
Use of Oleuropein and Hydroxytyrosol for Cancer Prevention and Treatment: Considerations about How Bioavailability and Metabolism Impact Their Adoption in Clinical Routine



Abstract
:1. Introduction
2. Effects of OLE and HT on Solid Tumors
2.1. Skin Carcinogenesis and Melanoma
2.2. Thyroid Cancer
2.3. Lung and Pleural Cancers
2.4. Breast Cancer
2.5. Liver and Bile Duct Cancers
2.6. Colorectal Cancer
2.7. Pancreatic Cancer
2.8. Cervical Cancer
2.9. Ovarian Cancer
2.10. Prostate Cancer
2.11. Osteosarcoma
2.12. Central Nervous System Cancers
2.13. Other Solid Tumors
3. Effects of OLE and HT on Hematological Malignancies
3.1. Acute Promyelocytic Leukemia
3.2. Other Hematological Malignancies
4. Discussion
4.1. Use of OLE and HT in Clinical Routine: Absorption, Bioavailability, and Safety
4.2. Effects of OLE and HT on Drug Action and Metabolism
4.3. Use of OLE and HT to Alleviate Cancer Risk Factors and Chemotherapy Toxicity: ROS Homeostasis and Inflammation
5. Future Perspectives and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Screening, P.D.Q.; Board, P.E. PDQ Screening and Prevention Editorial Board, Cancer Prevention Overview (PDQ®): Health Professional Version; National Cancer Institute: Bethesda, MD, USA, 2002. [Google Scholar]
- Castro-Espin, C.; Agudo, A. The Role of Diet in Prognosis among Cancer Survivors: A Systematic Review and Meta-Analysis of Dietary Patterns and Diet Interventions. Nutrients 2022, 14, 348. [Google Scholar] [CrossRef]
- Mittelman, S.D. The Role of Diet in Cancer Prevention and Chemotherapy Efficacy. Annu. Rev. Nutr. 2020, 40, 273–297. [Google Scholar] [CrossRef]
- Dayi, T.; Oniz, A. Effects of the Mediterranean diet polyphenols on cancer development. J. Prev. Med. Hyg. 2022, 63 (Suppl. S3), E74–E80. [Google Scholar] [CrossRef]
- Monllor-Tormos, A.; García-Vigara, A.; Morgan, O.; García-Pérez, M.-Á.; Mendoza, N.; Tarín, J.J.; Cano, A. Mediterranean diet for cancer prevention and survivorship. Maturitas 2023, 178, 107841. [Google Scholar] [CrossRef] [PubMed]
- Morze, J.; Danielewicz, A.; Przybyłowicz, K.; Zeng, H.; Hoffmann, G.; Schwingshackl, L. An updated systematic review and meta-analysis on adherence to mediterranean diet and risk of cancer. Eur. J. Nutr. 2021, 60, 1561–1586. [Google Scholar] [CrossRef]
- Mentella, M.C.; Scaldaferri, F.; Ricci, C.; Gasbarrini, A.; Miggiano, G.A.D. Cancer and Mediterranean Diet: A Review. Nutrients 2019, 11, 2059. [Google Scholar] [CrossRef] [PubMed]
- Mahmod, A.I.; Haif, S.K.; Kamal, A.; Al-Ataby, I.A.; Talib, W.H. Chemoprevention effect of the Mediterranean diet on colorectal cancer: Current studies and future prospects. Front. Nutr. 2022, 9, 924192. [Google Scholar] [CrossRef]
- Briguglio, G.; Costa, C.; Pollicino, M.; Giambò, F.; Catania, S.; Fenga, C. Polyphenols in cancer prevention: New insights (Review). Int. J. Funct. Nutr. 2020, 1, 9. [Google Scholar] [CrossRef]
- Fki, I.; Sayadi, S.; Mahmoudi, A.; Daoued, I.; Marrekchi, R.; Ghorbel, H. Comparative Study on Beneficial Effects of Hydroxytyrosol- and Oleuropein-Rich Olive Leaf Extracts on High-Fat Diet-Induced Lipid Metabolism Disturbance and Liver Injury in Rats. Biomed. Res. Int. 2020, 2020, 1315202. [Google Scholar] [CrossRef] [PubMed]
- Antoniou, C.; Hull, J. The Anti-cancer Effect of Olea europaea L. Products: A Review. Curr. Nutr. Rep. 2021, 10, 99–124. [Google Scholar] [CrossRef] [PubMed]
- Otero, D.M.; Oliveira, F.M.; Lorini, A.; da Fonseca, B.; Oliveira, R.M.; Zambiazi, R.C. Oleuropein: Methods for extraction, purifying and applying. Rev. Ceres 2020, 67, 315–329. [Google Scholar] [CrossRef]
- Vogel, P.; Machado, I.K.; Garavaglia, J.; Zani, V.T.; de Souza, D.; Morelo Dal Bosco, S. Polyphenols benefits of olive leaf (Olea europaea L.) to human health. Nutr. Hosp. 2014, 31, 1427–1433. [Google Scholar] [CrossRef]
- Catinella, G.; Donzella, S.; Borgonovo, G.; Dallavalle, S.; Contente, M.L.; Pinto, A. Efficient 2-Step Enzymatic Cascade for the Bioconversion of Oleuropein into Hydroxytyrosol. Antioxidants 2022, 11, 260. [Google Scholar] [CrossRef]
- Yuan, J.-J.; Wang, C.-Z.; Ye, J.-Z.; Tao, R.; Zhang, Y.-S. Enzymatic Hydrolysis of Oleuropein from Olea europea (Olive) Leaf Extract and Antioxidant Activities. Molecules 2015, 20, 2903–2921. [Google Scholar] [CrossRef]
- Omar, S.H. Oleuropein in Olive and its Pharmacological Effects. Sci. Pharm. 2010, 78, 133–154. [Google Scholar] [CrossRef]
- Hernández-Fernández, A.; Garrido, Y.; Iniesta-López, E.; de los Ríos, A.P.; Quesada-Medina, J.; Hernández-Fernández, F.J. Recovering Polyphenols in Aqueous Solutions from Olive Mill Wastewater and Olive Leaf for Biological Applications. Processes 2023, 11, 2668. [Google Scholar] [CrossRef]
- Rivas-Garcia, L.; Navarro-Hortal, M.D.; Romero-Marquez, J.M.; Llopis, J.; Forbes-Hernández, T.Y.; Xiao, J.; Quiles, J.L.; Sanchez-Gonzalez, C. Valorization of Olea europaea and olive oil processing by-products/wastes. Adv. Food Nutr. Res. 2023, 2023, 193–212. [Google Scholar] [CrossRef]
- Castejón, M.L.; Montoya, T.; Alarcón-de-la-Lastra, C.; Sánchez-Hidalgo, M. Potential Protective Role Exerted by Secoiridoids from Olea europaea L. in Cancer, Cardiovascular, Neurodegenerative, Aging-Related, and Immunoinflammatory Diseases. Antioxidants 2020, 9, 149. [Google Scholar] [CrossRef]
- Gorzynik-Debicka, M.; Przychodzen, P.; Cappello, F.; Kuban-Jankowska, A.; Marino Gammazza, A.; Knap, N.; Wozniak, M.; Gorska-Ponikowska, M. Potential Health Benefits of Olive Oil and Plant Polyphenols. Int. J. Mol. Sci. 2018, 19, 686. [Google Scholar] [CrossRef]
- Nediani, C.; Ruzzolini, J.; Romani, A.; Calorini, L. Oleuropein, a Bioactive Compound from Olea europaea L., as a Potential Preventive and Therapeutic Agent in Non-Communicable Diseases. Antioxidants 2019, 8, 578. [Google Scholar] [CrossRef] [PubMed]
- Karković Marković, A.; Torić, J.; Barbarić, M.; Jakobušić Brala, C. Hydroxytyrosol, Tyrosol and Derivatives and Their Potential Effects on Human Health. Molecules 2019, 24, 2001. [Google Scholar] [CrossRef] [PubMed]
- Lockyer, S.; Corona, G.; Yaqoob, P.; Spencer, J.P.E.; Rowland, I. Secoiridoids delivered as olive leaf extract induce acute improvements in human vascular function and reduction of an inflammatory cytokine: A randomised, double-blind, placebo-controlled, cross-over trial. Br. J. Nutr. 2015, 114, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Pojero, F.; Aiello, A.; Gervasi, F.; Caruso, C.; Ligotti, M.E.; Calabrò, A.; Procopio, A.; Candore, G.; Accardi, G.; Allegra, M. Effects of Oleuropein and Hydroxytyrosol on Inflammatory Mediators: Consequences on Inflammaging. Int. J. Mol. Sci. 2022, 24, 380. [Google Scholar] [CrossRef] [PubMed]
- Pojero, F.; Gervasi, F.; Fiore, S.D.; Aiello, A.; Bonacci, S.; Caldarella, R.; Attanzio, A.; Candore, G.; Caruso, C.; Ligotti, M.E.; et al. Anti-Inflammatory Effects of Nutritionally Relevant Concentrations of Oleuropein and Hydroxytyrosol on Peripheral Blood Mononuclear Cells: An Age-Related Analysis. Int. J. Mol. Sci. 2023, 24, 11029. [Google Scholar] [CrossRef] [PubMed]
- Imran, M.; Nadeem, M.; Gilani, S.A.; Khan, S.; Sajid, M.W.; Amir, R.M. Antitumor Perspectives of Oleuropein and Its Metabolite Hydroxytyrosol: Recent Updates. J. Food Sci. 2018, 83, 1781–1791. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Sumiyoshi, M. Olive Leaf Extract and Its Main Component Oleuropein Prevent Chronic Ultraviolet B Radiation-Induced Skin Damage and Carcinogenesis in Hairless Mice. J. Nutr. 2009, 139, 2079–2086. [Google Scholar] [CrossRef]
- Song, H.; Lim, D.Y.; Jung, J.I.; Cho, H.J.; Park, S.Y.; Kwon, G.T.; Kang, Y.-H.; Lee, K.W.; Choi, M.-S.; Park, J.H.Y. Dietary oleuropein inhibits tumor angiogenesis and lymphangiogenesis in the B16F10 melanoma allograft model: A mechanism for the suppression of high-fat diet-induced solid tumor growth and lymph node metastasis. Oncotarget 2017, 8, 32027–32042. [Google Scholar] [CrossRef]
- de Carvalho, A.L.M.B.; Caselli, F.; Rodrigues, V.; Paiva-Martins, F.; Marques, M.P.M. Antiproliferative Activity of Olive Oil Phenolics against Human Melan oma Cells. Lett. Drug Des. Discov. 2017, 14, 1053–1059. [Google Scholar] [CrossRef]
- Ruzzolini, J.; Peppicelli, S.; Andreucci, E.; Bianchini, F.; Scardigli, A.; Romani, A.; la Marca, G.; Nediani, C.; Calorini, L. Oleuropein, the Main Polyphenol of Olea europaea Leaf Extract, Has an Anti-Cancer Effect on Human BRAF Melanoma Cells and Potentiates the Cytotoxicity of Current Chemotherapies. Nutrients 2018, 10, 1950. [Google Scholar] [CrossRef]
- Brito, C.; Tomás, A.; Silva, S.; Bronze, M.R.; Serra, A.T.; Pojo, M. The Impact of Olive Oil Compounds on the Metabolic Reprogramming of Cutaneous Melanoma Cell Models. Molecules 2021, 26, 289. [Google Scholar] [CrossRef]
- Costantini, F.; Di Sano, C.; Barbieri, G. The Hydroxytyrosol Induces the Death for Apoptosis of Human Melanoma Cells. Int. J. Mol. Sci. 2020, 21, 8074. [Google Scholar] [CrossRef]
- Bulotta, S.; Corradino, R.; Celano, M.; Maiuolo, J.; D’Agostino, M.; Oliverio, M.; Procopio, A.; Filetti, S.; Russo, D. Antioxidant and antigrowth action of peracetylated oleuropein in thyroid cancer cells. J. Mol. Endocrinol. 2013, 51, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Toteda, G.; Lupinacci, S.; Vizza, D.; Bonofiglio, R.; Perri, E.; Bonofiglio, M.; Lofaro, D.; La Russa, A.; Leone, F.; Gigliotti, P.; et al. High doses of hydroxytyrosol induce apoptosis in papillary and follicular thyroid cancer cells. J. Endocrinol. Investig. 2017, 40, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Antognelli, C.; Frosini, R.; Santolla, M.F.; Peirce, M.J.; Talesa, V.N. Oleuropein-Induced Apoptosis Is Mediated by Mitochondrial Glyoxalase 2 in NSCLC A549 Cells: A Mechanistic Inside and a Possible Novel Nonenzymatic Role for an Ancient Enzyme. Oxid. Med. Cell Longev. 2019, 2019, 8576961. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wu, J.; Zhang, Q.; Li, X.; Zhu, X.; Wang, Q.; Cao, S.; Du, L. Mitochondria-mediated apoptosis was induced by oleuropein in H1299 cells involving activation of p38 MAP kinase. J. Cell Biochem. 2019, 120, 5480–5494. [Google Scholar] [CrossRef] [PubMed]
