
An In Silico and In Vitro Assessment of the Neurotoxicity of Mefloquine


Abstract
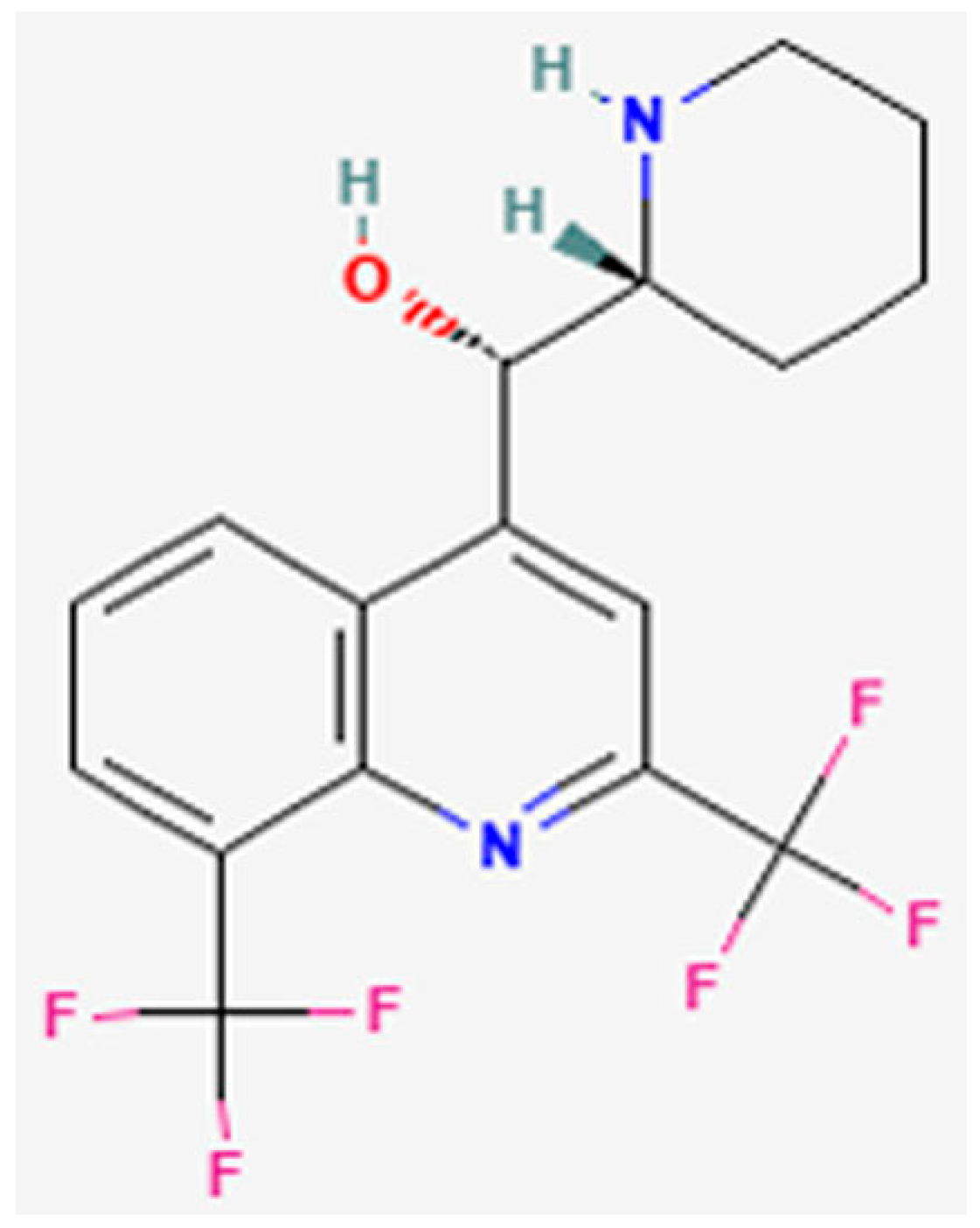
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
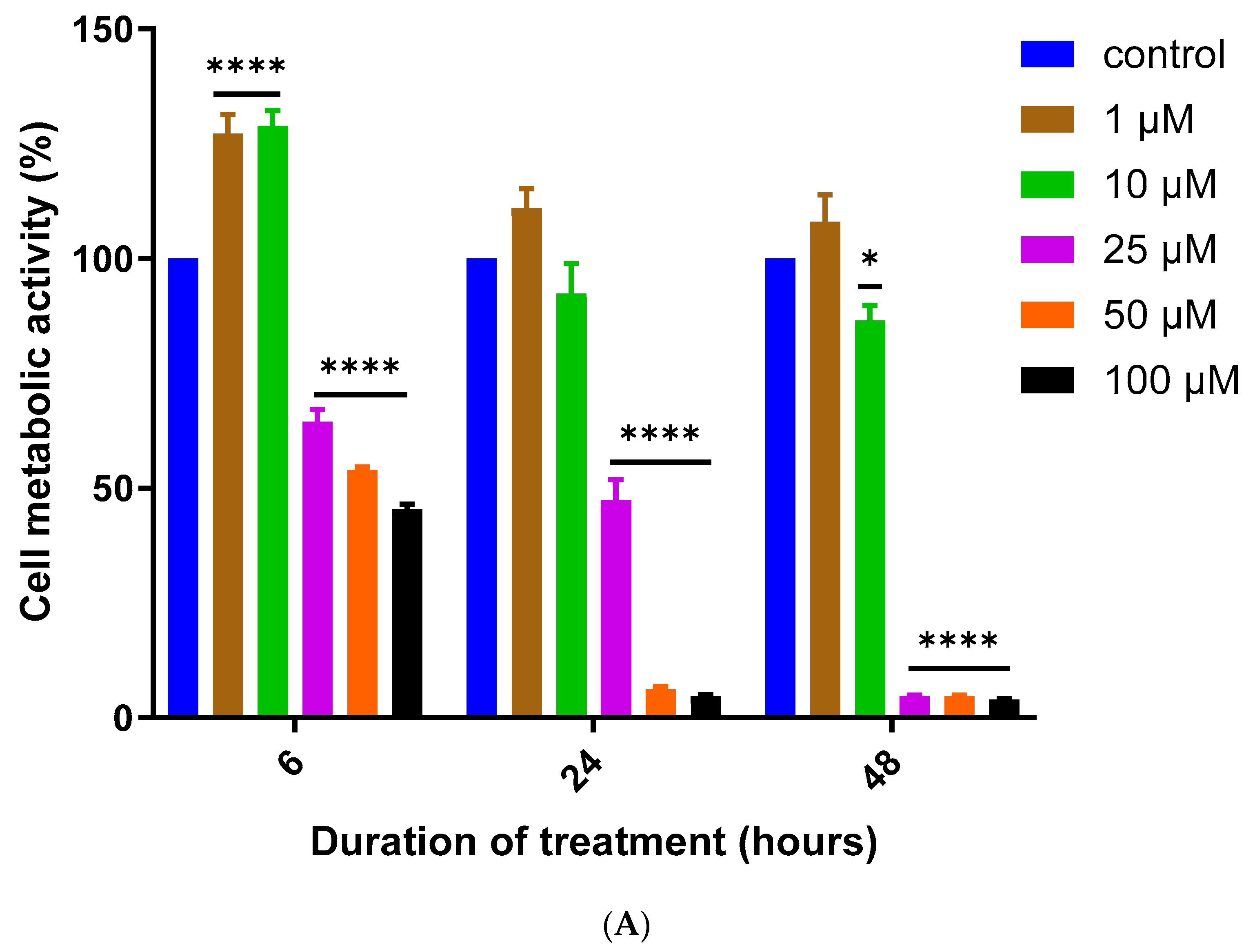
