH | Chest Hospital (subordinate to Ministry of Health and Welfare) |
| DLCO | diffusion capacity for carbon monoxide |
| ELISA | enzyme-linked immunosorbent assay |
| FVC | forced vital capacity |
| IPF | idiopathic pulmonary fibrosis |
| IQR | inter-quartile range |
| KL-6 | Krebs von den Lungen-6 |
| NA | not applicable |
| NCKUH | National Cheng Kung University Hospital |
| SAO | severe adverse outcomes |
| 95% CI | 95% confidence interval |
References
- Lederer, D.J.; Martinez, F.J. Idiopathic pulmonary fibrosis. N. Engl. J. Med. 2018, 378, 1811–1823. [Google Scholar] [CrossRef]
- Raghu, G.; Remy-Jardin, M.; Myers, J.L.; Richeldi, L.; Ryerson, C.J.; Lederer, D.J.; Behr, J.; Cottin, V.; Danoff, S.K.; Morell, F.; et al. Diagnosis of idiopathic pulmonary fibrosis an official ATS/ERS/JRS/ALAT clinical practice guideline. Am. J. Respir. Crit. Care Med. 2018, 198, e44–e68. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Remy-Jardin, M.; Richeldi, L.; Thomson, C.C.; Inoue, Y.; Johkoh, T.; Kreuter, M.; Lynch, D.A.; Maher, T.M.; Martinez, F.J.; et al. Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2022, 205, e18–e47. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.S. Acute exacerbations in patients with idiopathic pulmonary fibrosis. Respir. Res. 2013, 14, 86. [Google Scholar] [CrossRef] [PubMed]
- Collard, H.R.; Ryerson, C.J.; Corte, T.J.; Jenkins, G.; Kondoh, Y.; Lederer, D.J.; Lee, J.S.; Maher, T.M.; Wells, A.U.; Antoniou, K.M.; et al. Acute exacerbation of idiopathic pulmonary fibrosis—An international working group report. Am. J. Respir. Crit. Care Med. 2016, 194, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Ruwanpura, S.M.; Thomas, B.J.; Bardin, P.G. Pirfenidone: Molecular mechanisms and potential clinical applications in lung disease. Am. J. Respir. Cell Mol. Biol. 2020, 62, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Azuma, A.; Nukiwa, T.; Tsuboi, E.; Suga, M.; Abe, S.; Nakata, K.; Taguchi, Y.; Nagai, S.; Itoh, H.; Ohi, M.; et al. Double-blind, placebo-controlled trial of pirfenidone in patients with idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2005, 171, 1040–1047. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, H.; Ebina, M.; Kondoh, Y.; Ogura, T.; Azuma, A.; Suga, M.; Taguchi, Y.; Takahashi, H.; Nakata, K.; Sato, A.; et al. Pirfenidone Clinical Study Group in Japan. Pirfenidone in idiopathic pulmonary fibrosis. Eur. Respir. J. 2010, 35, 821–829. [Google Scholar] [CrossRef]
- King, T.E., Jr.; Bradford, W.Z.; Castro-Bernardini, S.; Fagan, E.A.; Glaspole, I.; Glassberg, M.K.; Gorina, E.; Hopkins, P.M.; Kardatzke, D.; Lancaster, L.; et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2083–2092. [Google Scholar] [CrossRef]
- Okuda, R.; Hagiwara, E.; Baba, T.; Kitamura, H.; Kato, T.; Ogura, T. Safety and efficacy of pirfenidone in idiopathic pulmonary fibrosis in clinical practice. Respir. Med. 2013, 107, 1431–1437. [Google Scholar] [CrossRef]
- Meyer, K.C.; Decker, C. Role of pirfenidone in the management of pulmonary fibrosis. Ther. Clin. Risk Manag. 2017, 13, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Alhamad, E.H. Pirfenidone treatment in idiopathic pulmonary fibrosis: A Saudi experience. Ann. Thorac. Med. 2015, 10, 38–43. [Google Scholar]