- Ahmed Wani, T.; Masoodi, F.A.; Akhter, R.; Akram, T.; Gani, A.; Shabir, N. Nanoencapsulation of hydroxytyrosol in chitosan crosslinked with sodium bisulfate tandem ultrasonication: Techno-characterization, release and antiproliferative properties. Ultrason. Sonochem. 2022, 82, 105900. [Google Scholar] [CrossRef]
- Calderón-Montaño, J.M.; Madrona, A.; Burgos-Morón, E.; Orta, M.L.; Mateos, S.; Espartero, J.L.; López-Lázaro, M. Selective Cytotoxic Activity of New Lipophilic Hydroxytyrosol Alkyl Ether Derivatives. J. Agric. Food Chem. 2013, 61, 5046–5053. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, C.; Clericuzio, M.; Borghesi, B.; Cornara, L.; Ribulla, S.; Gosetti, F.; Marengo, E.; Burlando, B. Oleuropein-Enriched Olive Leaf Extract Affects Calcium Dynamics and Impairs Viability of Malignant Mesothelioma Cells. Evid.-Based Complement. Altern. Med. 2015, 2015, 908493. [Google Scholar] [CrossRef]
- Elamin, M.H.; Elmahi, A.B.; Daghestani, M.H.; Al-Olayan, E.M.; Al-Ajmi, R.A.; Alkhuriji, A.F.; Hamed, S.S.; Elkhadragy, M.F. Synergistic Anti-Breast-Cancer Effects of Combined Treatment with Oleuropein and Doxorubicin In Vivo. Altern. Ther. Health Med. 2019, 25, 17–24. [Google Scholar]
- Junkins, K.; Rodgers, M.; Phelan, S.A. Oleuropein Induces Cytotoxicity and Peroxiredoxin Over-expression in MCF-7 Human Breast Cancer Cells. Anticancer Res. 2023, 43, 4333–4339. [Google Scholar] [CrossRef]
- Han, J.; Talorete, T.P.N.; Yamada, P.; Isoda, H. Anti-proliferative and apoptotic effects of oleuropein and hydroxytyrosol on human breast cancer MCF-7 cells. Cytotechnology 2009, 59, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Scicchitano, S.; Vecchio, E.; Battaglia, A.M.; Oliverio, M.; Nardi, M.; Procopio, A.; Costanzo, F.; Biamonte, F.; Faniello, M.C. The Double-Edged Sword of Oleuropein in Ovarian Cancer Cells: From Antioxidant Functions to Cytotoxic Effects. Int. J. Mol. Sci. 2023, 24, 842. [Google Scholar] [CrossRef] [PubMed]
- Hassan, Z.K.; Elamin, M.H.; Omer, S.A.; Daghestani, M.H.; Al-Olayan, E.S.; Elobeid, M.A.; Virk, P. Oleuropein Induces Apoptosis via the p53 Pathway in Breast Cancer Cells. Asian Pac. J. Cancer Prev. 2013, 14, 6739–6742. [Google Scholar] [CrossRef] [PubMed]
- Sirianni, R.; Chimento, A.; De Luca, A.; Casaburi, I.; Rizza, P.; Onofrio, A.; Iacopetta, D.; Puoci, F.; Andò, S.; Maggiolini, M.; et al. Oleuropein and hydroxytyrosol inhibit MCF-7 breast cancer cell proliferation interfering with ERK1/2 activation. Mol. Nutr. Food Res. 2010, 54, 833–840. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Guasch, M.; Medrano, M.; Costa, I.; Vela, E.; Grau, M.; Escrich, E.; Moral, R. Extra-Virgin Olive Oil and Its Minor Compounds Influence Apoptosis in Experimental Mammary Tumors and Human Breast Cancer Cell Lines. Cancers 2022, 14, 905. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.-Y.; Zhu, J.-S.; Xie, J.; Zhang, Z.; Zhu, J.; Jiang, S.; Shen, W.-J.; Wu, B.; Ding, T.; Wang, S.-L. Hydroxytyrosol and Oleuropein Inhibit Migration and Invasion via Induction of Autophagy in ER-Positive Breast Cancer Cell Lines (MCF7 and T47D). Nutr. Cancer 2021, 73, 350–360. [Google Scholar] [CrossRef]
- Abtin, M.; Alivand, M.R.; Khaniani, M.S.; Bastami, M.; Zaeifizadeh, M.; Derakhshan, S.M. Simultaneous downregulation of miR-21 and miR-155 through oleuropein for breast cancer prevention and therapy. J. Cell Biochem. 2018, 119, 7151–7165. [Google Scholar] [CrossRef]
- Bayat, S.; Mansoori Derakhshan, S.; Mansoori Derakhshan, N.; Shekari Khaniani, M.; Alivand, M.R. Downregulation of HDAC2 and HDAC3 via oleuropein as a potent prevention and therapeutic agent in MCF-7 breast cancer cells. J. Cell Biochem. 2019, 120, 9172–9180. [Google Scholar] [CrossRef]
- Karousi, P.; Kontos, C.K.; Papakotsi, P.; Kostakis, I.K.; Skaltsounis, A.-L.; Scorilas, A. Next-generation sequencing reveals altered gene expression and enriched pathways in triple-negative breast cancer cells treated with oleuropein and oleocanthal. Funct. Integr. Genom. 2023, 23, 299. [Google Scholar] [CrossRef]
- Liu, L.; Ahn, K.S.; Shanmugam, M.K.; Wang, H.; Shen, H.; Arfuso, F.; Chinnathambi, A.; Alharbi, S.A.; Chang, Y.; Sethi, G.; et al. Oleuropein induces apoptosis via abrogating NF-κB activation cascade in estrogen receptor–negative breast cancer cells. J. Cell Biochem. 2019, 120, 4504–4513. [Google Scholar] [CrossRef]
- Hassan, Z.K.; Elamin, M.H.; Daghestani, M.H.; Omer, S.A.; Al-Olayan, E.M.; Elobeid, M.A.; Virk, P.; Mohammed, O.B. Oleuropein Induces Anti-metastatic Effects in Breast Cancer. Asian Pac. J. Cancer Prev. 2012, 13, 4555–4559. [Google Scholar] [CrossRef]
- Granados-Principal, S.; Quiles, J.L.; Ramirez-Tortosa, C.; Camacho-Corencia, P.; Sanchez-Rovira, P.; Vera-Ramirez, L.; Ramirez-Tortosa, M.C. Hydroxytyrosol inhibits growth and cell proliferation and promotes high expression of sfrp4 in rat mammary tumours. Mol. Nutr. Food Res. 2011, 55, S117–S126. [Google Scholar] [CrossRef]
- Calahorra, J.; Martínez-Lara, E.; De Dios, C.; Siles, E. Hypoxia modulates the antioxidant effect of hydroxytyrosol in MCF-7 breast cancer cells. PLoS ONE 2018, 13, e0203892. [Google Scholar] [CrossRef]
- Calahorra, J.; Martínez-Lara, E.; Granadino-Roldán, J.M.; Martí, J.M.; Cañuelo, A.; Blanco, S.; Oliver, F.J.; Siles, E. Crosstalk between hydroxytyrosol, a major olive oil phenol, and HIF-1 in MCF-7 breast cancer cells. Sci. Rep. 2020, 10, 6361. [Google Scholar] [CrossRef]
- Rosignoli, P.; Fuccelli, R.; Sepporta, M.V.; Fabiani, R. In Vitro chemo-preventive activities of hydroxytyrosol: The main phenolic compound present in extra-virgin olive oil. Food Funct. 2016, 7, 301–307. [Google Scholar] [CrossRef]
- Fabiani, R.; Sepporta, M.V.; Rosignoli, P.; De Bartolomeo, A.; Crescimanno, M.; Morozzi, G. Anti-proliferative and pro-apoptotic activities of hydroxytyrosol on different tumour cells: The role of extracellular production of hydrogen peroxide. Eur. J. Nutr. 2012, 51, 455–464. [Google Scholar] [CrossRef]
- El-azem, N.; Pulido-Moran, M.; Ramirez-Tortosa, C.L.; Quiles, J.L.; Cara, F.E.; Sanchez-Rovira, P.; Granados-Principal, S.; Ramirez-Tortosa, M. Modulation by hydroxytyrosol of oxidative stress and antitumor activities of paclitaxel in breast cancer. Eur. J. Nutr. 2019, 58, 1203–1211. [Google Scholar] [CrossRef]
- Benot-Dominguez, R.; Tupone, M.G.; Castelli, V.; d’Angelo, M.; Benedetti, E.; Quintiliani, M.; Cinque, B.; Forte, I.M.; Cifone, M.G.; Ippoliti, R.; et al. Olive leaf extract impairs mitochondria by pro-oxidant activity in MDA-MB-231 and OVCAR-3 cancer cells. Biomed. Pharmacother. 2021, 134, 111139. [Google Scholar] [CrossRef] [PubMed]
- Sarsour, E.H.; Goswami, M.; Kalen, A.L.; Lafin, J.T.; Goswami, P.C. Hydroxytyrosol inhibits chemokine C-C motif ligand 5 mediated aged quiescent fibroblast-induced stimulation of breast cancer cell proliferation. Age 2014, 36, 9645. [Google Scholar] [CrossRef] [PubMed]
- Perta, N.; Torrieri Di Tullio, L.; Cugini, E.; Fattibene, P.; Rapanotti, M.C.; Borromeo, I.; Forni, C.; Malaspina, P.; Cacciamani, T.; Di Marino, D.; et al. Hydroxytyrosol Counteracts Triple Negative Breast Cancer Cell Dissemination via Its Copper Complexing Properties. Biology 2023, 12, 1437. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Vazquez-Martin, A.; Garcia-Villalba, R.; Carrasco-Pancorbo, A.; Oliveras-Ferraros, C.; Fernandez-Gutierrez, A.; Segura-Carretero, A. tabAnti-HER2 (erbB-2) oncogene effects of phenolic compounds directly isolated from commercial Extra-Virgin Olive Oil (EVOO). BMC Cancer 2008, 8, 377. [Google Scholar] [CrossRef]
- Yan, C.-M.; Chai, E.-Q.; Cai, H.-Y.; Miao, G.-Y.; Ma, W. Oleuropein induces apoptosis via activation of caspases and suppression of phosphatidylinositol 3-kinase/protein kinase B pathway in HepG2 human hepatoma cell line. Mol. Med. Rep. 2015, 11, 4617–4624. [Google Scholar] [CrossRef]
- Katsoulieris, E.N. The olive leaf extract oleuropein exerts protective effects against oxidant-induced cell death, concurrently displaying pro-oxidant activity in human hepatocarcinoma cells. Redox Rep. 2016, 21, 90–97. [Google Scholar] [CrossRef]
- Bossio, S.; Perri, A.; Malivindi, R.; Giordano, F.; Rago, V.; Mirabelli, M.; Salatino, A.; Brunetti, A.; Greco, E.A.; Aversa, A. Oleuropein Counteracts Both the Proliferation and Migration of Intra- and Extragonadal Seminoma Cells. Nutrients 2022, 14, 2323. [Google Scholar] [CrossRef]
- Tutino, V.; Caruso, M.G.; Messa, C.; Perri, E.; Notarnicola, M. Antiproliferative, antioxidant and anti-inflammatory effects of hydroxytyrosol on human hepatoma HepG2 and Hep3B cell lines. Anticancer Res. 2012, 32, 5371–5377. [Google Scholar] [PubMed]
- Zhao, B.; Ma, Y.; Xu, Z.; Wang, J.; Wang, F.; Wang, D.; Pan, S.; Wu, Y.; Pan, H.; Xu, D.; et al. Hydroxytyrosol, a natural molecule from olive oil, suppresses the growth of human hepatocellular carcinoma cells via inactivating AKT and nuclear factor-kappa B pathways. Cancer Lett. 2014, 347, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.-H.; Liao, W.-C.; Lin, R.-A.; Chen, I.-S.; Wang, J.-L.; Chien, J.-M.; Kuo, C.-C.; Hao, L.-J.; Chou, C.-T.; Jan, C.-R. Hydroxytyrosol [2-(3,4-Dihydroxyphenyl)-Ethanol], a Natural Phenolic Compound Found in the Olive, Alters Ca2+ Signaling and Viability in Human HepG2 Hepatoma Cells. Chin. J. Physiol. 2022, 65, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Han, Z.; Ma, Y.; Song, R.; Pei, T.; Zheng, T.; Wang, J.; Xu, D.; Fang, X.; Jiang, H.; et al. Hydroxytyrosol inhibits cholangiocarcinoma tumor growth: An in vivo and in vitro study. Oncol. Rep. 2014, 31, 145–152. [Google Scholar] [CrossRef]
- Giner, E.; Recio, M.C.; Ríos, J.L.; Cerdá-Nicolás, J.M.; Giner, R.M. Chemopreventive effect of oleuropein in colitis-associated colorectal cancer in c57bl/6 mice. Mol. Nutr. Food Res. 2016, 60, 242–255. [Google Scholar] [CrossRef] [PubMed]