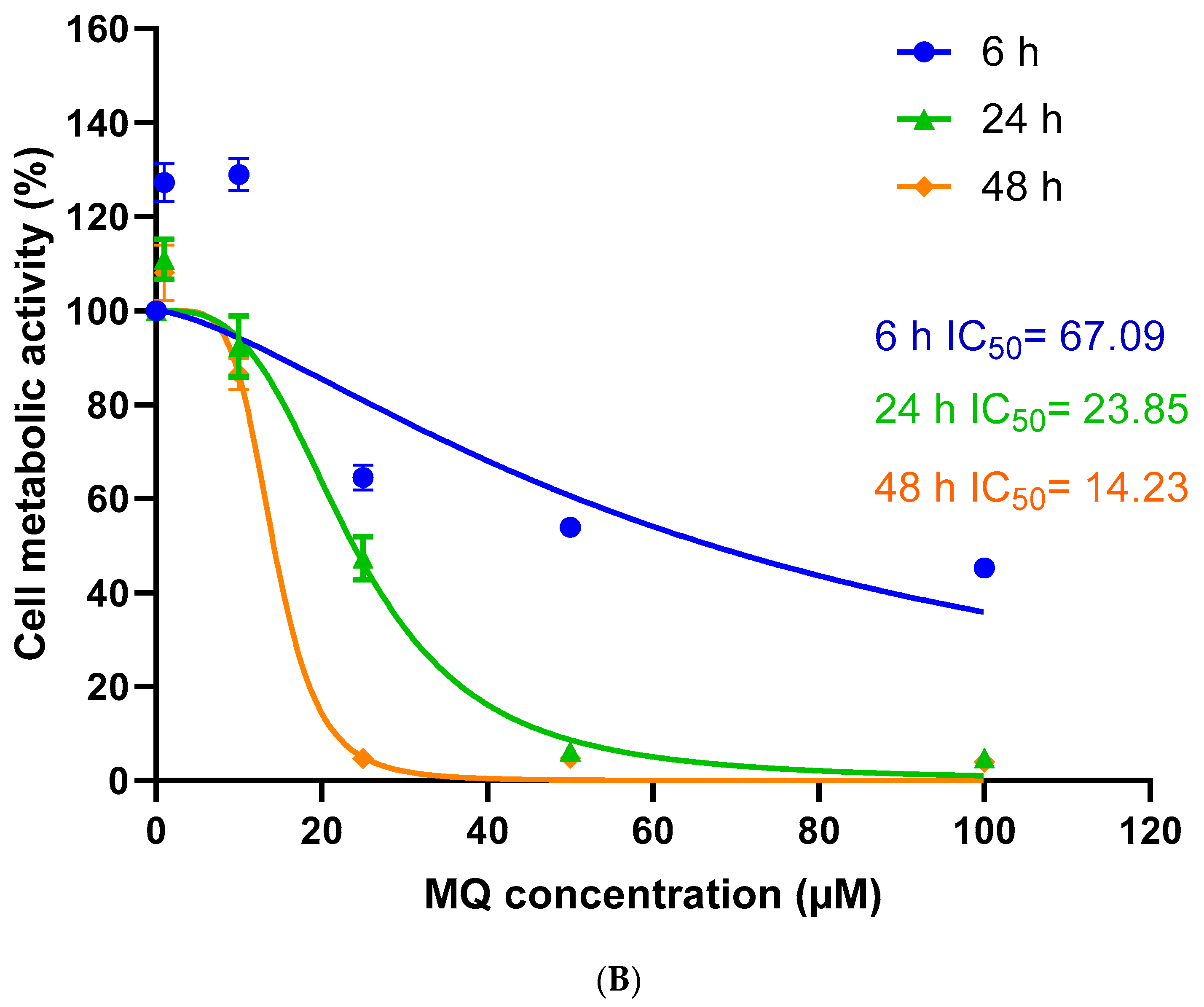
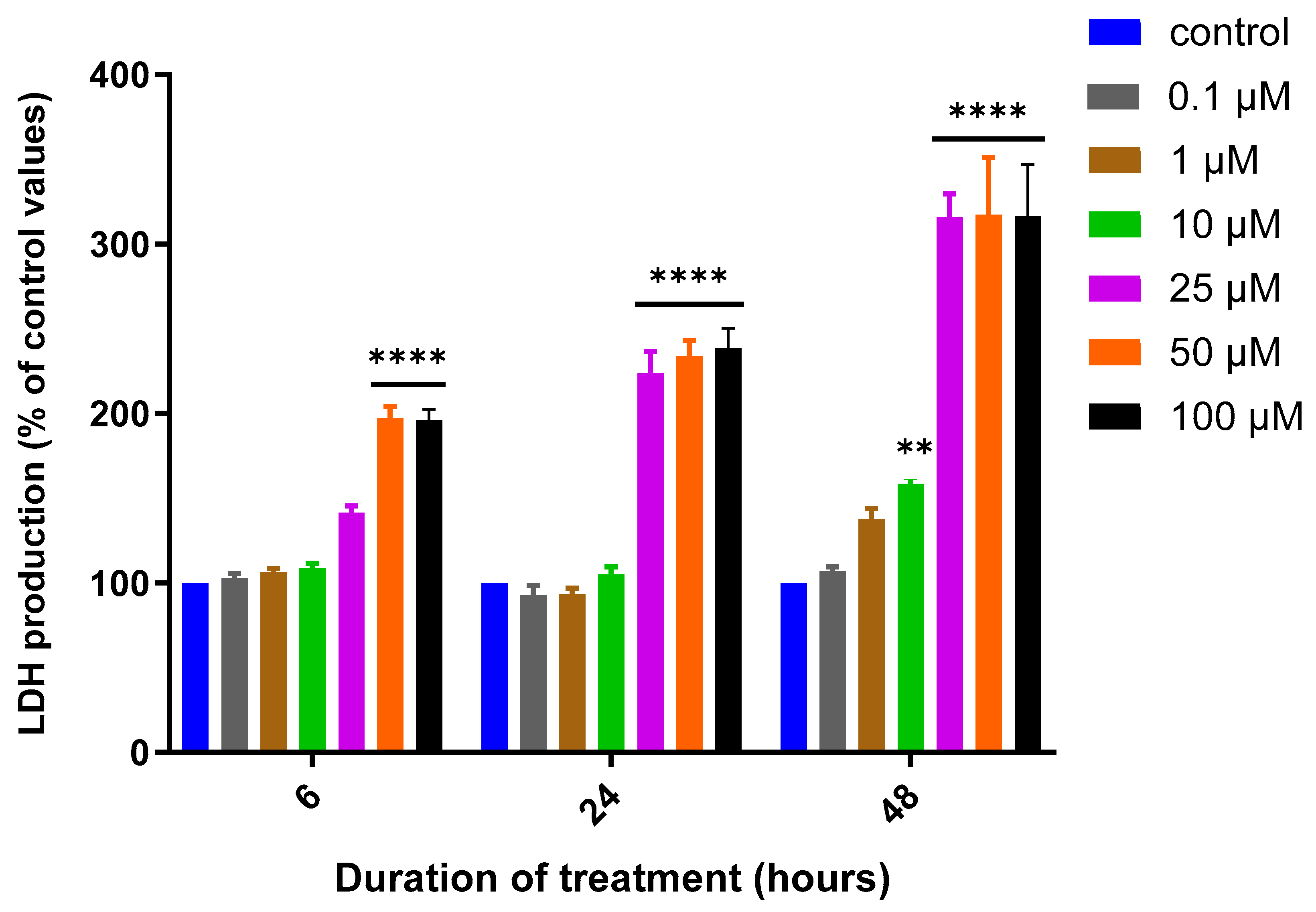
2.2. Cytotoxicity Assays
2.3. Cellular Bioenergetics Assay
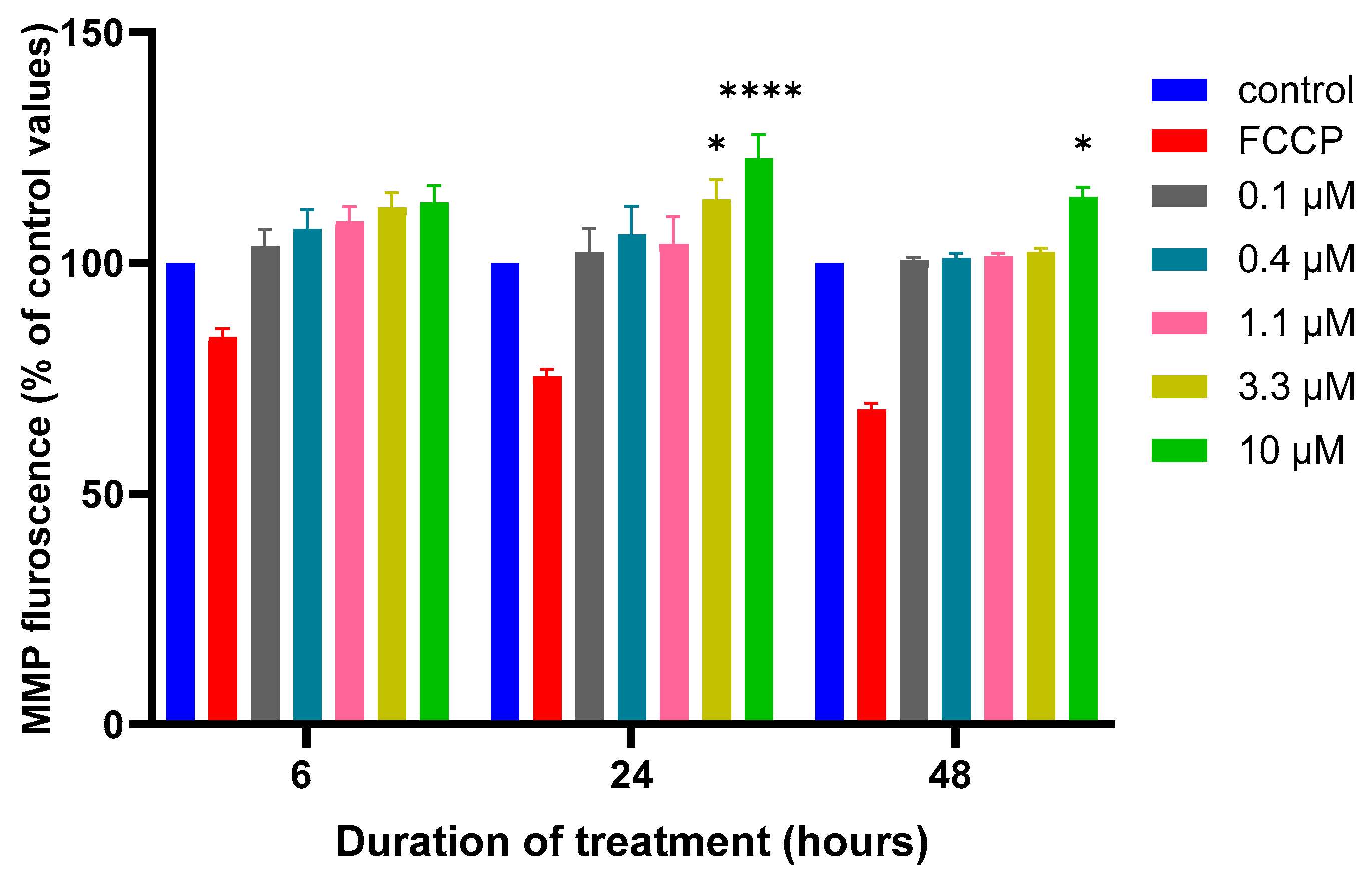