- Hughes, G.; Toellner, H.; Morris, H.; Leonard, C.; Chaudhuri, N. Real world experiences: Pirfenidone and nintedanib are effective and well tolerated treatments for idiopathic pulmonary fibrosis. J. Clin. Med. 2016, 5, 78. [Google Scholar] [CrossRef]
- Galli, J.A.; Pandya, A.; Vega-Olivo, M.; Dass, C.; Zhao, H.; Criner, G.J. Pirfenidone and nintedanib for pulmonary fibrosis in clinical practice: Tolerability and adverse drug reactions. Respirology 2017, 22, 1171–1178. [Google Scholar] [CrossRef]
- Zurkova, M.; Kriegova, E.; Kolek, V.; Lostakova, V.; Sterclova, M.; Bartos, V.; Doubkova, M.; Binkova, I.; Svoboda, M.; Strenkova, J.; et al. Effect of pirfenidone on lung function decline and survival: 5-yr experience from a real-life IPF cohort from the Czech EMPIRE registry. Respir. Res. 2019, 20, 16. [Google Scholar] [CrossRef]
- Feng, H.; Zhao, Y.; Li, Z.; Kang, J. Real-life experiences in a single center: Efficacy of pirfenidone in idiopathic pulmonary fibrosis and fibrotic idiopathic non-specific interstitial pneumonia patients. Ther. Adv. Respir. Dis. 2020, 14. [Google Scholar] [CrossRef]
- Cameli, P.; Refini, R.M.; Bergantini, L.; d’Alessandro, M.; Alonzi, V.; Magnoni, C.; Rottoli, P.; Sestini, P.; Bargagli, E. Long-term follow-up of patients with idio-pathic pulmonary fibrosis treated with pirfenidone or nintedanib: A Real-Life Comparison Study. Front. Mol. Biosci. 2020, 7, 581828. [Google Scholar] [CrossRef]
- Petnak, T.; Lertjitbanjong, P.; Thongprayoon, C.; Moua, T. Impact of anti-fibrotic therapy on mortality and acute exacerbation in idiopathic pulmonary fibrosis: A systematic review and meta-analysis. Chest 2021, 160, 1751–1763. [Google Scholar] [CrossRef]
- Ley, B.; Swigris, J.; Day, B.-M.; Stauffer, J.L.; Raimundo, K.; Chou, W.; Collard, H.R. Pirfenidone reduces respiratory-related hospitalizations in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2017, 196, 756–761. [Google Scholar] [CrossRef]
- Nathan, S.D.; Albera, C.; Bradford, W.Z.; Costabel, U.; Glaspole, I.; Glassberg, M.K.; Kardatzke, D.R.; Daigl, M.; Kirchgaessler, K.-U.; Lancaster, L.H.; et al. Effect of pirfenidone on mortality: Pooled analyses and meta-analyses of clinical trials in idiopathic pulmonary fibrosis. Lancet Respir. Med. 2017, 5, 33–41. [Google Scholar] [CrossRef]
- Furuya, K.; Sakamoto, S.; Shimizu, H.; Sekiya, M.; Kinoshita, A.; Isshiki, T.; Sugino, K.; Matsumoto, K.; Homma, S. Pirfenidone for acute exacerbation of idiopathic pulmonary fibrosis: A retrospective study. Respir. Med. 2017, 126, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Vianello, A.; Molena, B.; Turato, C.; Braccioni, F.; Arcaro, G.; Paladini, L.; Andretta, M.; Saetta, M. Pirfenidone improves the survival of patients with idiopathic pulmonary fibrosis hospitalized for acute exacerbation. Curr. Med. Res. Opin. 2019, 35, 1187–1190. [Google Scholar] [CrossRef] [PubMed]
- Wright, W.A.; Crowley, L.E.; Parekh, D.; Crawshaw, A.; Dosanjh, D.P.; Nightingale, P.; Thickett, D.R. Real-world retrospective observational study exploring the effectiveness and safety of antifibrotics in idiopathic pulmonary fibrosis. BMJ Open Respir. Res. 2021, 8, e000782. [Google Scholar] [CrossRef]