- Notarnicola, M.; Pisanti, S.; Tutino, V.; Bocale, D.; Rotelli, M.T.; Gentile, A.; Memeo, V.; Bifulco, M.; Perri, E.; Caruso, M.G. Effects of olive oil polyphenols on fatty acid synthase gene expression and activity in human colorectal cancer cells. Genes Nutr. 2011, 6, 63–69. [Google Scholar] [CrossRef]
- Cárdeno, A.; Sánchez-Hidalgo, M.; Rosillo, M.A.; de la Lastra, C.A. Oleuropein, a Secoiridoid Derived from Olive Tree, Inhibits the Proliferation of Human Colorectal Cancer Cell through Downregulation of HIF-1α. Nutr. Cancer 2013, 65, 147–156. [Google Scholar] [CrossRef]
- Guichard, C. Dihydroxyphenylethanol induces apoptosis by activating serine/threonine protein phosphatase PP2A and promotes the endoplasmic reticulum stress response in human colon carcinoma cells. Carcinogenesis 2006, 27, 1812–1827. [Google Scholar] [CrossRef]
- Leo, M.; Muccillo, L.; Dugo, L.; Bernini, R.; Santi, L.; Sabatino, L. Polyphenols Extracts from Oil Production Waste Products (OPWPs) Reduce Cell Viability and Exert Anti-Inflammatory Activity via PPARγ Induction in Colorectal Cancer Cells. Antioxidants 2022, 11, 624. [Google Scholar] [CrossRef]
- Zhang, S.-P.; Zhou, J.; Fan, Q.-Z.; Lv, X.-M.; Wang, T.; Wang, F.; Chen, Y.; Hong, S.-Y.; Liu, X.-P.; Xu, B.-S.; et al. Discovery of hydroxytyrosol as thioredoxin reductase 1 inhibitor to induce apoptosis and G1/S cell cycle arrest in human colorectal cancer cells via ROS generation. Exp. Ther. Med. 2021, 22, 829. [Google Scholar] [CrossRef]
- de las Hazas, M.-C.L.; Piñol, C.; Macià, A.; Motilva, M.-J. Hydroxytyrosol and the Colonic Metabolites Derived from Virgin Olive Oil Intake Induce Cell Cycle Arrest and Apoptosis in Colon Cancer Cells. J. Agric. Food Chem. 2017, 65, 6467–6476. [Google Scholar] [CrossRef] [PubMed]
- Sani, N.S.; Onsori, H.; Akrami, S.; Rahmati, M. A Comparison of the Anti-Cancer Effects of Free and PLGA-PAA Encapsulated Hydroxytyrosol on the HT-29 Colorectal Cancer Cell Line. Anticancer Agents Med. Chem. 2022, 22, 390–394. [Google Scholar] [CrossRef] [PubMed]
- Fabiani, R.; De Bartolomeo, A.; Rosignoli, P.; Servili, M.; Montedoro, G.F.; Morozzi, G. Cancer chemoprevention by hydroxytyrosol isolated from virgin olive oil through G1 cell cycle arrest and apoptosis. Eur. J. Cancer Prev. 2002, 11, 351–358. [Google Scholar] [CrossRef] [PubMed]
- del Saz-Lara, A.; Boughanem, H.; López de Las Hazas, M.-C.; Crespo, C.; Saz-Lara, A.; Visioli, F.; Macias-González, M.; Dávalos, A. Hydroxytyrosol decreases EDNRA expression through epigenetic modification in colorectal cancer cells. Pharmacol. Res. 2023, 187, 106612. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, C.; Bond, D.R.; Jankowski, H.; Weidenhofer, J.; Stathopoulos, C.E.; Roach, P.D.; Scarlett, C.J. The Olive Biophenols Oleuropein and Hydroxytyrosol Selectively Reduce Proliferation, Influence the Cell Cycle, and Induce Apoptosis in Pancreatic Cancer Cells. Int. J. Mol. Sci. 2018, 19, 1937. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Yang, L.; Liu, T.; Xun, J.; Zhuo, Y.; Zhang, L.; Zhang, Q.; Wang, X. Hydroxytyrosol Inhibits MDSCs and Promotes M1 Macrophages in Mice with Orthotopic Pancreatic Tumor. Front. Pharmacol. 2021, 12, 759172. [Google Scholar] [CrossRef]
- Jadid, M.F.S.; Shademan, B.; Chavoshi, R.; Seyyedsani, N.; Aghaei, E.; Taheri, E.; Goleij, P.; Hajazimian, S.; Karamad, V.; Behroozi, J.; et al. Enhanced anticancer potency of hydroxytyrosol and curcumin by PLGA-PAA nano-encapsulation on PANC -1 pancreatic cancer cell line. Environ. Toxicol. 2021, 36, 1043–1051. [Google Scholar] [CrossRef]
- Yao, J.; Wu, J.; Yang, X.; Yang, J.; Zhang, Y.; Du, L. Oleuropein Induced Apoptosis in HeLa Cells via a Mitochondrial Apoptotic Cascade Associated with Activation of the c-Jun NH2-Terminal Kinase. J. Pharmacol. Sci. 2014, 125, 300–311. [Google Scholar] [CrossRef]
- Amini-Farsani, Z.; Sheikhshabani, S.H.; Shaygan, N.; Asgharzade, S. The impact of oleuropein on miRNAs regulating cell death signaling pathway in human cervical cancer cells. Biotechnol. Appl. Biochem. 2023, 71, 61–71. [Google Scholar] [CrossRef]
- Acquaviva, R.; Di Giacomo, C.; Sorrenti, V.; Galvano, F.; Santangelo, R.; Cardile, V.; Gangia, S.; D’Orazio, N.; Abraham, N.G.; Vanella, L. Antiproliferative effect of oleuropein in prostate cell lines. Int. J. Oncol. 2012, 41, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Zubair, H.; Bhardwaj, A.; Ahmad, A.; Srivastava, S.K.; Khan, M.A.; Patel, G.K.; Singh, S.; Singh, A.P. Hydroxytyrosol Induces Apoptosis and Cell Cycle Arrest and Suppresses Multiple Oncogenic Signaling Pathways in Prostate Cancer Cells. Nutr. Cancer 2017, 69, 932–942. [Google Scholar] [CrossRef] [PubMed]
- León-González, A.J.; Sáez-Martínez, P.; Jiménez-Vacas, J.M.; Herrero-Aguayo, V.; Montero-Hidalgo, A.J.; Gómez-Gómez, E.; Madrona, A.; Castaño, J.P.; Espartero, J.L.; Gahete, M.D.; et al. Comparative Cytotoxic Activity of Hydroxytyrosol and Its Semisynthetic Lipophilic Derivatives in Prostate Cancer Cells. Antioxidants 2021, 10, 1348. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Li, Y.; Wang, H.; Cui, Y.; Feng, Z.; Li, H.; Li, Y.; Wang, Y.; Wurtz, K.; Weber, P.; et al. Hydroxytyrosol Promotes Superoxide Production and Defects in Autophagy Leading to Anti-proliferation and Apoptosis on Human Prostate Cancer Cells. Curr. Cancer Drug Targets 2013, 13, 625–639. [Google Scholar] [CrossRef] [PubMed]
- Przychodzen, P.; Wyszkowska, R.; Gorzynik-Debicka, M.; Kostrzewa, T.; Kuban-Jankowska, A.; Gorska-Ponikowska, M. Anticancer Potential of Oleuropein, the Polyphenol of Olive Oil, with 2-Methoxyestradiol, Separately or in Combination, in Human Osteosarcoma Cells. Anticancer Res. 2019, 39, 1243–1251. [Google Scholar] [CrossRef] [PubMed]
- Gioti, K.; Papachristodoulou, A.; Benaki, D.; Aligiannis, N.; Skaltsounis, A.-L.; Mikros, E.; Tenta, R. Assessment of the Nutraceutical Effects of Oleuropein and the Cytotoxic Effects of Adriamycin, When Administered Alone and in Combination, in MG-63 Human Osteosarcoma Cells. Nutrients 2021, 13, 354. [Google Scholar] [CrossRef] [PubMed]
- Moran, J.M.; Leal-Hernande, O.; Canal-Macías, M.L.; Roncero-Martin, R.; Guerrero-Bonmatty, R.; Aliaga, I.; Zamorano, J.D. Antiproliferative Properties of Oleuropein in Human Osteosarcoma Cells. Nat. Prod. Commun. 2016, 11, 1934578X1601100. [Google Scholar] [CrossRef]
- Seçme, M.; Eroğlu, C.; Dodurga, Y.; Bağcı, G. Investigation of anticancer mechanism of oleuropein via cell cycle and apoptotic pathways in SH-SY5Y neuroblastoma cells. Gene 2016, 585, 93–99. [Google Scholar] [CrossRef]
- Laghezza Masci, V.; Bernini, R.; Villanova, N.; Clemente, M.; Cicaloni, V.; Tinti, L.; Salvini, L.; Taddei, A.R.; Tiezzi, A.; Ovidi, E. In Vitro Anti-Proliferative and Apoptotic Effects of Hydroxytyrosyl Oleate on SH-SY5Y Human Neuroblastoma Cells. Int. J. Mol. Sci. 2022, 23, 12348. [Google Scholar] [CrossRef]
- Liu, M.; Wang, J.; Huang, B.; Chen, A.; Li, X. Oleuropein inhibits the proliferation and invasion of glioma cells via suppression of the AKT signaling pathway. Oncol. Rep. 2016, 36, 2009–2016. [Google Scholar] [CrossRef]
- Tezcan, G.; Aksoy, S.A.; Tunca, B.; Bekar, A.; Mutlu, M.; Cecener, G.; Egeli, U.; Kocaeli, H.; Demirci, H.; Taskapilioglu, M.O. Oleuropein modulates glioblastoma miRNA pattern different from Olea europaea leaf extract. Hum. Exp. Toxicol. 2019, 38, 1102–1110. [Google Scholar] [CrossRef]
- Xu, T.; Liu, X. Oleuropein inhibits invasion of squamous cell carcinoma of the head and neck through TGF-β1 signaling pathway. BMC Cancer 2022, 22, 942. [Google Scholar] [CrossRef] [PubMed]
- Türkdoğan, M.K.; Koçyiğit, A.; Güler, E.M.; Özer, Ö.F.; Demir, K.; Uğur, H. Oleuropein exhibits anticarcinogen effects against gastric cancer cell lines. Mol. Biol. Rep. 2023, 50, 9099–9105. [Google Scholar] [CrossRef] [PubMed]
- Fulco, N.; Fagan, S.; Phelan, S.A. Oleuropein Reduces Prdx1 Expression, Cell Proliferation and Viability in K562 Human Leukemia Cells. ARC J. Cancer Sci. 2019, 5, 1872. [Google Scholar] [CrossRef]
- Della Ragione, F.; Cucciolla, V.; Borriello, A.; Pietra, V.D.; Pontoni, G.; Racioppi, L.; Manna, C.; Galletti, P.; Zappia, V. Hydroxytyrosol, a Natural Molecule Occurring in Olive Oil, Induces Cytochrome c-Dependent Apoptosis. Biochem. Biophys. Res. Commun. 2000, 278, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Fabiani, R.; Rosignoli, P.; De Bartolomeo, A.; Fuccelli, R.; Morozzi, G. Inhibition of Cell Cycle Progression by Hydroxytyrosol Is Associated with Upregulation of Cyclin-Dependent Protein Kinase Inhibitors p21WAF1/Cip1 and p27Kip1 and with Induction of Differentiation in HL60 Cells. J. Nutr. 2008, 138, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Parra-Perez, A.M.; Pérez-Jiménez, A.; Gris-Cárdenas, I.; Bonel-Pérez, G.C.; Carrasco-Díaz, L.M.; Mokhtari, K.; García-Salguero, L.; Lupiáñez, J.A.; Rufino-Palomares, E.E. Involvement of the PI3K/AKT Intracellular Signaling Pathway in the AntiCancer Activity of Hydroxytyrosol, a Polyphenol from Olea europaea, in Hematological Cells and Implication of HSP60 Levels in Its Anti-Inflammatory Activity. Int. J. Mol. Sci. 2022, 23, 7053. [Google Scholar] [CrossRef] [PubMed]
- Fabiani, R.; Fuccelli, R.; Pieravanti, F.; De Bartolomeo, A.; Morozzi, G. Production of hydrogen peroxide is responsible for the induction of apoptosis by hydroxytyrosol on HL60 cells. Mol. Nutr. Food Res. 2009, 53, 887–896. [Google Scholar] [CrossRef]
- Rafehi, H.; Smith, A.J.; Balcerczyk, A.; Ziemann, M.; Ooi, J.; Loveridge, S.J.; Baker, E.K.; El-Osta, A.; Karagiannis, T.C. Investigation into the biological properties of the olive polyphenol, hydroxytyrosol: Mechanistic insights by genome-wide mRNA-Seq analysis. Genes Nutr. 2012, 7, 343–355. [Google Scholar] [CrossRef] [PubMed]
- Ricelli, A.; Gionfra, F.; Percario, Z.; De Angelis, M.; Primitivo, L.; Bonfantini, V.; Antonioletti, R.; Bullitta, S.M.; Saso, L.; Incerpi, S.; et al. Antioxidant and Biological Activities of Hydroxytyrosol and Homovanillic Alcohol Obtained from Olive Mill Wastewaters of Extra-Virgin Olive Oil Production. J. Agric. Food Chem. 2020, 68, 15428–15439. [Google Scholar] [CrossRef] [PubMed]