2.4. Mitochondrial Membrane Potential (MMP) Assay
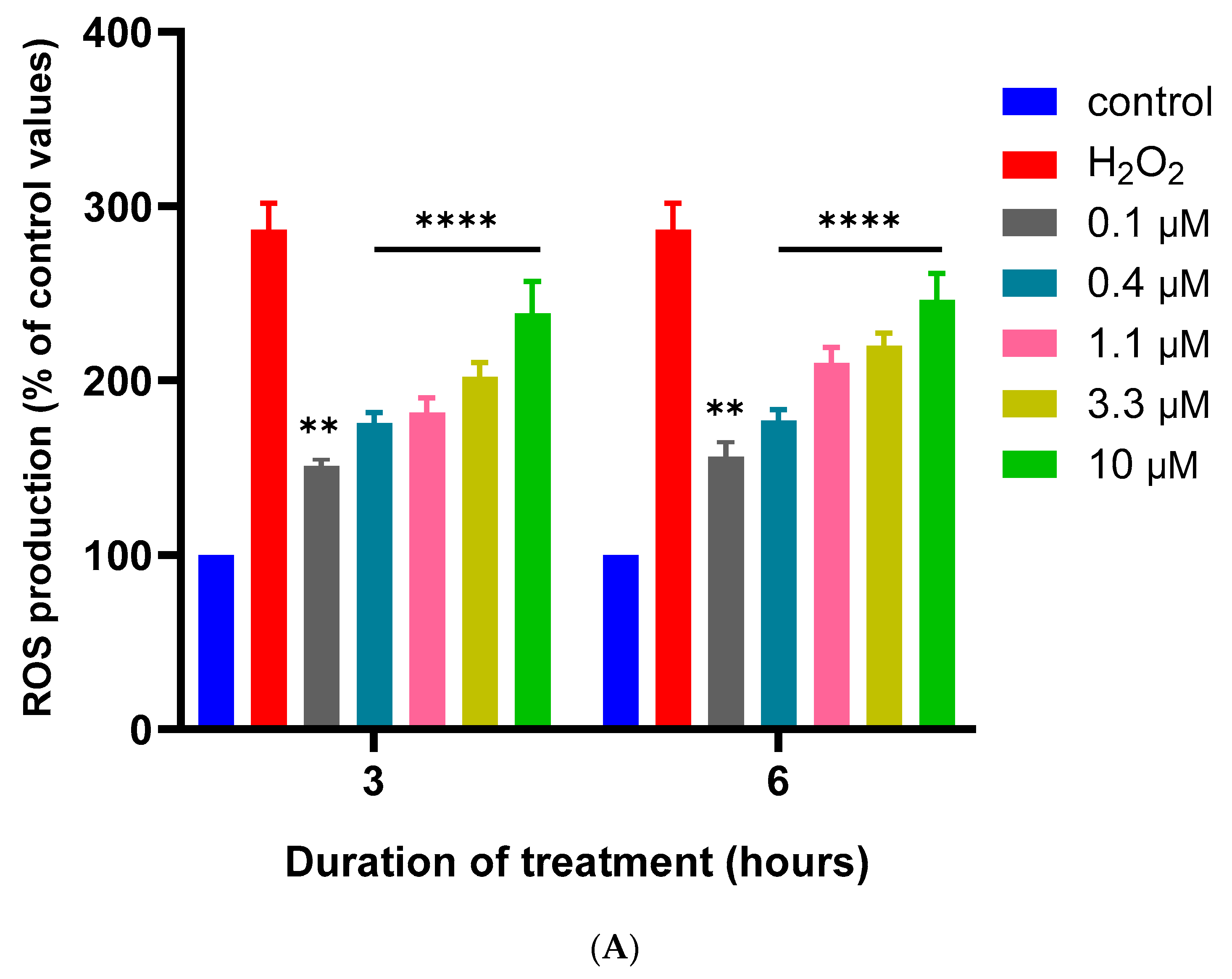
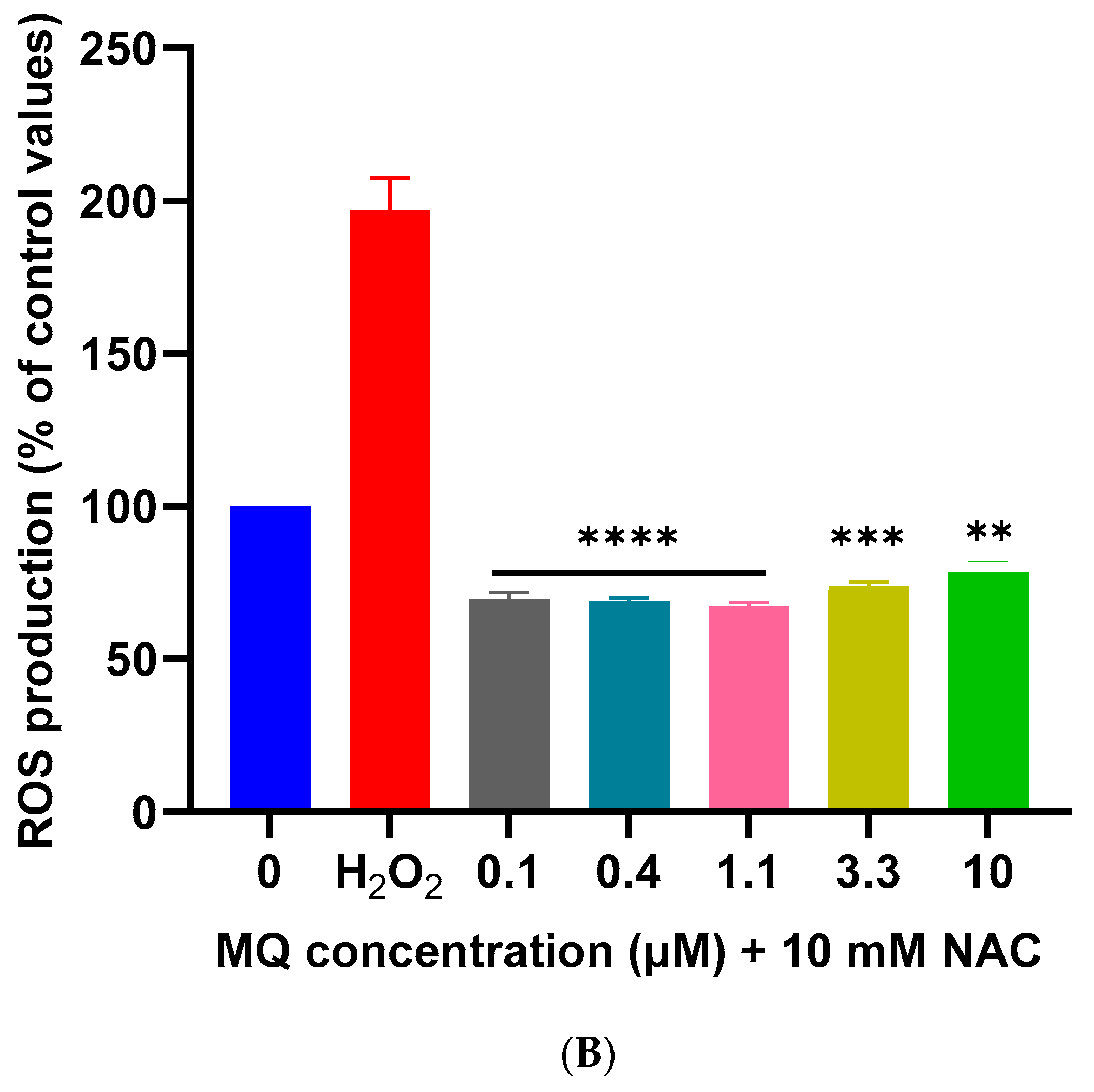
2.5. Measurement of Reactive Oxygen Species Generation
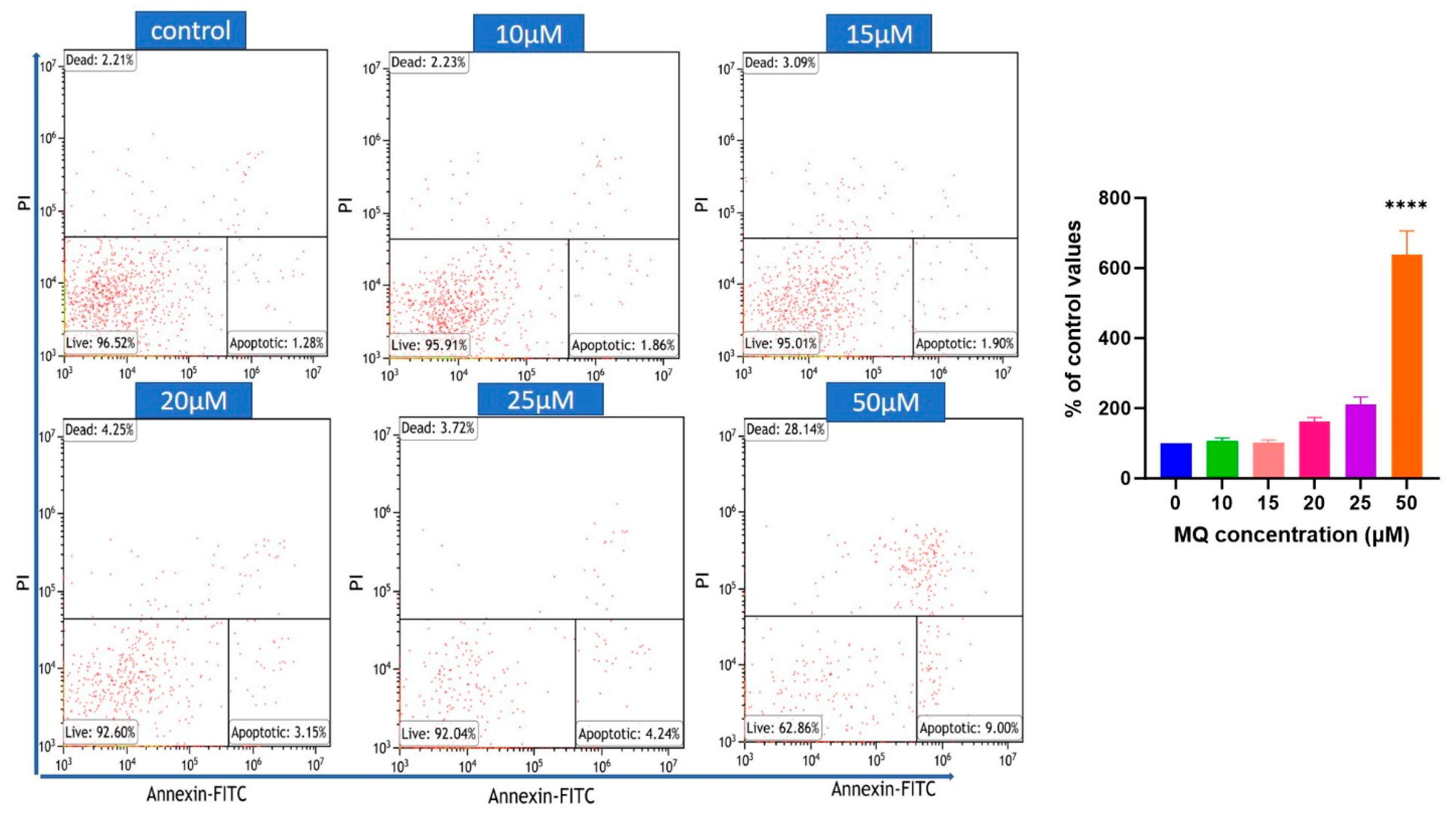