- Oltmanns, U.; Kahn, N.; Palmowski, K.; Träger, A.; Wenz, H.; Heussel, C.P.; Schnabel, P.A.; Puderbach, M.; Wiebel, M.; Ehlers-Tenenbaum, S.; et al. Pirfenidone in Idiopathic Pulmonary Fibrosis: Real-Life Experience from a German Tertiary Referral Center for Interstitial Lung Diseases. Respiration 2014, 88, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Biondini, D.; Balestro, E.; Lacedonia, D.; Cerri, S.; Milaneschi, R.; Luppi, F.; Cocconcelli, E.; Bazzan, E.; Clini, E.; Barbaro, M.P.F.; et al. Pretreatment rate of decay in forced vital capacity predicts long-term response to pirfenidone in patients with idiopathic pulmonary fibrosis. Sci. Rep. 2018, 8, 5961. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zhang, Z.; Zhang, S.; Zhu, P.; Ko, J.K.-S.; Yung, K.K.-L. MUC1: Structure, Function, and Clinic Application in Epithelial Cancers. Int. J. Mol. Sci. 2021, 22, 6567. [Google Scholar] [CrossRef] [PubMed]
- Ballester, B.; Milara, J.; Cortijo, J. The role of mucin 1 in respiratory diseases. Eur. Respir. Rev. 2021, 30, 200149. [Google Scholar] [CrossRef] [PubMed]
- Hirasawa, Y.; Kohno, N.; Yokoyama, A.; Inoue, Y.; Abe, M.; Hiwada, K. KL-6, a human MUC1 mucin, is chemotactic for human fibroblasts. Am. J. Respir. Cell Mol. Biol. 1997, 17, 501–507. [Google Scholar] [CrossRef]
- Ohshimo, S.; Yokoyama, A.; Hattori, N.; Ishikawa, N.; Hirasawa, Y.; Kohno, N. KL-6, a human MUC1 mucin, promotes proliferation and survival of lung fibroblasts. Biochem. Biophys. Res. Commun. 2005, 338, 1845–1852. [Google Scholar] [CrossRef]
- Inoue, Y.; Barker, E.; Daniloff, E.; Kohno, N.; Hiwada, K.; Newman, L.S. Pulmonary Epithelial Cell Injury and Alveolar–Capillary Permeability in Berylliosis. Am. J. Respir. Crit. Care Med. 1997, 156, 109–115. [Google Scholar] [CrossRef]
- Stainer, A.; Faverio, P.; Busnelli, S.; Catalano, M.; Della Zoppa, M.; Marruchella, A.; Pesci, A.; Luppi, F. Molecular Biomarkers in Idiopathic Pulmonary Fibrosis: State of the Art and Future Directions. Int. J. Mol. Sci. 2021, 22, 6255. [Google Scholar] [CrossRef] [PubMed]
- Kohno, N.; Kyoizumi, S.; Awaya, Y.; Fukuhara, H.; Yamakido, M.; Akiyama, M. New serum indicator of interstitial pneumonitis activity. Sialylated carbohydrate antigen KL-6. Chest 1989, 96, 68–73. [Google Scholar] [PubMed]
- Yokoyama, A.; Kondo, K.; Nakajima, M.; Matsushima, T.; Takahashi, T.; Nishimura, M.; Bando, M.; Sugiyama, Y.; Totani, Y.; Ishizaki, T.; et al. Prognostic value of circulating KL-6 in idiopathic pulmonary fibrosis. Respirology 2006, 11, 164–168. [Google Scholar] [CrossRef] [PubMed]
- Ohshimo, S.; Ishikawa, N.; Horimasu, Y.; Hattori, N.; Hirohashi, N.; Tanigawa, K.; Kohno, N.; Bonella, F.; Guzman, J.; Costabel, U. Baseline KL-6 predicts increased risk for acute exacerbation of idiopathic pulmonary fibrosis. Respir. Med. 2014, 108, 1031–1039. [Google Scholar] [CrossRef]
- Ley, B.; Brown, K.K.; Collard, H.R. Molecular biomarkers in idiopathic pulmonary fibrosis. Am. J. Physiol. Cell Mol. Physiol. 2014, 307, L681–L691. [Google Scholar] [CrossRef] [PubMed]
- D’Alessandro, M.; Bergantini, L.; Cameli, P.; Vietri, L.; Lanzarone, N.; Alonzi, V.; Pieroni, M.; Refini, R.M.; Sestini, P.; Bonella, F.; et al. Krebs von den Lungen-6 as a biomarker for disease severity assessment in interstitial lung disease: A comprehensive review. Respiration 2023, 102, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Shen, P.; Duan, C.; Gao, L. KL-6 as an Immunological Biomarker Predicts the Severity, Progression, Acute Exacerbation, and Poor Outcomes of Interstitial Lung Disease: A Systematic Review and Meta-Analysis. Front. Immunol. 2021, 12, 745233. [Google Scholar] [CrossRef]