- Granados-Principal, S.; Quiles, J.L.; Ramirez-Tortosa, C.L.; Ochoa-Herrera, J.; Perez-Lopez, P.; Pulido-Moran, M.; Ramirez-Tortosa, M.C. Squalene ameliorates atherosclerotic lesions through the reduction of CD 36 scavenger receptor expression in macrophages. Mol. Nutr. Food Res. 2012, 56, 733–740. [Google Scholar] [CrossRef]
- Eddy, K.; Shah, R.; Chen, S. Decoding Melanoma Development and Progression: Identification of Therapeutic Vulnerabilities. Front. Oncol. 2021, 10, 626129. [Google Scholar] [CrossRef] [PubMed]
- Rabbie, R.; Ferguson, P.; Molina-Aguilar, C.; Adams, D.J.; Robles-Espinoza, C.D. Melanoma subtypes: Genomic profiles, prognostic molecular markers and therapeutic possibilities. J. Pathol. 2019, 247, 539–551. [Google Scholar] [CrossRef]
- Chacón, M.; Pfluger, Y.; Angel, M.; Waisberg, F.; Enrico, D. Uncommon Subtypes of Malignant Melanomas: A Review Based on Clinical and Molecular Perspectives. Cancers 2020, 12, 2362. [Google Scholar] [CrossRef] [PubMed]
- Saginala, K.; Barsouk, A.; Aluru, J.S.; Rawla, P.; Barsouk, A. Epidemiology of Melanoma. Med. Sci. 2021, 9, 63. [Google Scholar] [CrossRef] [PubMed]
- Cabral-Pacheco, G.A.; Garza-Veloz, I.; Castruita-De la Rosa, C.; Ramirez-Acuña, J.M.; Perez-Romero, B.A.; Guerrero-Rodriguez, J.F.; Martinez-Avila, N.; Martinez-Fierro, M.L. The Roles of Matrix Metalloproteinases and Their Inhibitors in Human Diseases. Int. J. Mol. Sci. 2020, 21, 9739. [Google Scholar] [CrossRef]
- Sun, X.; Kaufman, P.D. Ki-67: More than a proliferation marker. Chromosoma 2018, 127, 175–186. [Google Scholar] [CrossRef]
- Feng, Y.-M.; Chen, X.-H.; Zhang, X. Roles of PECAM-1 in cell function and disease progression. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 4082–4088. [Google Scholar] [PubMed]
- Apte, R.S.; Chen, D.S.; Ferrara, N. VEGF in Signaling and Disease: Beyond Discovery and Development. Cell 2019, 176, 1248–1264. [Google Scholar] [CrossRef] [PubMed]
- Montalto, F.I.; De Amicis, F. Cyclin D1 in Cancer: A Molecular Connection for Cell Cycle Control, Adhesion and Invasion in Tumor and Stroma. Cells 2020, 9, 2648. [Google Scholar] [CrossRef]
- Łukasik, P.; Załuski, M.; Gutowska, I. Cyclin-Dependent Kinases (CDK) and Their Role in Diseases Development–Review. Int. J. Mol. Sci. 2021, 22, 2935. [Google Scholar] [CrossRef]
- Nitulescu, G.; Van De Venter, M.; Nitulescu, G.; Ungurianu, A.; Juzenas, P.; Peng, Q.; Olaru, O.T.; Grădinaru, D.; Tsatsakis, A.; Tsoukalas, D.; et al. The Akt pathway in oncology therapy and beyond (Review). Int. J. Oncol. 2018, 53, 2319–2331. [Google Scholar] [CrossRef] [PubMed]
- Markert, C.L.; Shaklee, J.B.; Whitt, G.S. Evolution of a Gene. Science (1979) 1975, 189, 102–114. [Google Scholar] [CrossRef]
- Claps, G.; Faouzi, S.; Quidville, V.; Chehade, F.; Shen, S.; Vagner, S.; Robert, C. The multiple roles of LDH in cancer. Nat. Rev. Clin. Oncol. 2022, 19, 749–762. [Google Scholar] [CrossRef]
- Castegna, A.; Menga, A. Glutamine Synthetase: Localization Dictates Outcome. Genes 2018, 9, 108. [Google Scholar] [CrossRef]
- Mackenzie, B.; Erickson, J.D. Sodium-coupled neutral amino acid (System N/A) transporters of the SLC38 gene family. Pflügers Arch. Eur. J. Physiol. 2004, 447, 784–795. [Google Scholar] [CrossRef]
- Pérez-Escuredo, J.; Van Hée, V.F.; Sboarina, M.; Falces, J.; Payen, V.L.; Pellerin, L.; Sonveaux, P. Monocarboxylate transporters in the brain and in cancer. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2016, 1863, 2481–2497. [Google Scholar] [CrossRef]
- Malik, A.R.; Willnow, T.E. Excitatory Amino Acid Transporters in Physiology and Disorders of the Central Nervous System. Int. J. Mol. Sci. 2019, 20, 5671. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Lozada, Z.; Ortega, A. Milestone Review: Excitatory amino acid transporters—Beyond their expected function. J. Neurochem. 2023, 165, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Wang, W.; Yang, Y.; Gu, C. Exploring the role of glucose-6-phosphate dehydrogenase in cancer (Review). Oncol. Rep. 2020, 44, 2325–2336. [Google Scholar] [CrossRef]
- Marei, H.E.; Althani, A.; Afifi, N.; Hasan, A.; Caceci, T.; Pozzoli, G.; Morrione, A.; Giordano, A.; Cenciarelli, C. p53 signaling in cancer progression and therapy. Cancer Cell Int. 2021, 21, 703. [Google Scholar] [CrossRef]
- Jan, R.; Chaudhry, G.-S. Understanding Apoptosis and Apoptotic Pathways Targeted Cancer Therapeutics. Adv. Pharm. Bull. 2019, 9, 205–218. [Google Scholar] [CrossRef]
- Cavalcante, G.C.; Schaan, A.P.; Cabral, G.F.; Santana-da-Silva, M.N.; Pinto, P.; Vidal, A.F.; Ribeiro-Dos-Santos, Â. A Cell’s Fate: An Overview of the Molecular Biology and Genetics of Apoptosis. Int. J. Mol. Sci. 2019, 20, 4133. [Google Scholar] [CrossRef] [PubMed]
- Richard, I.A.; Burgess, J.T.; O’Byrne, K.J.; Bolderson, E. Beyond PARP1: The Potential of Other Members of the Poly (ADP-Ribose) Polymerase Family in DNA Repair and Cancer Therapeutics. Front. Cell Dev. Biol. 2022, 9, 801200. [Google Scholar] [CrossRef]
- Stope, M. Phosphorylation of histone H2A.X as a DNA-associated biomarker (Review). World Acad. Sci. J. 2021, 3, 31. [Google Scholar] [CrossRef]
- Aggarwal, V.; Tuli, H.S.; Varol, A.; Thakral, F.; Yerer, M.B.; Sak, K.; Varol, M.; Jain, A.; Khan, M.A.; Sethi, G. Role of Reactive Oxygen Species in Cancer Progression: Molecular Mechanisms and Recent Advancements. Biomolecules 2019, 9, 735. [Google Scholar] [CrossRef]
- Hu, J.; Yuan, I.J.; Mirshahidi, S.; Simental, A.; Lee, S.C.; Yuan, X. Thyroid Carcinoma: Phenotypic Features, Underlying Biology and Potential Relevance for Targeting Therapy. Int. J. Mol. Sci. 2021, 22, 1950. [Google Scholar] [CrossRef]
- Filetti, S.; Durante, C.; Hartl, D.; Leboulleux, S.; Locati, L.D.; Newbold, K.; Papotti, M.G.; Berruti, A.; ESMO Guidelines Committee. Thyroid cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2019, 30, 1856–1883. [Google Scholar] [CrossRef]
- Capdevila, J.; Awada, A.; Führer-Sakel, D.; Leboulleux, S.; Pauwels, P. Molecular diagnosis and targeted treatment of advanced follicular cell-derived thyroid cancer in the precision medicine era. Cancer Treat. Rev. 2022, 106, 102380. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Pan, W.; Liu, S.; Shen, Z.; Xu, Y.; Hu, L. ERK/MAPK signalling pathway and tumorigenesis (Review). Exp. Ther. Med. 2020, 19, 1997–2007. [Google Scholar] [CrossRef] [PubMed]
- Al Bitar, S.; Gali-Muhtasib, H. The Role of the Cyclin Dependent Kinase Inhibitor p21cip1/waf1 in Targeting Cancer: Molecular Mechanisms and Novel Therapeutics. Cancers 2019, 11, 1475. [Google Scholar] [CrossRef] [PubMed]
- Thai, A.A.; Solomon, B.J.; Sequist, L.V.; Gainor, J.F.; Heist, R.S. Lung cancer. Lancet 2021, 398, 535–554. [Google Scholar] [CrossRef] [PubMed]
- Alduais, Y.; Zhang, H.; Fan, F.; Chen, J.; Chen, B. Non-small cell lung cancer (NSCLC): A review of risk factors, diagnosis, and treatment. Medicine 2023, 102, e32899. [Google Scholar] [CrossRef] [PubMed]
- Shakeri, R.; Kheirollahi, A.; Davoodi, J. Apaf-1: Regulation and function in cell death. Biochimie 2017, 135, 111–125. [Google Scholar] [CrossRef] [PubMed]
- Vlahopoulos, S.A.; Logotheti, S.; Mikas, D.; Giarika, A.; Gorgoulis, V.; Zoumpourlis, V. The role of ATF-2 in oncogenesis. BioEssays 2008, 30, 314–327. [Google Scholar] [CrossRef] [PubMed]
- Huebner, K.; Procházka, J.; Monteiro, A.C.; Mahadevan, V.; Schneider-Stock, R. The activating transcription factor 2: An influencer of cancer progression. Mutagenesis 2019, 34, 375–389. [Google Scholar] [CrossRef]
- Watson, G.; Ronai, Z.A.; Lau, E. ATF2, a paradigm of the multifaceted regulation of transcription factors in biology and disease. Pharmacol. Res. 2017, 119, 347–357. [Google Scholar] [CrossRef]
- Han, J.; Wu, J.; Silke, J. An overview of mammalian p38 mitogen-activated protein kinases, central regulators of cell stress and receptor signaling. F1000Research 2020, 9, 653. [Google Scholar] [CrossRef] [PubMed]
- Canovas, B.; Nebreda, A.R. Diversity and versatility of p38 kinase signalling in health and disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 346–366. [Google Scholar] [CrossRef] [PubMed]
- Asciak, R.; George, V.; Rahman, N.M. Update on biology and management of mesothelioma. Eur. Respir. Rev. 2021, 30, 200226. [Google Scholar] [CrossRef] [PubMed]
- Smolarz, B.; Nowak, A.Z.; Romanowicz, H. Breast Cancer—Epidemiology, Classification, Pathogenesis and Treatment (Review of Literature). Cancers 2022, 14, 2569. [Google Scholar] [CrossRef] [PubMed]
- Mayrovitz, H.N. (Ed.) Breast Cancer; Exon Publications: Brisbane City, Australia, 2022. [Google Scholar] [CrossRef]
- Zinatizadeh, M.R.; Schock, B.; Chalbatani, G.M.; Zarandi, P.K.; Jalali, S.A.; Miri, S.R. The Nuclear Factor Kappa B (NF-kB) signaling in cancer development and immune diseases. Genes Dis. 2021, 8, 287–297. [Google Scholar] [CrossRef]
- Taniguchi, K.; Karin, M. NF-κB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef]
- Holliday, D.L.; Speirs, V. Choosing the right cell line for breast cancer research. Breast Cancer Res. 2011, 13, 215. [Google Scholar] [CrossRef]
- Lee, A.V.; Oesterreich, S.; Davidson, N.E. MCF-7 Cells—Changing the Course of Breast Cancer Research and Care for 45 Years. JNCI J. Natl. Cancer Inst. 2015, 107, djv073. [Google Scholar] [CrossRef]
- Rhee, S.G. Overview on Peroxiredoxin. Mol. Cells 2016, 39, 1–5. [Google Scholar] [CrossRef]
- Perkins, A.; Nelson, K.J.; Parsonage, D.; Poole, L.B.; Karplus, P.A. Peroxiredoxins: Guardians against oxidative stress and modulators of peroxide signaling. Trends Biochem. Sci. 2015, 40, 435–445. [Google Scholar] [CrossRef]
- Nicolussi, A.; D’Inzeo, S.; Capalbo, C.; Giannini, G.; Coppa, A. The role of peroxiredoxins in cancer. Mol. Clin. Oncol. 2017, 6, 139–153. [Google Scholar] [CrossRef] [PubMed]