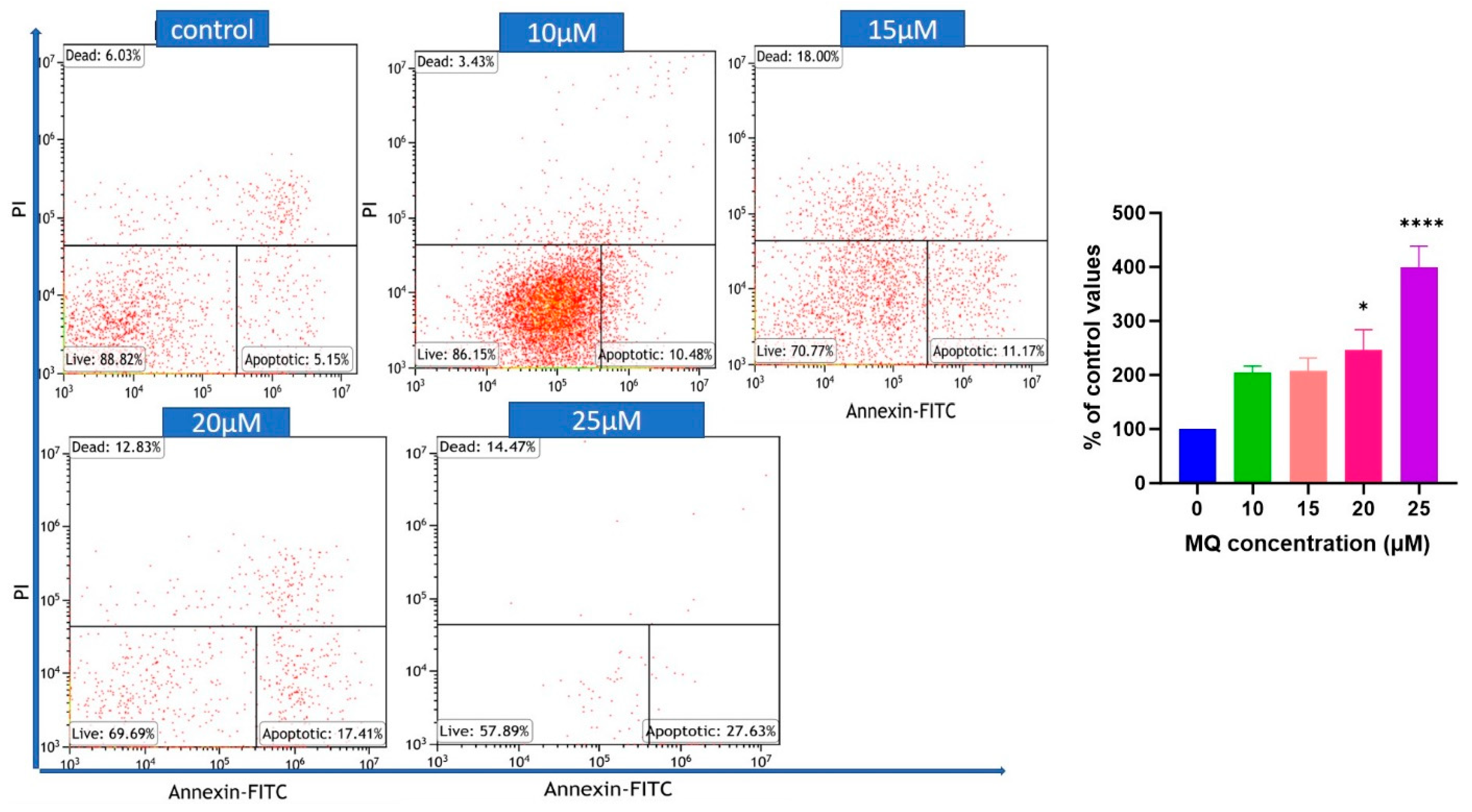
2.6. Flow Cytometric Assessment of Cell Death
2.7. In Silico Analysis
2.7.1. Structure Preparation
2.7.2. Ligand Preparation
2.7.3. Molecular Docking