- Yoshikawa, T.; Otsuka, M.; Chiba, H.; Ikeda, K.; Mori, Y.; Umeda, Y.; Nishikiori, H.; Kuronuma, K.; Takahashi, H. Surfactant protein A as a biomarker of outcomes of anti-fibrotic drug therapy in patients with idiopathic pulmonary fibrosis. BMC Pulm. Med. 2020, 20, 27. [Google Scholar]
- Majewski, S.; Szewczyk, K.; Żal, A.; Białas, A.J.; Miłkowska-Dymanowska, J.; Piotrowski, W.J. Serial Measurements of Circulating KL-6, SP-D, MMP-7, CA19-9, CA-125, CCL18, and Periostin in Patients with Idiopathic Pulmonary Fibrosis Receiving Antifi-brotic Therapy: An Exploratory Study. J. Clin. Med. 2021, 10, 3864. [Google Scholar] [CrossRef]
- Bergantini, L.; Bargagli, E.; Cameli, P.; Cekorja, B.; Lanzarone, N.; Pianigiani, L.; Vietri, L.; Bennett, D.; Sestini, P.; Rottoli, P. Serial KL-6 analysis in patients with idiopathic pulmonary fibrosis treated with nintedanib. Respir. Investig. 2019, 57, 290–291. [Google Scholar] [CrossRef]
- D’Alessandro, M.; Bergantini, L.; Cameli, P.; Pieroni, M.; Refini, R.M.; Sestini, P.; Bargagli, E. Serum Concentrations of KL-6 in Patients with IPF and Lung Cancer and Serial Measurements of KL-6 in IPF Patients Treated with Antifibrotic Therapy. Cancers 2021, 13, 689. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.G.; Choi, S.M.; Lee, J.H.; Kim, J.-Y.; Song, J.W. Blood Krebs von den Lungen-6 levels predict treatment response to antifibrotic therapy in patients with idiopathic pulmonary fibrosis. Respir. Res. 2022, 23, 334. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.H.; Kuo, C.W.; Chen, C.W.; Tseng, Y.L.; Wu, C.L.; Lin, S.H. Baseline plasma KL-6 level predicts adverse outcomes in patients with idiopathic pulmonary fibrosis receiving nintedanib: A retrospective real-world cohort study. BMC Pulm. Med. 2021, 21, 165. [Google Scholar]
- Raghu, G.; Collard, H.R.; Egan, J.J.; Martinez, F.J.; Behr, J.; Brown, K.K.; Colby, T.V.; Cordier, J.-F.; Flaherty, K.R.; Lasky, J.A.; et al. An official ATS/ERS/JRS/ALAT statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med. 2011, 183, 788–824. [Google Scholar] [CrossRef] [PubMed]
- Mukerjee, D.; George, D.S.; Knight, C.; Davar, J.; Wells, A.U.; Du Bois, R.M.; Black, C.M.; Coghlan, J.G. Echocardiography and pulmonary function as screening tests for pulmonary arterial hypertension in systemic sclerosis. Rheumatology 2004, 43, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Wakamatsu, K.; Nagata, N.; Kumazoe, H.; Oda, K.; Ishimoto, H.; Yoshimi, M.; Takata, S.; Hamada, M.; Koreeda, Y.; Takakura, K.; et al. Prognostic value of serial serum KL-6 measurements in patients with idiopathic pulmonary fibrosis. Respir. Investig. 2017, 55, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Vancheri, C.; Sebastiani, A.; Tomassetti, S.; Pesci, A.; Rogliani, P.; Tavanti, L.; Luppi, F.; Harari, S.; Rottoli, P.; Ghirardini, A.; et al. Pirfenidone in real life: A retrospective observational multicentre study in Italian patients with idiopathic pulmonary fibrosis. Respir. Med. 2019, 156, 78–84. [Google Scholar] [CrossRef]
- Song, M.J.; Moon, S.W.; Choi, J.S.; Lee, S.H.; Lee, S.H.; Chung, K.S.; Jung, J.Y.; Kang, Y.A.; Park, M.S.; Kim, Y.S.; et al. Efficacy of low dose pirfenidone in idiopathic pulmonary fibrosis: Real world experience from a tertiary university hospital. Sci. Rep. 2020, 10, 21218. [Google Scholar] [CrossRef]