- Bolduc, J.; Koruza, K.; Luo, T.; Malo Pueyo, J.; Vo, T.N.; Ezeriņa, D.; Messens, J. Peroxiredoxins wear many hats: Factors that fashion their peroxide sensing personalities. Redox Biol. 2021, 42, 101959. [Google Scholar] [CrossRef] [PubMed]
- Ning, L.; Rui, X.; Bo, W.; Qing, G. The critical roles of histone deacetylase 3 in the pathogenesis of solid organ injury. Cell Death Dis. 2021, 12, 734. [Google Scholar] [CrossRef] [PubMed]
- Jo, H.; Shim, K.; Kim, H.-U.; Jung, H.S.; Jeoung, D. HDAC2 as a target for developing anti-cancer drugs. Comput. Struct. Biotechnol. J. 2023, 21, 2048–2057. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.-H.; Tsao, C.-J. Emerging role of microRNA-21 in cancer. Biomed. Rep. 2016, 5, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Mattiske, S.; Suetani, R.J.; Neilsen, P.M.; Callen, D.F. The Oncogenic Role of miR-155 in Breast Cancer. Cancer Epidemiol. Biomark. Prev. 2012, 21, 1236–1243. [Google Scholar] [CrossRef] [PubMed]
- Aman, Y.; Schmauck-Medina, T.; Hansen, M.; Morimoto, R.I.; Simon, A.K.; Bjedov, I.; Palikaras, K.; Simonsen, A.; Johansen, T.; Tavernarakis, N.; et al. Autophagy in healthy aging and disease. Nat. Aging 2021, 1, 634–650. [Google Scholar] [CrossRef] [PubMed]
- Bell, E.S.; Coelho, P.P.; Ratcliffe, C.D.H.; Rajadurai, C.V.; Peschard, P.; Vaillancourt, R.; Zuo, D.; Park, M. LC3C-Mediated Autophagy Selectively Regulates the Met RTK and HGF-Stimulated Migration and Invasion. Cell Rep. 2019, 29, 4053–4068.e6. [Google Scholar] [CrossRef]
- Wu, Y.-T.; Tan, H.-L.; Shui, G.; Bauvy, C.; Huang, Q.; Wenk, M.R.; Ong, C.-N.; Codogno, P.; Shen, H.-M. Dual Role of 3-Methyladenine in Modulation of Autophagy via Different Temporal Patterns of Inhibition on Class I and III Phosphoinositide 3-Kinase. J. Biol. Chem. 2010, 285, 10850–10861. [Google Scholar] [CrossRef]
- Ngo, V.; Duennwald, M.L. Nrf2 and Oxidative Stress: A General Overview of Mechanisms and Implications in Human Disease. Antioxidants 2022, 11, 2345. [Google Scholar] [CrossRef]
- Chaltel-Lima, L.; Domínguez, F.; Domínguez-Ramírez, L.; Cortes-Hernandez, P. The Role of the Estrogen-Related Receptor Alpha (ERRa) in Hypoxia and Its Implications for Cancer Metabolism. Int. J. Mol. Sci. 2023, 24, 7983. [Google Scholar] [CrossRef]
- Yun, S.-H.; Han, S.-H.; Park, J.-I. Peroxisome Proliferator-Activated Receptor γ and PGC-1 α in Cancer: Dual Actions as Tumor Promoter and Suppressor. PPAR Res. 2018, 2018, 6727421. [Google Scholar] [CrossRef]
- Zhang, J.; Xiang, H.; Liu, J.; Chen, Y.; He, R.-R.; Liu, B. Mitochondrial Sirtuin 3: New emerging biological function and therapeutic target. Theranostics 2020, 10, 8315–8342. [Google Scholar] [CrossRef]
- Hoang, K.N.L.; Anstee, J.E.; Arnold, J.N. The Diverse Roles of Heme Oxygenase-1 in Tumor Progression. Front. Immunol. 2021, 12, 658315. [Google Scholar] [CrossRef] [PubMed]
- Ryter, S.W. Heme Oxygenase-1: An Anti-Inflammatory Effector in Cardiovascular, Lung, and Related Metabolic Disorders. Antioxidants 2022, 11, 555. [Google Scholar] [CrossRef] [PubMed]
- Mazari, A.M.A.; Zhang, L.; Ye, Z.-W.; Zhang, J.; Tew, K.D.; Townsend, D.M. The Multifaceted Role of Glutathione S-Transferases in Health and Disease. Biomolecules 2023, 13, 688. [Google Scholar] [CrossRef] [PubMed]
- Townsend, D.M.; Tew, K.D. The role of glutathione-S-transferase in anti-cancer drug resistance. Oncogene 2003, 22, 7369–7375. [Google Scholar] [CrossRef] [PubMed]
- Lowndes, S.A.; Harris, A.L. The Role of Copper in Tumour Angiogenesis. J. Mammary Gland. Biol. Neoplasia 2005, 10, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.; Avadhani, N.G. Cytochrome c oxidase dysfunction in oxidative stress. Free Radic. Biol. Med. 2012, 53, 1252–1263. [Google Scholar] [CrossRef] [PubMed]
- Masoud, G.N.; Li, W. HIF-1α pathway: Role, regulation and intervention for cancer therapy. Acta Pharm. Sin. B 2015, 5, 378–389. [Google Scholar] [CrossRef]
- Taylor, C.T.; Scholz, C.C. The effect of HIF on metabolism and immunity. Nat. Rev. Nephrol. 2022, 18, 573–587. [Google Scholar] [CrossRef]
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- Calderon-Martinez, E.; Landazuri-Navas, S.; Vilchez, E.; Cantu-Hernandez, R.; Mosquera-Moscoso, J.; Encalada, S.; Al Lami, Z.; Zevallos-Delgado, C.; Cinicola, J. Prognostic Scores and Survival Rates by Etiology of Hepatocellular Carcinoma: A Review. J. Clin. Med. Res. 2023, 15, 200–207. [Google Scholar] [CrossRef]
- Thulasiram, H.V.; Poulter, C.D. Farnesyl diphosphate synthase: The art of compromise between substrate selectivity and stereoselectivity. J. Am. Chem. Soc. 2006, 128, 15819–15823. [Google Scholar] [CrossRef] [PubMed]
- Fhu, C.W.; Ali, A. Fatty Acid Synthase: An Emerging Target in Cancer. Molecules 2020, 25, 3935. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.; Gao, J.-H.; Zhao, C.; Tong, H.; Zheng, S.-P.; Huang, Z.-Y.; Liu, R.; Tang, C.-W.; Li, J. SK-Hep1: Not hepatocellular carcinoma cells but a cell model for liver sinusoidal endothelial cells. Int. J. Clin. Exp. Pathol. 2018, 11, 2931–2938. [Google Scholar] [PubMed]
- Xue, R.; Meng, H.; Yin, J.; Xia, J.; Hu, Z.; Liu, H. The Role of Calmodulin vs. Synaptotagmin in Exocytosis. Front. Mol. Neurosci. 2021, 14, 691363. [Google Scholar] [CrossRef] [PubMed]
- Hessvik, N.P.; Llorente, A. Current knowledge on exosome biogenesis and release. Cell. Mol. Life Sci. 2018, 75, 193–208. [Google Scholar] [CrossRef] [PubMed]
- Brindley, P.J.; Bachini, M.; Ilyas, S.I.; Khan, S.A.; Loukas, A.; Sirica, A.E.; Teh, B.T.; Wongkham, S.; Gores, G.J. Cholangiocarcinoma. Nat. Rev. Dis. Primers 2021, 7, 65. [Google Scholar] [CrossRef] [PubMed]
- Halder, R.; Amaraneni, A.; Shroff, R.T. Cholangiocarcinoma: A review of the literature and future directions in therapy. Hepatobiliary Surg. Nutr. 2022, 11, 555–566. [Google Scholar] [CrossRef]
- Okumura, K.; Gogna, S.; Gachabayov, M.; Felsenreich, D.M.; McGuirk, M.; Rojas, A.; Quintero, L.; Seshadri, R.; Gu, K.; Dong, X.D. Gallbladder cancer: Historical treatment and new management options. World J. Gastrointest. Oncol. 2021, 13, 1317–1335. [Google Scholar] [CrossRef]
- Roa, J.C.; García, P.; Kapoor, V.K.; Maithel, S.K.; Javle, M.; Koshiol, J. Gallbladder cancer. Nat. Rev. Dis. Primers 2022, 8, 69. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.S.; Karuniawati, H.; Jairoun, A.A.; Urbi, Z.; Ooi, J.; John, A.; Lim, Y.C.; Kibria, K.M.K.; Mohiuddin, A.K.M.; Ming, L.C.; et al. Colorectal Cancer: A Review of Carcinogenesis, Global Epidemiology, Current Challenges, Risk Factors, Preventive and Treatment Strategies. Cancers 2022, 14, 1732. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Gautam, V.; Sandhu, A.; Rawat, K.; Sharma, A.; Saha, L. Current and emerging therapeutic approaches for colorectal cancer: A comprehensive review. World J. Gastrointest. Surg. 2023, 15, 495–519. [Google Scholar] [CrossRef] [PubMed]
- Banias, L.; Jung, I.; Chiciudean, R.; Gurzu, S. From Dukes-MAC Staging System to Molecular Classification: Evolving Concepts in Colorectal Cancer. Int. J. Mol. Sci. 2022, 23, 9455. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-Q.; Man, Q.-W.; Huo, F.-Y.; Gao, X.; Lin, H.; Li, S.-R.; Wang, J.; Su, F.-C.; Cai, L.; Shi, Y.; et al. STAT3 pathway in cancers: Past, present, and future. MedComm (Beijing) 2022, 3, e124. [Google Scholar] [CrossRef]
- Lee, Y.-J.; Jung, E.; Choi, J.; Hwang, J.-S.; Jeong, E.-J.; Roh, Y.; Ban, H.S.; Kim, S.; Kim, S.-K.; Kim, S.-Y.; et al. The EDN1/EDNRA/β-arrestin axis promotes colorectal cancer progression by regulating STAT3 phosphorylation. Int. J. Oncol. 2022, 62, 13. [Google Scholar] [CrossRef] [PubMed]
- Bjørklund, G.; Zou, L.; Wang, J.; Chasapis, C.T.; Peana, M. Thioredoxin reductase as a pharmacological target. Pharmacol. Res. 2021, 174, 105854. [Google Scholar] [CrossRef] [PubMed]
- Razavipour, S.F.; Harikumar, K.B.; Slingerland, J.M. p27 as a Transcriptional Regulator: New Roles in Development and Cancer. Cancer Res. 2020, 80, 3451–3458. [Google Scholar] [CrossRef]
- Chen, S.; Chen, J.; Hua, X.; Sun, Y.; Cui, R.; Sha, J.; Zhu, X. The emerging role of XBP1 in cancer. Biomed. Pharmacother. 2020, 127, 110069. [Google Scholar] [CrossRef]
- Vermot, A.; Petit-Härtlein, I.; Smith, S.M.E.; Fieschi, F. NADPH Oxidases (NOX): An Overview from Discovery, Molecular Mechanisms to Physiology and Pathology. Antioxidants 2021, 10, 890. [Google Scholar] [CrossRef]
- Akinyemi, A.O.; Simpson, K.E.; Oyelere, S.F.; Nur, M.; Ngule, C.M.; Owoyemi, B.C.D.; Ayarick, V.A.; Oyelami, F.F.; Obaleye, O.; Esoe, D.-P.; et al. Unveiling the dark side of glucose-regulated protein 78 (GRP78) in cancers and other human pathology: A systematic review. Mol. Med. 2023, 29, 112. [Google Scholar] [CrossRef]
- Rozpedek, W.; Pytel, D.; Mucha, B.; Leszczynska, H.; Diehl, J.A.; Majsterek, I. The Role of the PERK/eIF2α/ATF4/CHOP Signaling Pathway in Tumor Progression during Endoplasmic Reticulum Stress. Curr. Mol. Med. 2016, 16, 533–544. [Google Scholar] [CrossRef]
- Dhanasekaran, D.N.; Reddy, E.P. JNK-signaling: A multiplexing hub in programmed cell death. Genes Cancer 2017, 8, 682–694. [Google Scholar] [CrossRef] [PubMed]
- Hammouda, M.; Ford, A.; Liu, Y.; Zhang, J. The JNK Signaling Pathway in Inflammatory Skin Disorders and Cancer. Cells 2020, 9, 857. [Google Scholar] [CrossRef]
- Brennan, A.; Leech, J.T.; Kad, N.M.; Mason, J.M. Selective antagonism of cJun for cancer therapy. J. Exp. Clin. Cancer Res. 2020, 39, 184. [Google Scholar] [CrossRef] [PubMed]
- Park, W.; Chawla, A.; O’Reilly, E.M. Pancreatic Cancer. JAMA 2021, 326, 851. [Google Scholar] [CrossRef] [PubMed]
- Kolbeinsson, H.M.; Chandana, S.; Wright, G.P.; Chung, M. Pancreatic Cancer: A Review of Current Treatment and Novel Therapies. J. Investig. Surg. 2023, 36, 2129884. [Google Scholar] [CrossRef]
- Burmeister, C.A.; Khan, S.F.; Schäfer, G.; Mbatani, N.; Adams, T.; Moodley, J.; Prince, S. Cervical cancer therapies: Current challenges and future perspectives. Tumour. Virus Res. 2022, 13, 200238. [Google Scholar] [CrossRef]