2.7.4. Prime Molecular Mechanics-Generalised Born and Surface Area Solvation (MM-GBSA)
2.7.5. Molecular Dynamics Simulations
2.8. Cholinesterase (ChE) Activity Assessments
2.9. Statistical Analysis
3. Results
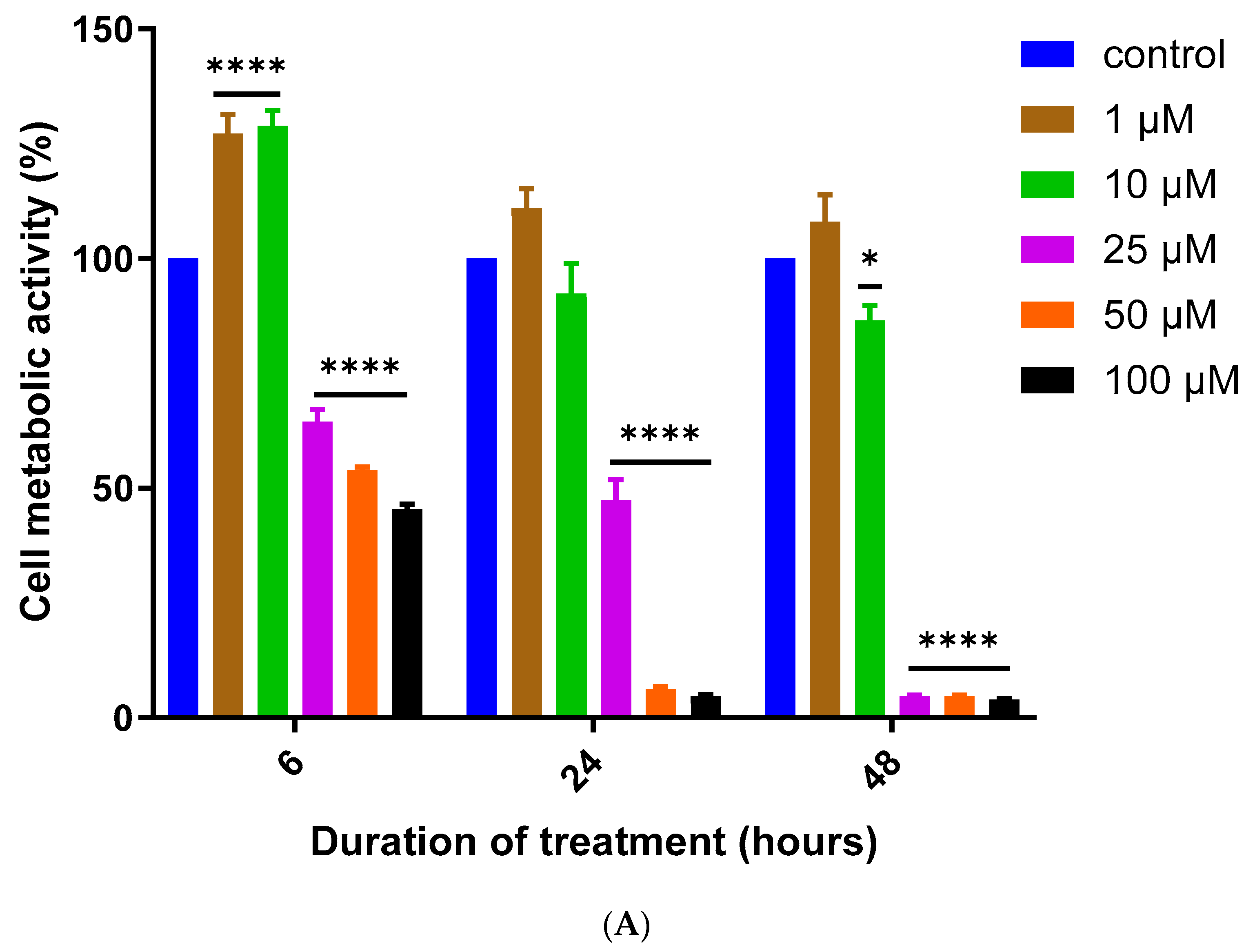
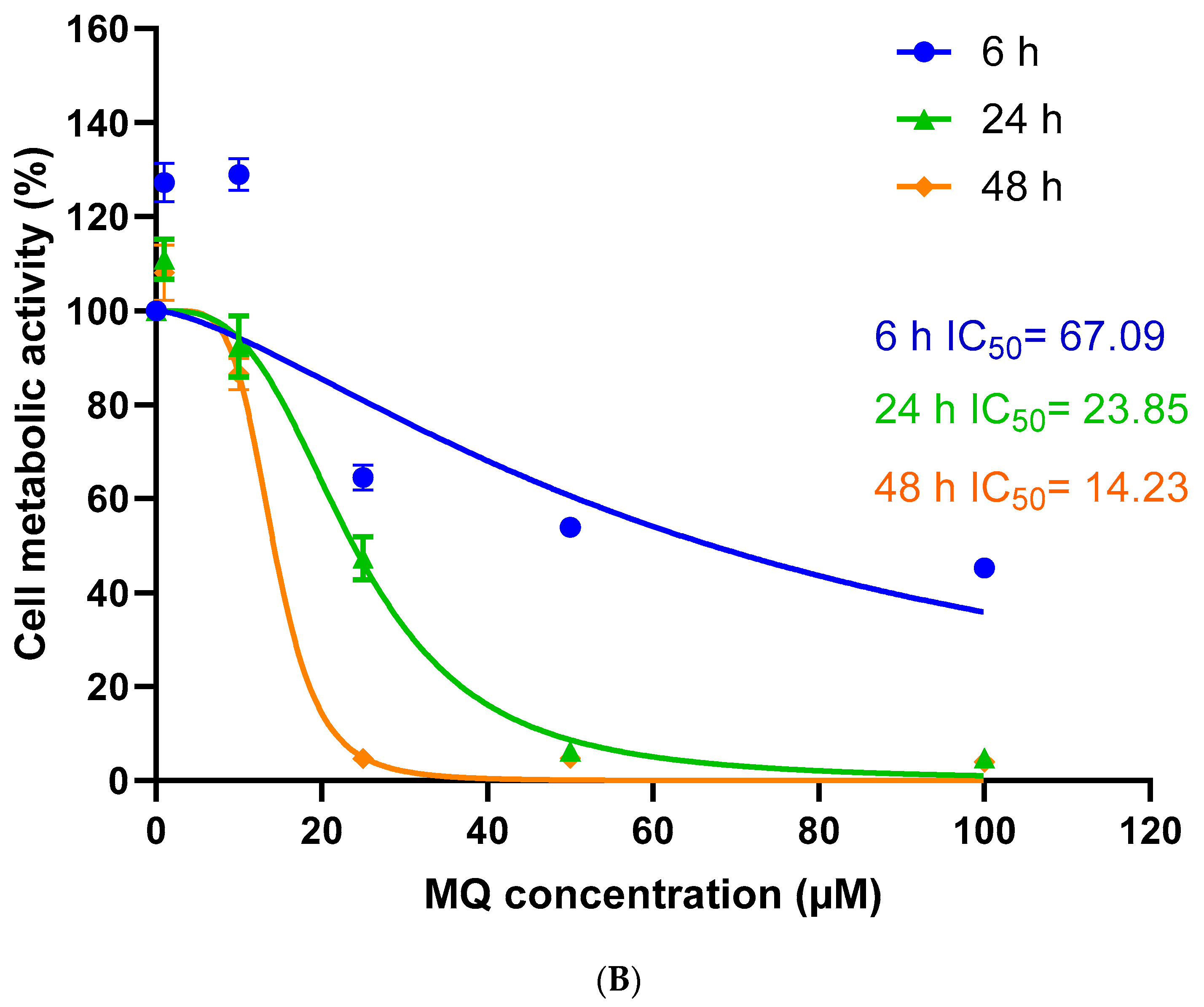
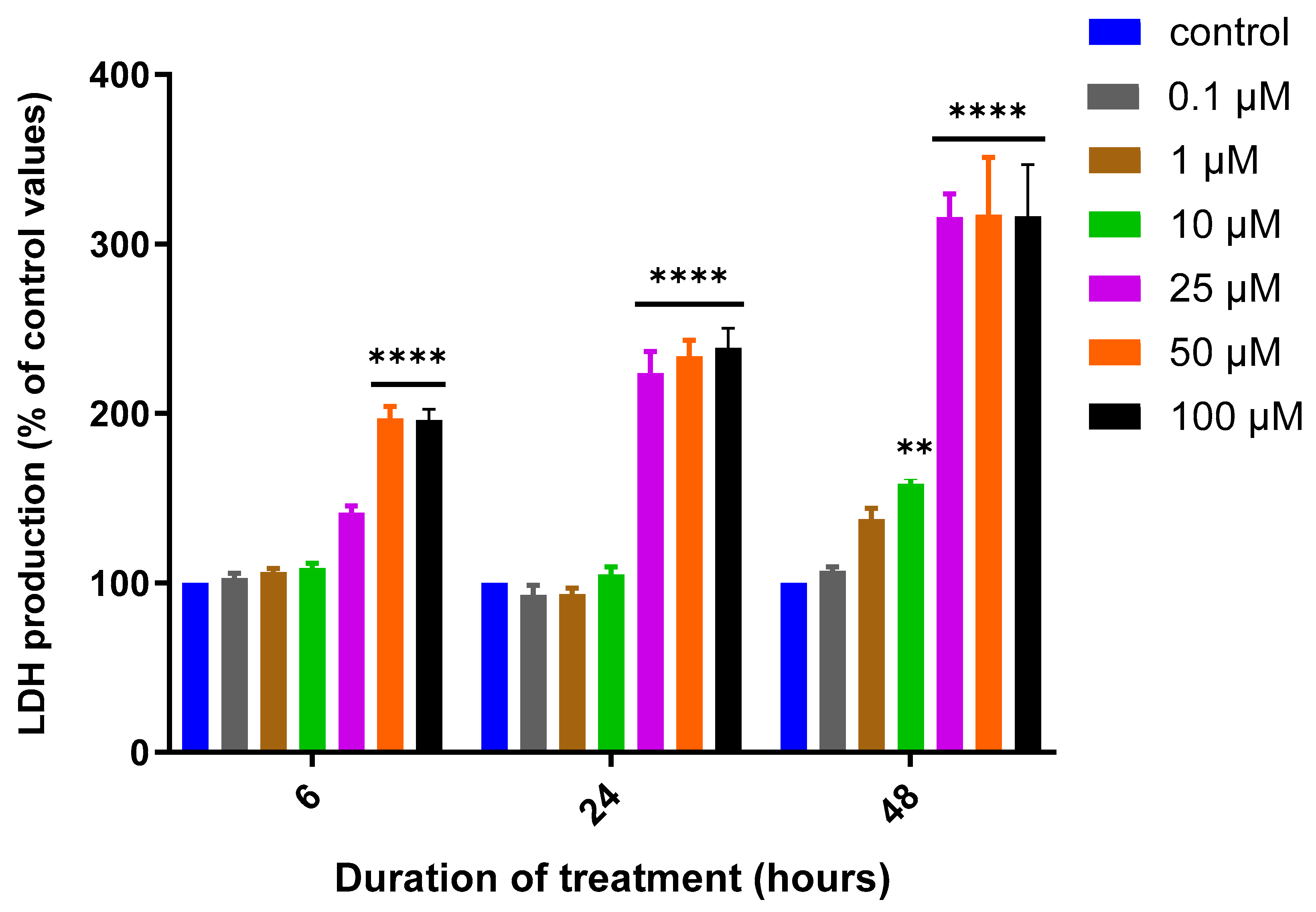
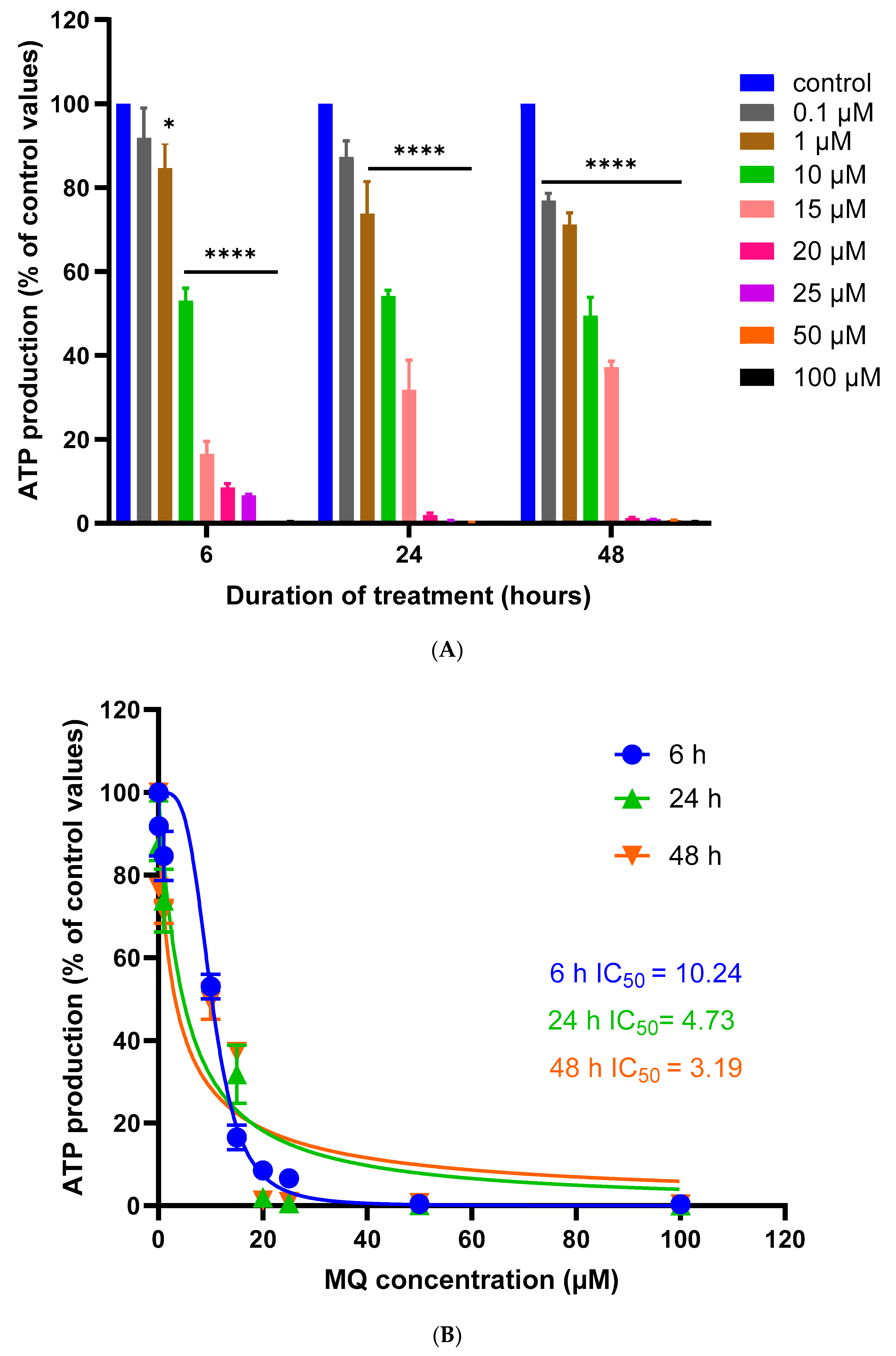
3.1. Assessment of MQ Cytotoxicity