- Song, J.W.; Hong, S.-B.; Lim, C.-M.; Koh, Y.; Kim, D.S. Acute exacerbation of idiopathic pulmonary fibrosis: Incidence, risk factors and outcome. Eur. Respir. J. 2011, 37, 356–363. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Baseline Characteristics and Outcome Events | Results |
|---|---|
| Age, years | 75.2 (± 9.5) |
| Sex | |
| Female, n (%) | 12 (17) |
| Male, n (%) | 58 (83) |
| Body mass index, kg/m2 | 23.5 (±3.5) |
| Body surface area, m2 | 1.66 (±0.16) |
| Smoking status | |
| Never smoker, n (%) | 25 (36) |
| Current smoker, n (%) | 7 (10) |
| Former smoker, n (%) | 38 (54) |
| Charlson comorbidity index | 5 (4–6) |
| Echocardiographic evidence of pulmonary hypertension, n (%) | 35 (50) |
| Baseline plasma mucin-1 level, ng/mL | 1.64 (0.75–3.43) |
| Baseline FVC, L | 1.98 (±0.50) |
| Baseline FVC, % predicted | 66 (±11) |
| Baseline DLCO, mmol/min/kPa | 2.72 (±1.23) |
| Baseline DLCO, % predicted | 53 (±23) |
| Stages based on the GAP index | |
| Stage 1, n (%) | 13 (19) |
| Stage 2, n (%) | 45 (64) |
| Stage 3, n (%) | 12 (17) |
| Dosing: | |
| Low (1200 mg/day), n (%) | 40 (57) |
| High (1800 mg/day), n (%) | 30 (43) |
| On-treatment AE-IPF, n (%) | 20 (29) |
| On-treatment SAO: | |
| Anytime during the follow-up period, n (%) | 31 (44) |
| All-cause mortality, n (%) | 30 (43) |
| Lung transplantation, n (%) | 1 (1) |
| Within 2 years following pirfenidone initiation, n (%) | 22 (31) |
| All-cause mortality, n (%) | 21 (30) |
| Lung transplantation, n (%) | 1 (1) |
| Duration of pirfenidone therapy, weeks | 55.9 (23.3–123.3) |
| Time to first on-treatment AE-IPF, weeks | 14.9 (7.9–50.6) |
| Time to on-treatment mortality, weeks | 52.6 (15.0–112.6) |
| Time to on-treatment lung transplantation, weeks | 68.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, T.-H.; Wei, S.-H.; Kuo, H.-I.; Hou, H.-Y.; Kuo, C.-W.; Tseng, Y.-L.; Lin, S.-H.; Wu, C.-L. Baseline Blood Levels of Mucin-1 Are Associated with Crucial On-Treatment Adverse Outcomes in Patients with Idiopathic Pulmonary Fibrosis Receiving Antifibrotic Pirfenidone. Biomedicines 2024, 12, 402. https://doi.org/10.3390/biomedicines12020402
Huang T-H, Wei S-H, Kuo H-I, Hou H-Y, Kuo C-W, Tseng Y-L, Lin S-H, Wu C-L. Baseline Blood Levels of Mucin-1 Are Associated with Crucial On-Treatment Adverse Outcomes in Patients with Idiopathic Pulmonary Fibrosis Receiving Antifibrotic Pirfenidone. Biomedicines. 2024; 12(2):402. https://doi.org/10.3390/biomedicines12020402
Chicago/Turabian StyleHuang, Tang-Hsiu, Sheng-Huan Wei, Hung-I Kuo, Hsin-Yu Hou, Chin-Wei Kuo, Yau-Lin Tseng, Sheng-Hsiang Lin, and Chao-Liang Wu. 2024. "Baseline Blood Levels of Mucin-1 Are Associated with Crucial On-Treatment Adverse Outcomes in Patients with Idiopathic Pulmonary Fibrosis Receiving Antifibrotic Pirfenidone" Biomedicines 12, no. 2: 402. https://doi.org/10.3390/biomedicines12020402
APA StyleHuang, T.-H., Wei, S.-H., Kuo, H.-I., Hou, H.-Y., Kuo, C.-W., Tseng, Y.-L., Lin, S.-H., & Wu, C.-L. (2024). Baseline Blood Levels of Mucin-1 Are Associated with Crucial On-Treatment Adverse Outcomes in Patients with Idiopathic Pulmonary Fibrosis Receiving Antifibrotic Pirfenidone. Biomedicines, 12(2), 402. https://doi.org/10.3390/biomedicines12020402