- Duiker, E.W.; Mom, C.H.; de Jong, S.; Willemse, P.H.; Gietema, J.A.; van der Zee, A.G.; de Vries, E.G. The clinical trail of TRAIL. Eur. J. Cancer 2006, 42, 2233–2240. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, W.; Yan, L.; Zheng, P.; Li, J. miR-29a-3p directly targets Smad nuclear interacting protein 1 and inhibits the migration and proliferation of cervical cancer HeLa cells. PeerJ 2020, 8, e10148. [Google Scholar] [CrossRef]
- Wang, J.; Chen, L. The role of miRNAs in the invasion and metastasis of cervical cancer. Biosci. Rep. 2019, 39, BSR20181377. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, W.; Wu, X.; Liu, W.; Ding, F. miR-16-5p modulates the radiosensitivity of cervical cancer cells via regulating coactivator-associated arginine methyltransferase 1. Pathol. Int. 2020, 70, 12–20. [Google Scholar] [CrossRef]
- Veneziani, A.C.; Gonzalez-Ochoa, E.; Alqaisi, H.; Madariaga, A.; Bhat, G.; Rouzbahman, M.; Sneha, S.; Oza, A.M. Heterogeneity and treatment landscape of ovarian carcinoma. Nat. Rev. Clin. Oncol. 2023, 20, 820–842. [Google Scholar] [CrossRef]
- Lino-Silva, L.S. Ovarian carcinoma: Pathology review with an emphasis in their molecular characteristics. Chin. Clin. Oncol. 2020, 9, 45. [Google Scholar] [CrossRef]
- Sekhoacha, M.; Riet, K.; Motloung, P.; Gumenku, L.; Adegoke, A.; Mashele, S. Prostate Cancer Review: Genetics, Diagnosis, Treatment Options, and Alternative Approaches. Molecules 2022, 27, 5730. [Google Scholar] [CrossRef]
- Fujita, K.; Nonomura, N. Role of Androgen Receptor in Prostate Cancer: A Review. World J. Mens. Health 2019, 37, 288. [Google Scholar] [CrossRef] [PubMed]
- Sayegh, N.; Swami, U.; Agarwal, N. Recent Advances in the Management of Metastatic Prostate Cancer. JCO Oncol. Pract. 2022, 18, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Higashiyama, Y.; Goto, S.; Iida, T.; Cho, S.; Iwanaga, M.; Mori, K.; Tani, M.; Urata, Y. Regulation of γ -glutamycysteine synthetase expression in response to oxidative stress. Free Radic. Res. 1999, 31, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Forman, H.J.; Zhang, H.; Rinna, A. Glutathione: Overview of its protective roles, measurement, and biosynthesis. Mol. Aspects Med. 2009, 30, 1–12. [Google Scholar] [CrossRef]
- Chu, C.; Geng, Y.; Zhou, Y.; Sicinski, P. Cyclin E in normal physiology and disease states. Trends Cell Biol. 2021, 31, 732–746. [Google Scholar] [CrossRef]
- Misaghi, A.; Goldin, A.; Awad, M.; Kulidjian, A.A. Osteosarcoma: A comprehensive review. SICOT J. 2018, 4, 12. [Google Scholar] [CrossRef]
- Van Arendonk, K.; Chung, D. Neuroblastoma: Tumor Biology and Its Implications for Staging and Treatment. Children 2019, 6, 12. [Google Scholar] [CrossRef]
- Qiu, B.; Matthay, K.K. Advancing therapy for neuroblastoma. Nat. Rev. Clin. Oncol. 2022, 19, 515–533. [Google Scholar] [CrossRef]
- Saleban, M.; Harris, E.L.; Poulter, J.A. D-Type Cyclins in Development and Disease. Genes 2023, 14, 1445. [Google Scholar] [CrossRef]
- Besser, A.; Slingerland, J. CDK Inhibitors in Normal and Malignant Cells. In Encyclopedia of Cell Biology; Elsevier: Amsterdam, The Netherlands, 2023; pp. 243–253. [Google Scholar] [CrossRef]
- Schaff, L.R.; Mellinghoff, I.K. Glioblastoma and Other Primary Brain Malignancies in Adults. JAMA 2023, 329, 574. [Google Scholar] [CrossRef]
- Jovčevska, I.; Kočevar, N.; Komel, R. Glioma and glioblastoma—How much do we (not) know? Mol. Clin. Oncol. 2013, 1, 935–941. [Google Scholar] [CrossRef] [PubMed]
- Hanif, F.; Muzaffar, K.; Perveen, K.; Malhi, S.M.; Simjee, S.U. Glioblastoma Multiforme: A Review of its Epidemiology and Pathogenesis through Clinical Presentation and Treatment. Asian Pac. J. Cancer Prev. 2017, 18, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Oronsky, B.; Reid, T.R.; Oronsky, A.; Sandhu, N.; Knox, S.J. A Review of Newly Diagnosed Glioblastoma. Front. Oncol. 2021, 10, 574012. [Google Scholar] [CrossRef] [PubMed]
- Lei, Q.; Yang, Y.; Zhou, W.; Liu, W.; Li, Y.; Qi, N.; Li, Q.; Wen, Z.; Ding, L.; Huang, X.; et al. MicroRNA-based therapy for glioblastoma: Opportunities and challenges. Eur. J. Pharmacol. 2023, 938, 175388. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.P.; Castresana, J.S.; Shahi, M.H. Glioblastoma and MiRNAs. Cancers 2021, 13, 1581. [Google Scholar] [CrossRef]
- Buonfiglioli, A.; Efe, I.E.; Guneykaya, D.; Ivanov, A.; Huang, Y.; Orlowski, E.; Krüger, C.; Deisz, R.A.; Markovic, D.; Flüh, C.; et al. let-7 MicroRNAs Regulate Microglial Function and Suppress Glioma Growth through Toll-Like Receptor 7. Cell. Rep. 2019, 29, 3460–3471.e7. [Google Scholar] [CrossRef]
- Johnson, D.E.; Burtness, B.; Leemans, C.R.; Lui, V.W.Y.; Bauman, J.E.; Grandis, J.R. Head and neck squamous cell carcinoma. Nat. Rev. Dis. Primers 2020, 6, 92. [Google Scholar] [CrossRef]
- Parmar, K.; Mohamed, A.; Vaish, E.; Thawani, R.; Cetnar, J.; Thein, K.Z. Immunotherapy in head and neck squamous cell carcinoma: An updated review. Cancer Treat. Res. Commun. 2022, 33, 100649. [Google Scholar] [CrossRef] [PubMed]
- Baba, A.B.; Rah, B.; Bhat, G.R.; Mushtaq, I.; Parveen, S.; Hassan, R.; Hameed Zargar, M.; Afroze, D. Transforming Growth Factor-Beta (TGF-β) Signaling in Cancer-A Betrayal Within. Front. Pharmacol. 2022, 13, 791272. [Google Scholar] [CrossRef]
- Ajani, J.A.; Lee, J.; Sano, T.; Janjigian, Y.Y.; Fan, D.; Song, S. Gastric adenocarcinoma. Nat. Rev. Dis. Primers 2017, 3, 17036. [Google Scholar] [CrossRef]
- Sexton, R.E.; Al Hallak, M.N.; Diab, M.; Azmi, A.S. Gastric cancer: A comprehensive review of current and future treatment strategies. Cancer Metastasis Rev. 2020, 39, 1179–1203. [Google Scholar] [CrossRef]
- Oldenburg, J.; Berney, D.M.; Bokemeyer, C.; Climent, M.A.; Daugaard, G.; Gietema, J.A.; De Giorgi, U.; Haugnes, H.S.; Huddart, R.A.; Leão, R.; et al. Testicular seminoma and non-seminoma: ESMO-EURACAN Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann. Oncol. 2022, 33, 362–375. [Google Scholar] [CrossRef]
- Zhang, N.; Wu, J.; Wang, Q.; Liang, Y.; Li, X.; Chen, G.; Ma, L.; Liu, X.; Zhou, F. Global burden of hematologic malignancies and evolution patterns over the past 30 years. Blood Cancer J. 2023, 13, 82. [Google Scholar] [CrossRef]
- Tawfik, E.A.; Aldrak, N.A.; Albrahim, S.H.; Alzahrani, D.A.; Alfassam, H.A.; Alkoblan, S.M.; Almalik, A.M.; Chen, K.-S.; Abou-Khalil, R.; Shah, K.; et al. Immunotherapy in hematological malignancies: Recent advances and open questions. Immunotherapy 2021, 13, 1215–1229. [Google Scholar] [CrossRef] [PubMed]
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef]
- Liquori, A.; Ibañez, M.; Sargas, C.; Sanz, M.; Barragán, E.; Cervera, J. Acute Promyelocytic Leukemia: A Constellation of Molecular Events around a Single PML-RARA Fusion Gene. Cancers 2020, 12, 624. [Google Scholar] [CrossRef]
- Leak, S.; Horne, G.A.; Copland, M. Targeting BCR-ABL1-positive leukaemias: A review article. Camb. Prism. Precis. Med. 2023, 1, e21. [Google Scholar] [CrossRef]
- Haznedaroğlu, İ.C.; Kuzu, I.; İlhan, O. Who 2016 Definition of Chronic Myeloid Leukemia and Tyrosine Kinase Inhibitors. Turk. J. Hematol. 2019, 37, 42. [Google Scholar] [CrossRef]
- Rinaldi, I.; Winston, K. Chronic Myeloid Leukemia, from Pathophysiology to Treatment-Free Remission: A Narrative Literature Review. J. Blood Med. 2023, 14, 261–277. [Google Scholar] [CrossRef]
- Varotto, E.; Munaretto, E.; Stefanachi, F.; Della Torre, F.; Buldini, B. Diagnostic challenges in acute monoblastic/monocytic leukemia in children. Front. Pediatr. 2022, 10, 911093. [Google Scholar] [CrossRef]
- Chanput, W.; Peters, V.; Wichers, H. THP-1 and U937 Cells. In The Impact of Food Bioactives on Health; Springer International Publishing: Cham, Switzerland, 2015; pp. 147–159. [Google Scholar] [CrossRef]
- DuVall, A.S.; Sheade, J.; Anderson, D.; Yates, S.J.; Stock, W. Updates in the Management of Relapsed and Refractory Acute Lymphoblastic Leukemia: An Urgent Plea for New Treatments Is Being Answered! JCO Oncol. Pract. 2022, 18, 479–487. [Google Scholar] [CrossRef]
- Cordo, V.; van der Zwet, J.C.G.; Canté-Barrett, K.; Pieters, R.; Meijerink, J.P.P. T-cell Acute Lymphoblastic Leukemia: A Roadmap to Targeted Therapies. Blood Cancer Discov. 2021, 2, 19–31. [Google Scholar] [CrossRef]
- Xu, J.; Zhu, H.-H. Targeted treatment of T-cell acute lymphoblastic leukemia: Latest updates from the 2022 ASH Annual Meeting. Exp. Hematol. Oncol. 2023, 12, 30. [Google Scholar] [CrossRef]
- Singh, K.; Kumari, S.; Singh, B.; Choubey, R.B.; Mitra, D.K.; Rai, A.K. Jurkat T Cells are Immunophenotypically Distinct from T-Cell Acute Lymphoblastic Leukemia Cells Due to High-Level Surface Expression of CD5. Cancer Investig. 2022, 40, 675–679. [Google Scholar] [CrossRef]
- Gikas, E.; Papadopoulos, N.; Tsarbopoulos, A. Kinetic Study of the Acidic Hydrolysis of Oleuropein, the Major Bioactive Metabolite of Olive Oil. J. Liq. Chromatogr. Relat. Technol. 2006, 29, 497–508. [Google Scholar] [CrossRef]
- Corona, G.; Tzounis, X.; Assunta Dessì, M.; Deiana, M.; Debnam, E.S.; Visioli, F.; Spencer, J.P. The fate of olive oil polyphenols in the gastrointestinal tract: Implications of gastric and colonic microflora-dependent biotransformation. Free Radic. Res. 2006, 40, 647–658. [Google Scholar] [CrossRef] [PubMed]
- López de las Hazas, M.-C.; Piñol, C.; Macià, A.; Romero, M.-P.; Pedret, A.; Solà, R.; Rubió, L.; Motilva, M.-J. Differential absorption and metabolism of hydroxytyrosol and its precursors oleuropein and secoiridoids. J. Funct. Foods 2016, 22, 52–63. [Google Scholar] [CrossRef]
- Edgecombe, S.C.; Stretch, G.L.; Hayball, P.J. Oleuropein, an Antioxidant Polyphenol from Olive Oil, Is Poorly Absorbed from Isolated Perfused Rat Intestine. J. Nutr. 2000, 130, 2996–3002. [Google Scholar] [CrossRef]
- Vissers, M.N.; Zock, P.L.; Roodenburg, A.J.C.; Leenen, R.; Katan, M.B. Olive Oil Phenols Are Absorbed in Humans. J. Nutr. 2002, 132, 409–417. [Google Scholar] [CrossRef]