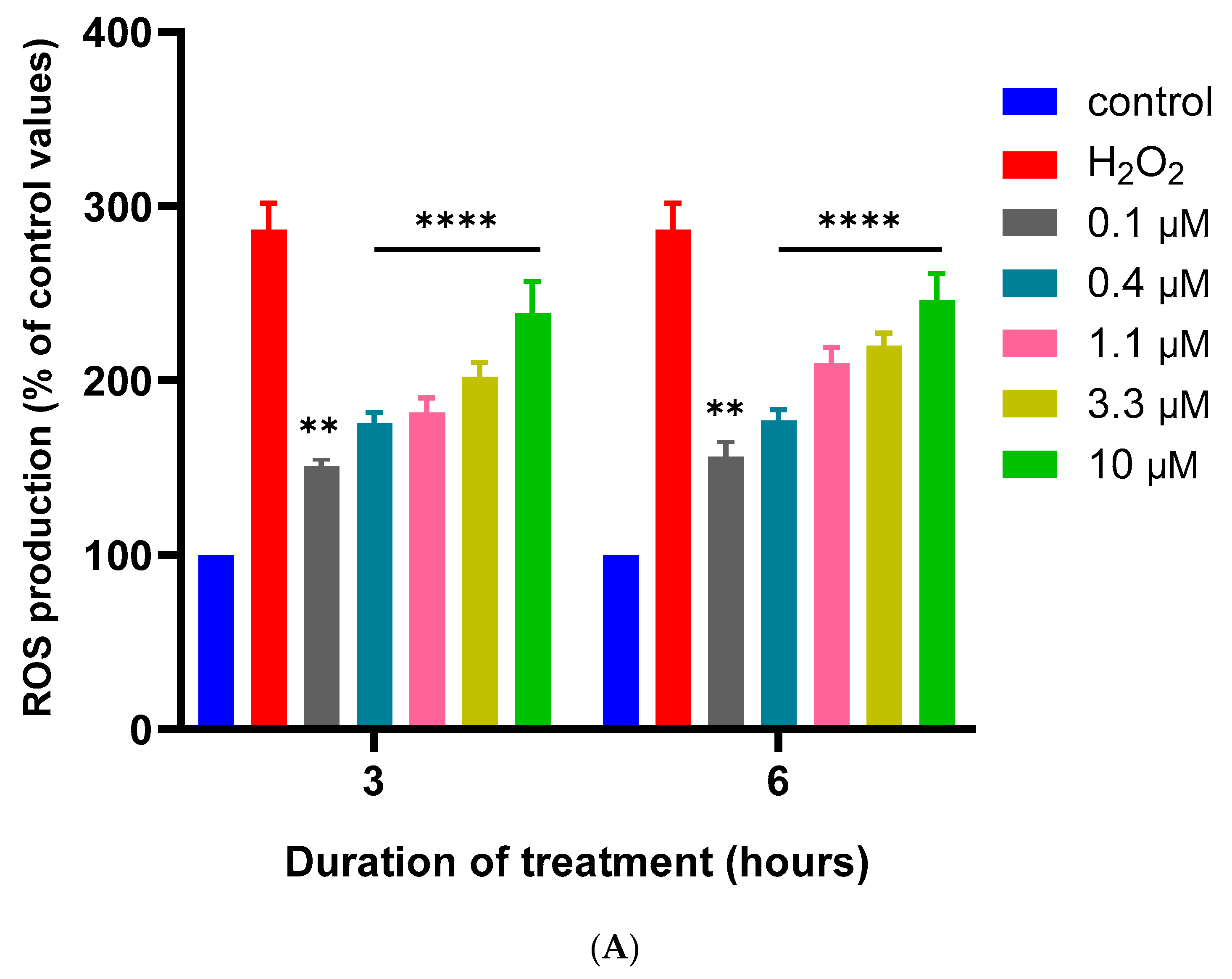
3.2. Effects of MQ on the MMP and Production of ROS
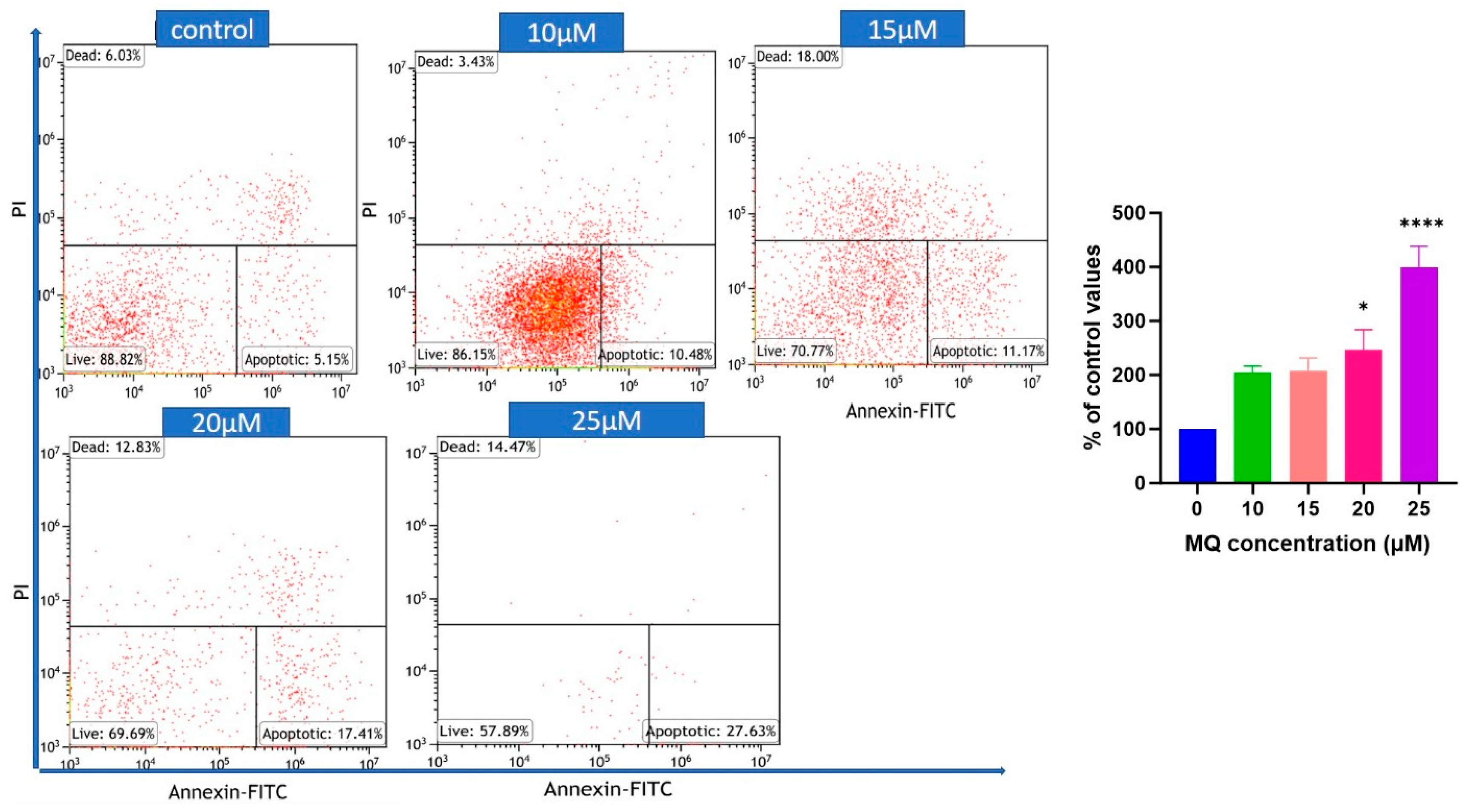
3.3. MQ-Induced Apoptosis
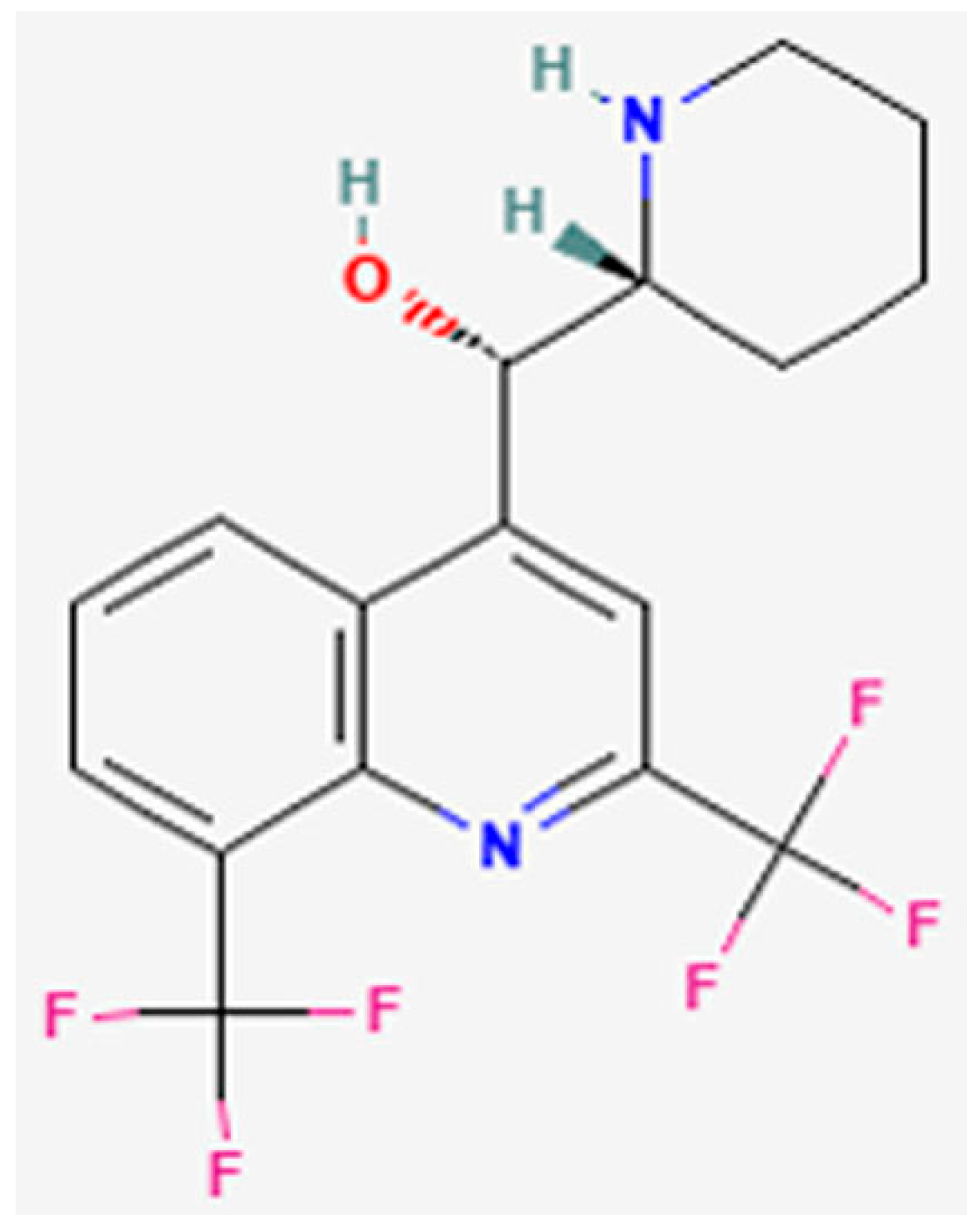