- García-Villalba, R.; Larrosa, M.; Possemiers, S.; Tomás-Barberán, F.A.; Espín, J.C. Bioavailability of phenolics from an oleuropein-rich olive (Olea europaea) leaf extract and its acute effect on plasma antioxidant status: Comparison between pre- and postmenopausal women. Eur. J. Nutr. 2014, 53, 1015–1027. [Google Scholar] [CrossRef]
- García-Villalba, R.; Carrasco-Pancorbo, A.; Nevedomskaya, E.; Mayboroda, O.A.; Deelder, A.M.; Segura-Carretero, A.; Fernández-Gutiérrez, A. Exploratory analysis of human urine by LC–ESI-TOF MS after high intake of olive oil: Understanding the metabolism of polyphenols. Anal. Bioanal. Chem. 2010, 398, 463–475. [Google Scholar] [CrossRef]
- de la Torre-Carbot, K.; Chávez-Servín, J.L.; Jaúregui, O.; Castellote, A.I.; Lamuela-Raventós, R.M.; Nurmi, T.; Poulsen, H.E.; Gaddi, A.V.; Kaikkonen, J.; Zunft, H.-F.; et al. Elevated Circulating LDL Phenol Levels in Men Who Consumed Virgin Rather Than Refined Olive Oil Are Associated with Less Oxidation of Plasma LDL. J. Nutr. 2010, 140, 501–508. [Google Scholar] [CrossRef]
- de Bock, M.; Thorstensen, E.B.; Derraik, J.G.B.; Henderson, H.V.; Hofman, P.L.; Cutfield, W.S. Human absorption and metabolism of oleuropein and hydroxytyrosol ingested as olive (Olea europaea L.) leaf extract. Mol. Nutr. Food Res. 2013, 57, 2079–2085. [Google Scholar] [CrossRef]
- Delboccio, P.; Dideo, A.; Decurtis, A.; Celli, N.; Iacoviello, L.; Rotilio, D. Liquid chromatography–tandem mass spectrometry analysis of oleuropein and its metabolite hydroxytyrosol in rat plasma and urine after oral administration. J. Chromatogr. B 2003, 785, 47–56. [Google Scholar] [CrossRef]
- Rishmawi, S.; Haddad, F.; Dokmak, G.; Karaman, R. A Comprehensive Review on the Anti-Cancer Effects of Oleuropein. Life 2022, 12, 1140. [Google Scholar] [CrossRef]
- Tan, H.; Tuck, K.; Stupans, I.; Hayball, P. Simultaneous determination of oleuropein and hydroxytyrosol in rat plasma using liquid chromatography with fluorescence detection. J. Chromatogr. B 2003, 785, 187–191. [Google Scholar] [CrossRef]
- Manna, C.; Galletti, P.; Maisto, G.; Cucciolla, V.; D’Angelo, S.; Zappia, V. Transport mechanism and metabolism of olive oil hydroxytyrosol in Caco-2 cells. FEBS Lett. 2000, 470, 341–344. [Google Scholar] [CrossRef]
- Oliveras-López, M.-J.; Berná, G.; Carneiro, E.M.; de la Serrana, H.L.-G.; Martín, F.; López, M.C. An Extra-Virgin Olive Oil Rich in Polyphenolic Compounds Has Antioxidant Effects in Of1 Mice. J. Nutr. 2008, 138, 1074–1078. [Google Scholar] [CrossRef] [PubMed]
- Kano, S.; Komada, H.; Yonekura, L.; Sato, A.; Nishiwaki, H.; Tamura, H. Absorption, Metabolism, and Excretion by Freely Moving Rats of 3,4-DHPEA-EDA and Related Polyphenols from Olive Fruits (Olea europaea). J. Nutr. Metab. 2016, 2016, 9104208. [Google Scholar] [CrossRef] [PubMed]
- Suárez, M.; Valls, R.M.; Romero, M.-P.; Macià, A.; Fernández, S.; Giralt, M.; Solà, R.; Motilva, M.-J. Bioavailability of phenols from a phenol-enriched olive oil. Br. J. Nutr. 2011, 106, 1691–1701. [Google Scholar] [CrossRef] [PubMed]
- Visioli, F.; Galli, C.; Grande, S.; Colonnelli, K.; Patelli, C.; Galli, G.; Caruso, D. Hydroxytyrosol Excretion Differs between Rats and Humans and Depends on the Vehicle of Administration. J. Nutr. 2003, 133, 2612–2615. [Google Scholar] [CrossRef] [PubMed]
- Miro-Casas, E.; Covas, M.-I.; Farre, M.; Fito, M.; Ortuño, J.; Weinbrenner, T.; Roset, P.; de la Torre, R. Hydroxytyrosol Disposition in Humans. Clin. Chem. 2003, 49, 945–952. [Google Scholar] [CrossRef]
- Tuck, K.L.; Hayball, P.J.; Stupans, I. Structural Characterization of the Metabolites of Hydroxytyrosol, the Principal Phenolic Component in Olive Oil, in Rats. J. Agric. Food Chem. 2002, 50, 2404–2409. [Google Scholar] [CrossRef]
- Alemán-Jiménez, C.; Domínguez-Perles, R.; Medina, S.; Prgomet, I.; López-González, I.; Simonelli-Muñoz, A.; Campillo-Cano, M.; Auñón, D.; Ferreres, F.; Gil-Izquierdo, Á. Pharmacokinetics and bioavailability of hydroxytyrosol are dependent on the food matrix in humans. Eur. J. Nutr. 2021, 60, 905–915. [Google Scholar] [CrossRef] [PubMed]
- Mateos, R.; Martínez-López, S.; Arévalo, G.B.; Amigo-Benavent, M.; Sarriá, B.; Bravo-Clemente, L. Hydroxytyrosol in functional hydroxytyrosol-enriched biscuits is highly bioavailable and decreases oxidised low density lipoprotein levels in humans. Food Chem. 2016, 205, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Bender, C.; Strassmann, S.; Golz, C. Oral Bioavailability and Metabolism of Hydroxytyrosol from Food Supplements. Nutrients 2023, 15, 325. [Google Scholar] [CrossRef] [PubMed]
- Covas, M.-I.; de la Torre, K.; Farré-Albaladejo, M.; Kaikkonen, J.; Fitó, M.; López-Sabater, C.; Pujadas-Bastardes, M.A.; Joglar, J.; Weinbrenner, T.; Lamuela-Raventós, R.M.; et al. Postprandial LDL phenolic content and LDL oxidation are modulated by olive oil phenolic compounds in humans. Free Radic. Biol. Med. 2006, 40, 608–616. [Google Scholar] [CrossRef] [PubMed]
- Pastor, A.; Rodríguez-Morató, J.; Olesti, E.; Pujadas, M.; Pérez-Mañá, C.; Khymenets, O.; Fitó, M.; Covas, M.-I.; Solá, R.; Motilva, M.-J.; et al. Analysis of free hydroxytyrosol in human plasma following the administration of olive oil. J. Chromatogr. A 2016, 1437, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Weinbrenner, T.; Fitó, M.; de la Torre, R.; Saez, G.T.; Rijken, P.; Tormos, C.; Coolen, S.; Albaladejo, M.F.; Abanades, S.; Schroder, H.; et al. Olive Oils High in Phenolic Compounds Modulate Oxidative/Antioxidative Status in Men. J. Nutr. 2004, 134, 2314–2321. [Google Scholar] [CrossRef] [PubMed]
- Babich, H.; Visioli, F. In Vitro cytotoxicity to human cells in culture of some phenolics from olive oil. Il Farmaco 2003, 58, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Katsiki, M.; Chondrogianni, N.; Chinou, I.; Rivett, A.J.; Gonos, E.S. The Olive Constituent Oleuropein Exhibits Proteasome Stimulatory Properties In Vitro and Confers Life Span Extension of Human Embryonic Fibroblasts. Rejuvenation Res. 2007, 10, 157–172. [Google Scholar] [CrossRef]
- Sarsour, E.H.; Kumar, M.G.; Kalen, A.L.; Goswami, M.; Buettner, G.R.; Goswami, P.C. MnSOD activity regulates hydroxytyrosol-induced extension of chronological lifespan. Age 2012, 34, 95–109. [Google Scholar] [CrossRef]
- Torić, J.; Marković, A.K.; Brala, C.J.; Barbarić, M. Anticancer effects of olive oil polyphenols and their combinations with anticancer drugs. Acta Pharm. 2019, 69, 461–482. [Google Scholar] [CrossRef]
- Ramirez-Tortosa, C.; Sanchez, A.; Perez-Ramirez, C.; Quiles, J.L.; Robles-Almazan, M.; Pulido-Moran, M.; Sanchez-Rovira, P.; Ramirez-Tortosa, M. Hydroxytyrosol Supplementation Modifies Plasma Levels of Tissue Inhibitor of Metallopeptidase 1 in Women with Breast Cancer. Antioxidants 2019, 8, 393. [Google Scholar] [CrossRef] [PubMed]
- Terzuoli, E.; Nannelli, G.; Frosini, M.; Giachetti, A.; Ziche, M.; Donnini, S. Inhibition of cell cycle progression by the hydroxytyrosol-cetuximab combination yields enhanced chemotherapeutic efficacy in colon cancer cells. Oncotarget 2017, 8, 83207–83224. [Google Scholar] [CrossRef] [PubMed]
- Sirangelo, I.; Liccardo, M.; Iannuzzi, C. Hydroxytyrosol Prevents Doxorubicin-Induced Oxidative Stress and Apoptosis in Cardiomyocytes. Antioxidants 2022, 11, 1087. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Xiao, D. Oleuropein enhances radiation sensitivity of nasopharyngeal carcinoma by downregulating PDRG1 through HIF1α-repressed microRNA-519d. J. Exp. Clin. Cancer Res. 2017, 36, 3. [Google Scholar] [CrossRef] [PubMed]
- Stupans, I.; Murray, M.; Kirlich, A.; Tuck, K.L.; Hayball, P.J. Inactivation of cytochrome P450 by the food-derived complex phenol oleuropein. Food Chem. Toxicol. 2001, 39, 1119–1124. [Google Scholar] [CrossRef]
- Stupans, I.; Stretch, G.; Hayball, P. Olive Oil Phenols Inhibit Human Hepatic Microsomal Activity. J. Nutr. 2000, 130, 2367–2370. [Google Scholar] [CrossRef]
- Malliou, F.; Andriopoulou, C.E.; Gonzalez, F.J.; Kofinas, A.; Skaltsounis, A.-L.; Konstandi, M. Oleuropein-Induced Acceleration of Cytochrome P450–Catalyzed Drug Metabolism: Central Role for Nuclear Receptor Peroxisome Proliferator-Activated Receptor α. Drug Metab. Dispos. 2021, 49, 833–843. [Google Scholar] [CrossRef]
- Bekhet, O.H.; Eid, M.E. The interplay between reactive oxygen species and antioxidants in cancer progression and therapy: A narrative review. Transl. Cancer Res. 2021, 10, 4196–4206. [Google Scholar] [CrossRef]
- Singh, R.; Manna, P.P. Reactive oxygen species in cancer progression and its role in therapeutics. Explor. Med. 2022, 3, 43–57. [Google Scholar] [CrossRef]
- George, S.; Abrahamse, H. Redox Potential of Antioxidants in Cancer Progression and Prevention. Antioxidants 2020, 9, 1156. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Zhou, L.; Huang, Z.; Li, B.; Nice, E.C.; Xu, J.; Huang, C. Antioxidant Therapy in Cancer: Rationale and Progress. Antioxidants 2022, 11, 1128. [Google Scholar] [CrossRef]
- Khan, A.Q.; Rashid, K.; AlAmodi, A.A.; Agha, M.V.; Akhtar, S.; Hakeem, I.; Raza, S.S.; Uddin, S. Reactive oxygen species (ROS) in cancer pathogenesis and therapy: An update on the role of ROS in anticancer action of benzophenanthridine alkaloids. Biomed. Pharmacother. 2021, 143, 112142. [Google Scholar] [CrossRef]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef]
- Galadari, S.; Rahman, A.; Pallichankandy, S.; Thayyullathil, F. Reactive oxygen species and cancer paradox: To promote or to suppress? Free Radic. Biol. Med. 2017, 104, 144–164. [Google Scholar] [CrossRef]
- Gurnari, C.; De Bellis, E.; Divona, M.; Ottone, T.; Lavorgna, S.; Voso, M.T. When Poisons Cure: The Case of Arsenic in Acute Promyelocytic Leukemia. Chemotherapy 2019, 64, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Shen, X.; Zhi, F.; Wen, Z.; Gao, Y.; Xu, J.; Yang, B.; Bai, Y. An overview of arsenic trioxide-involved combined treatment algorithms for leukemia: Basic concepts and clinical implications. Cell Death Discov. 2023, 9, 266. [Google Scholar] [CrossRef] [PubMed]
- Romo-González, M.; Ijurko, C.; Hernández-Hernández, Á. Reactive Oxygen Species and Metabolism in Leukemia: A Dangerous Liaison. Front. Immunol. 2022, 13, 889875. [Google Scholar] [CrossRef] [PubMed]
- Çeriğ, S.; Geyikoglu, F.; BAKIR, M.; Çolak, S.; Sönmez, M.; Koç, K. Hepatoprotective Effect of Oleuropein against Cisplatin-Induced Liver Damage in Rat. Int. J. Med. Health Sci. 2016, 10, 260–267. [Google Scholar] [CrossRef]
- Bakir, M.; Geyikoglu, F.; Koc, K.; Cerig, S. Therapeutic effects of oleuropein on cisplatin-induced pancreas injury in rats. J. Cancer Res. Ther. 2018, 14, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Geyikoglu, F.; Isikgoz, H.; Onalan, H.; Colak, S.; Cerig, S.; Bakir, M.; Hosseinigouzdagani, M.; Koc, K.; Erol, H.S.; Saglam, Y.S.; et al. Impact of high-dose oleuropein on cisplatin-induced oxidative stress, genotoxicity and pathological changes in rat stomach and lung. J. Asian Nat. Prod. Res. 2017, 19, 1214–1231. [Google Scholar] [CrossRef]
- Geyikoğlu, F.; Çolak, S.; Türkez, H.; Bakır, M.; Koç, K.; Hosseinigouzdagani, M.K.; Çeriğ, S.; Sönmez, M. Oleuropein Ameliorates Cisplatin-induced Hematological Damages Via Restraining Oxidative Stress and DNA Injury. Indian J. Hematol. Blood Transfus. 2017, 33, 348–354. [Google Scholar] [CrossRef]
- Karakoç, M.D.; Sekkin, S. Effects of Oleuropein on Epirubicin and Cyclophosphamide Combination Treatment in Rats. Turk. J. Pharm. Sci. 2021, 18, 420–429. [Google Scholar] [CrossRef]
- Shan, C.; Miao, F. Immunomodulatory and antioxidant effects of hydroxytyrosol in cyclophosphamide-induced immunosuppressed broilers. Poult. Sci. 2022, 101, 101516. [Google Scholar] [CrossRef] [PubMed]
- Fabiani, R.; Rosignoli, P.; De Bartolomeo, A.; Fuccelli, R.; Servili, M.; Montedoro, G.F.; Morozzi, G. Oxidative DNA Damage Is Prevented by Extracts of Olive Oil, Hydroxytyrosol, and Other Olive Phenolic Compounds in Human Blood Mononuclear Cells and HL60 Cells. J. Nutr. 2008, 138, 1411–1416. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, Y.; Zhang, R.; Wang, F.; Wang, T.; Jiao, Y. The Role of Erastin in Ferroptosis and Its Prospects in Cancer Therapy. Onco Targets Ther. 2020, 13, 5429–5441. [Google Scholar] [CrossRef] [PubMed]
- Nie, Q.; Hu, Y.; Yu, X.; Li, X.; Fang, X. Induction and application of ferroptosis in cancer therapy. Cancer Cell Int. 2022, 22, 12. [Google Scholar] [CrossRef]
- Sayin, V.I.; Ibrahim, M.X.; Larsson, E.; Nilsson, J.A.; Lindahl, P.; Bergo, M.O. Antioxidants Accelerate Lung Cancer Progression in Mice. Sci. Transl. Med. 2014, 6, 221. [Google Scholar] [CrossRef]
- Le Gal, K.; Ibrahim, M.X.; Wiel, C.; Sayin, V.I.; Akula, M.K.; Karlsson, C.; Dalin, M.G.; Akyürek, L.M.; Lindahl, P.; Nilsson, J.; et al. Antioxidants can increase melanoma metastasis in mice. Sci. Transl. Med. 2015, 7, 308. [Google Scholar] [CrossRef]
- Zhao, H.; Wu, L.; Yan, G.; Chen, Y.; Zhou, M.; Wu, Y.; Li, Y. Inflammation and tumor progression: Signaling pathways and targeted intervention. Signal Transduct. Target. Ther. 2021, 6, 263. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Baby, D.; Rajguru, J.P.; Patil, P.B.; Thakkannavar, S.S.; Pujari, V.B. Inflammation and cancer. Ann. Afr. Med. 2019, 18, 121–126. [Google Scholar] [CrossRef]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Potočnjak, I.; Škoda, M.; Pernjak-Pugel, E.; Peršić, M.P.; Domitrović, R. Oral administration of oleuropein attenuates cisplatin-induced acute renal injury in mice through inhibition of ERK signaling. Mol. Nutr. Food Res. 2016, 60, 530–541. [Google Scholar] [CrossRef] [PubMed]
- Bulotta, S.; Corradino, R.; Celano, M.; D’Agostino, M.; Maiuolo, J.; Oliverio, M.; Procopio, A.; Iannone, M.; Rotiroti, D.; Russo, D. Antiproliferative and antioxidant effects on breast cancer cells of oleuropein and its semisynthetic peracetylated derivatives. Food Chem. 2011, 127, 1609–1614. [Google Scholar] [CrossRef]
- Niyaki, Z.M.; Salehzadeh, A.; Peymani, M.; Zaefizadeh, M. Exploring the Therapeutic Potential of Fe3O4@Glu-Oleuropein Nanoparticles in Targeting KRAS Pathway-Regulating lncRNAs in Colorectal Cancer Cells. Biol. Trace Elem. Res. 2023, 2023, 1–13. [Google Scholar] [CrossRef]
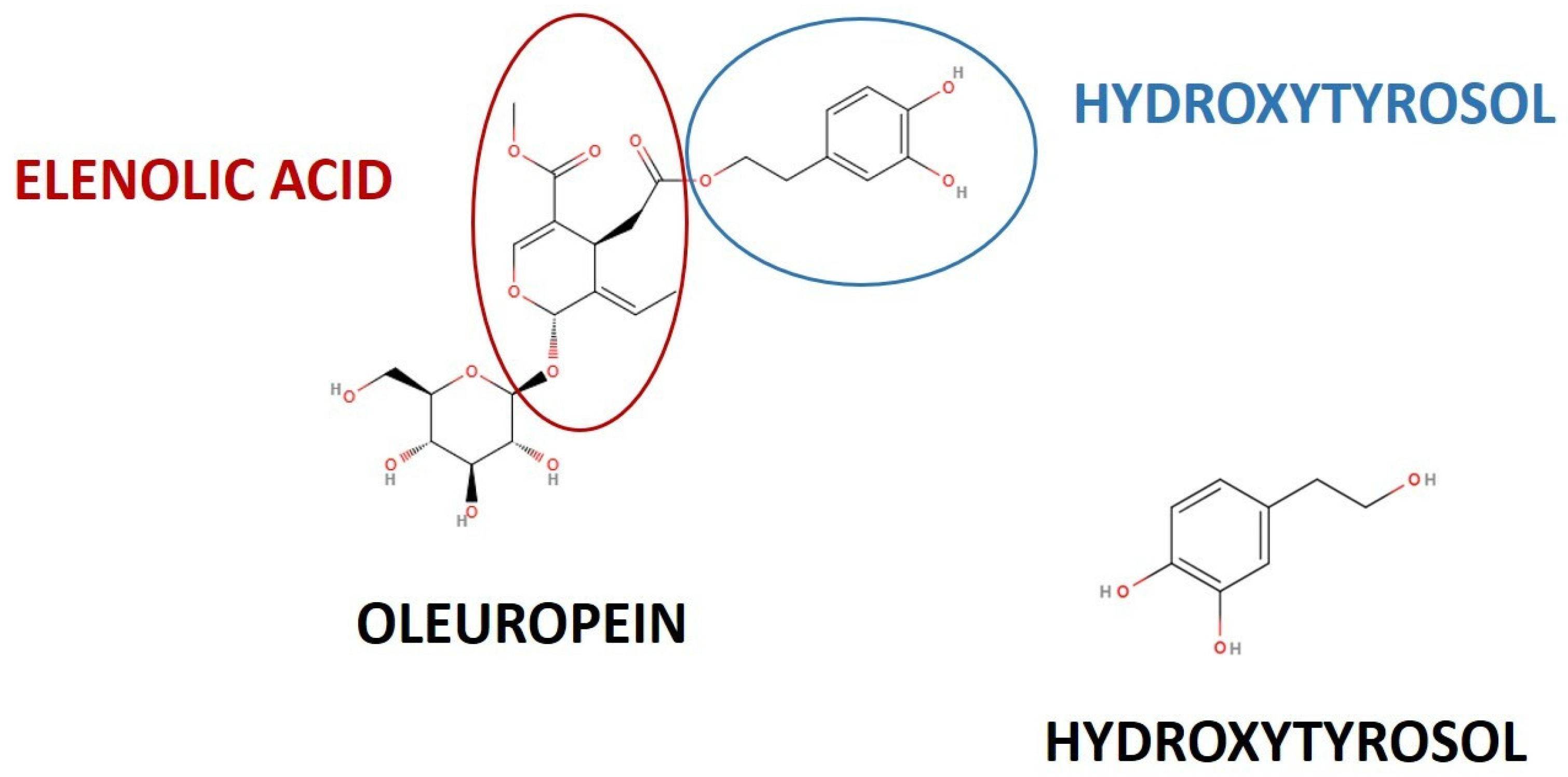
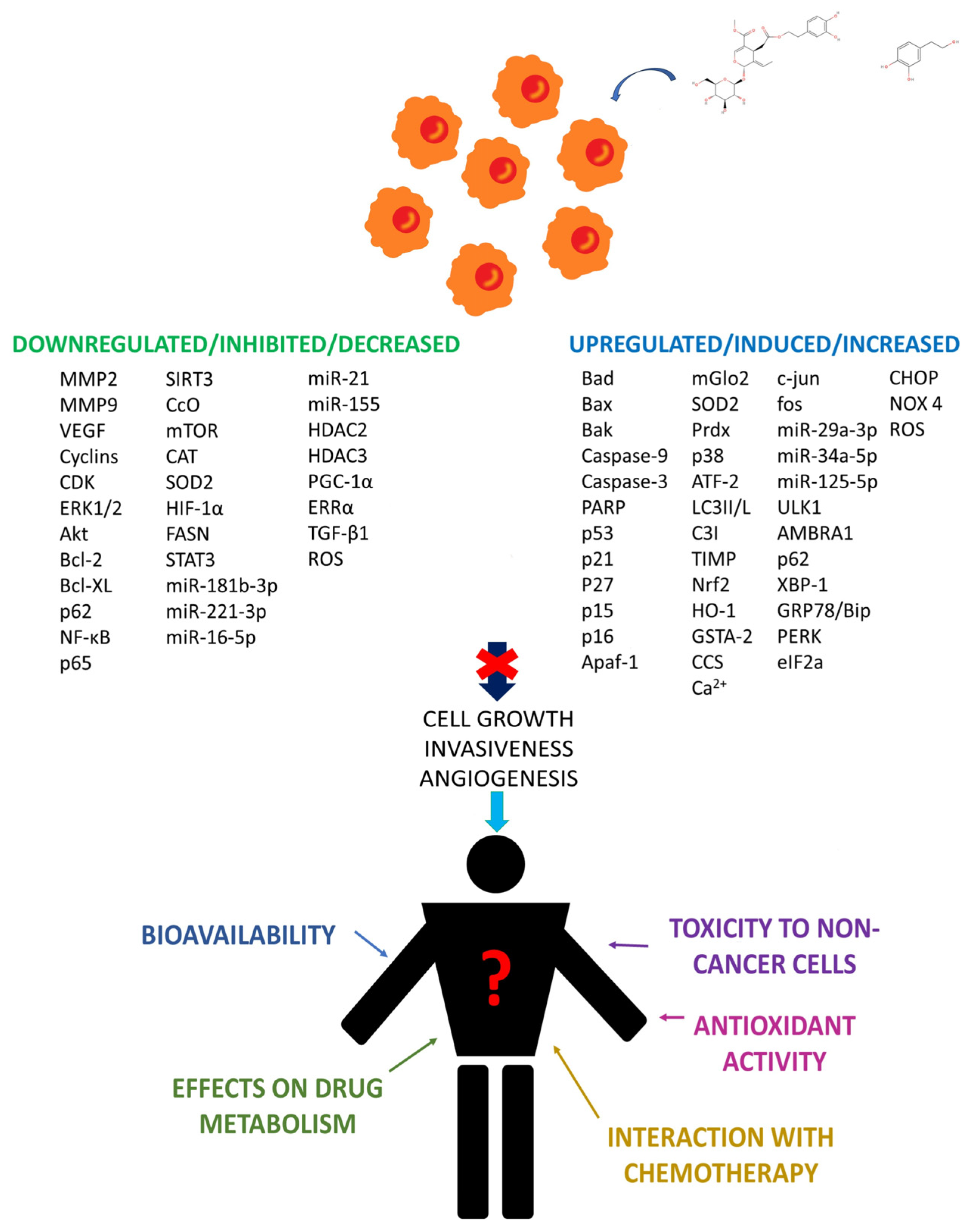

{kind=link}
{kind=link}
| Malignancy | Model | [C] | Ref. | |
|---|---|---|---|---|
| Melanoma | ||||
| OLE | UVB-irradiated albino hairless HOS: HR-1 mouse | 10 mg/kg 25 mg/kg | [27] | |
| B16F10 allograft HFD-induced melanoma progression in C57BL/6N mouse | 0.02% HFD 0.04% HFD | [28] | ||
| C32 | None | [29] | ||
| A375 | 250, 500 and 800 μM | [30] | ||
| WM266-4 | 250, 500 and 800 μM | [30] | ||
| M21 | 250, 500 and 800 μM | [30] | ||
| HT | C32 | 400 and 1000 μM | [29] | |
| A375 | 50–500 μM | [31,32] | ||
| MNT1 | None | [31] | ||
| HT-144 | 50–450 μM | [32] | ||
| M74 | 50–250 μM | [32] | ||
| Thyroid cancer | ||||
| OLE | TPC-1 | 50–100 μM | [33] | |
| BCPAP | 50–100 μM | [33] | ||
| HT | TPC-1 | 324–973 μM | [34] | |
| FB-2 | 324–973 μM | [34] | ||
| WRO | 65–973 μM | [34] | ||
| Lung cancer | ||||
| OLE | A549 | 50 and 150 μM | [35] | |
| H1299 | 50–200 μM | [36] | ||
| HT | A549 | 147–230.60 μM | [37,38] | |
| Malignant pleural mesothelioma | ||||
| OLE | REN | 25 μg/mL | [39] | |
| Breast cancer | ||||
| OLE | Tumor xenograft (MDA-MB-231) in BALB/c OlaHsd-foxn1 mouse | 50 mg/kg | [40] | |
| OLE | MCF-7 | 10–1100 μM | [41,42,43,44,45,46,47,48,49] | |
| MDA-MB-231 | 12.5–500 μM | [46,50,51] | ||
| MDA-MB-468 | 500 μM | [50] | ||
| MDA (unknown) | 200 μg/mL | [52] | ||
| T47D | 150 μM | [47] | ||
| HT | Dimethylbenz[α]anthracene-induced mammary tumors in Sprague–Dawley rat | 0.5 mg/kg | [53] | |
| MCF-7 | 10–600 μM | [42,45,46,47,54,55,56,57,58] | ||
| MDA-MB-231 | 10–400 μM | [37,46,58,59,60,61] | ||
| MDA-MB-468 | 100–300 μM | [61] | ||
| SKBR3 | 100 μM | [62] | ||
| MDA (unknown) | 25–100 μM | [56,57] | ||
| SUM159 | 300 μM | [61] | ||
| T47D | 100–150 μM | [47] | ||
| Hepatocellular carcinoma | ||||
| OLE | Huh7 | 20–80 μM | [63] | |
| HepG2 | 20–80 μM | [63] | ||
| None | [64,65] | |||
| HT | HepG2 | 30–400 μM | [66,67,68] | |
| Hep3B | 30–400 μM | [66,67] | ||
| SK-HEP-1 | 200–400 μM | [67] | ||
| Huh7 | 200–400 μM | [67] | ||
| Cholangiocarcinoma | ||||
| HT | KMBC | 25–200 μM | [69] | |
| TFK-1 | 25–200 μM | [69] | ||
| TFK-1 xeonograft in BALB/c mouse | 500 mg/kg | [69] | ||
| Gallbladder carcinoma | ||||
| HT | GBC-SD | 25–200 μM | [69] | |
| Colorectal cancer | OLE | AOM/DSS-induced colorectal cancer in C57BL/6 mouse | 50 and 100 mg/kg | [70] |
| RKO | 20–80 μM | [63] | ||
| HT-29 | 100, 400 and 800 μM | [71,72] | ||
| SW620 | 10–100 μM | [71] | ||
| HT | SW480 | 100–400 μM | [56,57,73] | |
| HCT116 | 5–300 μM | [56,57,73,74,75] | ||
| HT-29 | 100–800 μM | [71,72,73,76,77,78] | ||
| SW620 | 5–100 μM | [71,75] | ||
| Caco-2 | 10 *, 100 and 150 μM | [76,79] | ||
| LoVo | 100–400 μM | [73,74] | ||
| Pancreatic cancer | ||||
| OLE | BxPC-3 | None | [80] | |
| CFPAC-1 | None | [80] | ||
| MIA PaCa-2 | 150.1 μM | [80] | ||
| HT | Orthotopic model of pancreatic cancer (Panc02) in C57BL/6 mouse | 200 mg/kg | [81] | |
| BxPC-3 | None | [80] | ||
| CFPAC-1 | None | [80] | ||
| MIA PaCa-2 | 75.1 μM | [80] | ||
| PANC-1 | 10, 32, 100, and 320 μM | [82] | ||
| Panc02 | 50, 150, and 200 μM | [81] | ||
| Cervical cancer | ||||
| OLE | HeLa | 50–2220 μM | [83,84] | |
| Ovarian cancer | ||||
| OLE | HEY | 200 and 400 μM | [43] | |
| OVCAR3 | 200 μg/mL | [59] | ||
| Prostate cancer | ||||
| OLE | LNCaP | 100 and 500 μM | [85] | |
| DU145 | 100 and 500 μM | [85] | ||
| HT | LNCap | 30–400 μM | [56,57,86,87] | |
| C4–2 | 50–400 μM | [86] | ||
| PC3 | 30–300 μM | [56,57,88] | ||
| 22Rv1 | 30–300 μM | [87] | ||
| Osteosarcoma | ||||
| OLE | 143B | 1–250 μM | [89] | |
| MG-63 | 5–400 μM | [90,91] | ||
| Saos2 | 100, 200, and 400 μM | [91] | ||
| Neuroblastoma | ||||
| OLE | SH-SY5Y | 350 μM | [92] | |
| HT | SH-SY5Y | 25–200 μM | [93] | |
| Glioma | ||||
| OLE | A-172 | 200 and 400 μM | [94] | |
| U-251 | 200 and 400 μM | [94] | ||
| T-98G | 277.5 and 555 μM | [95] | ||
| Head and neck squamous cell carcinoma | ||||
| OLE | Tu686 | 50–200 μg/mL | [96] | |
| CAL-27 | 50–200 μg/mL | [96] | ||
| Tumor xenograft (686LN-M2) in BALB/c nude mouse | 1200 μg/mL | [96] | ||
| Gastric adenocarcinoma | ||||
| OLE | CRL-1739 | 50–500 μM | [97] | |
| Seminoma | ||||
| OLE | SEM-1 | 15–200 μM | [65] | |
| TCAM-2 | 15–200 μM | [65] | ||
| Hematological malignancies | ||||
| OLE | K562 | 200 and 400 μg/mL | [98] | |
| HT | HL-60 | 50–100 μM | [57,78,99,100,101,102] | |
| K562 | 100–1000 μM | [103] | ||
| THP-1 | 20 μM | [104] | ||
| U937 | 75 and 200 μM | [105] | ||
| CCRF-CEM | 50–1000 μM | [103] | ||
| Jurkat | 10–100 μg/mL § | [101] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gervasi, F.; Pojero, F. Use of Oleuropein and Hydroxytyrosol for Cancer Prevention and Treatment: Considerations about How Bioavailability and Metabolism Impact Their Adoption in Clinical Routine. Biomedicines 2024, 12, 502. https://doi.org/10.3390/biomedicines12030502
Gervasi F, Pojero F. Use of Oleuropein and Hydroxytyrosol for Cancer Prevention and Treatment: Considerations about How Bioavailability and Metabolism Impact Their Adoption in Clinical Routine. Biomedicines. 2024; 12(3):502. https://doi.org/10.3390/biomedicines12030502
Chicago/Turabian StyleGervasi, Francesco, and Fanny Pojero. 2024. "Use of Oleuropein and Hydroxytyrosol for Cancer Prevention and Treatment: Considerations about How Bioavailability and Metabolism Impact Their Adoption in Clinical Routine" Biomedicines 12, no. 3: 502. https://doi.org/10.3390/biomedicines12030502
APA StyleGervasi, F., & Pojero, F. (2024). Use of Oleuropein and Hydroxytyrosol for Cancer Prevention and Treatment: Considerations about How Bioavailability and Metabolism Impact Their Adoption in Clinical Routine. Biomedicines, 12(3), 502. https://doi.org/10.3390/biomedicines12030502