3.4. In Silico Assessment of Cholinesterase Inhibitor Potential
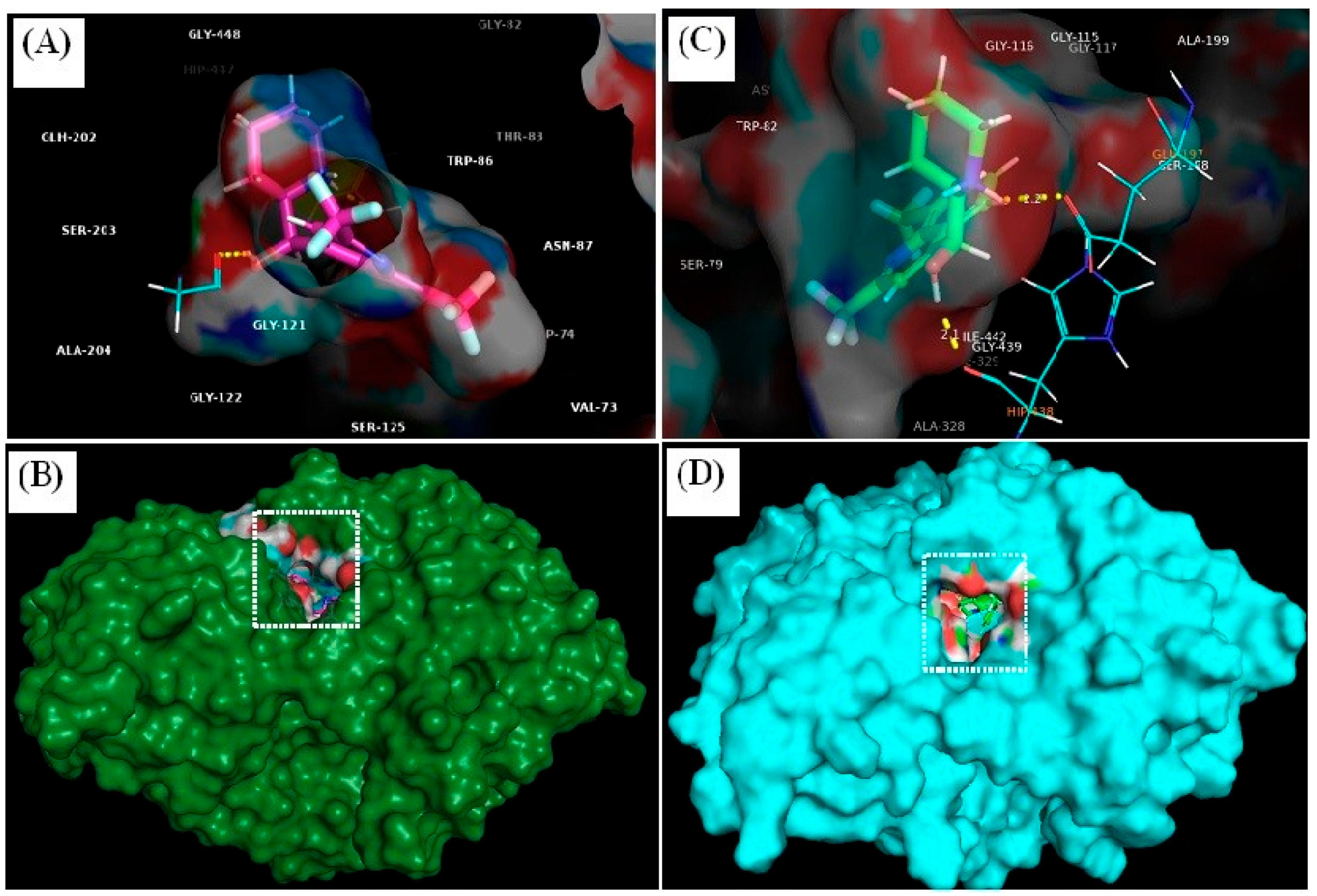
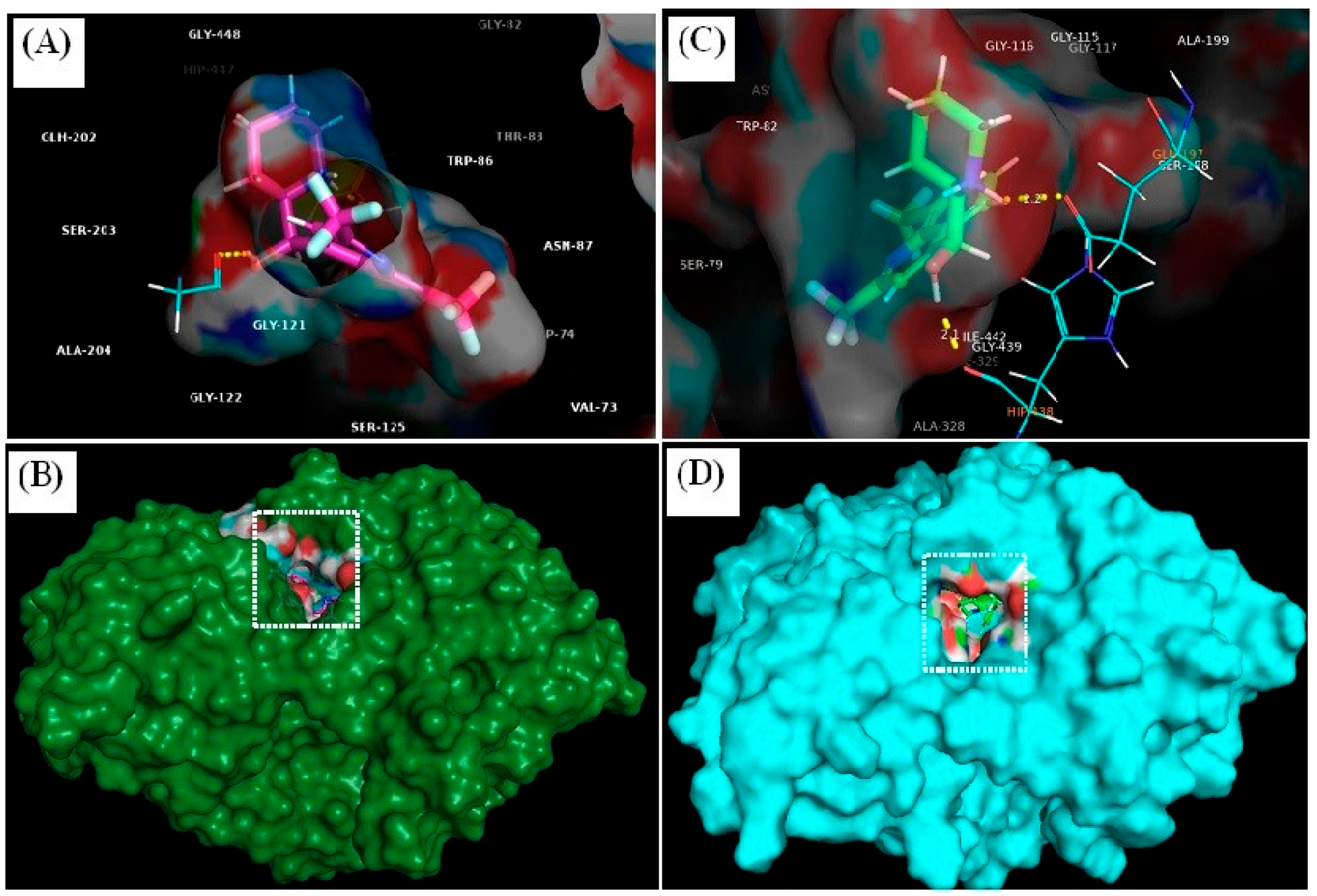
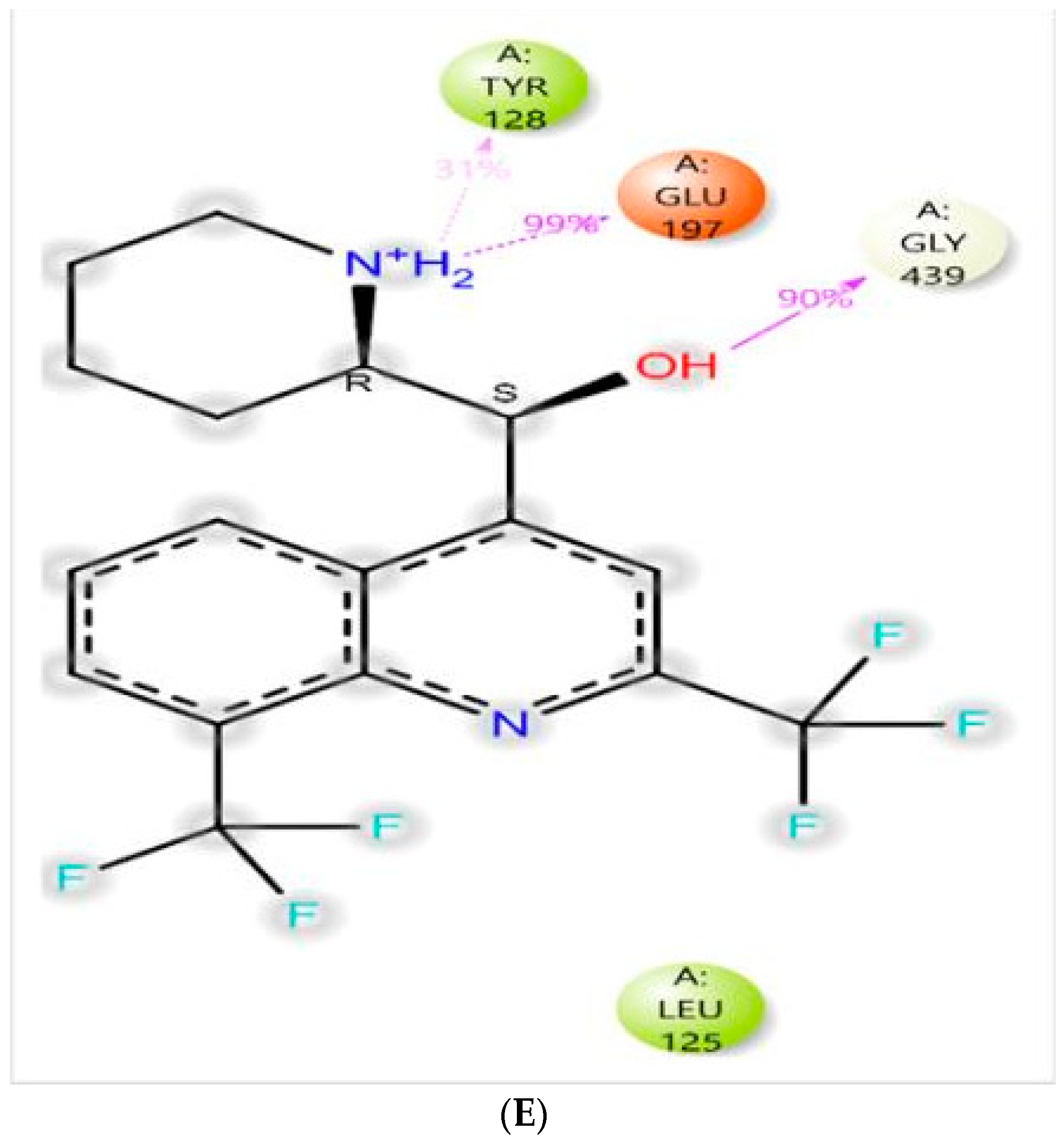
3.4.1. Molecular Docking
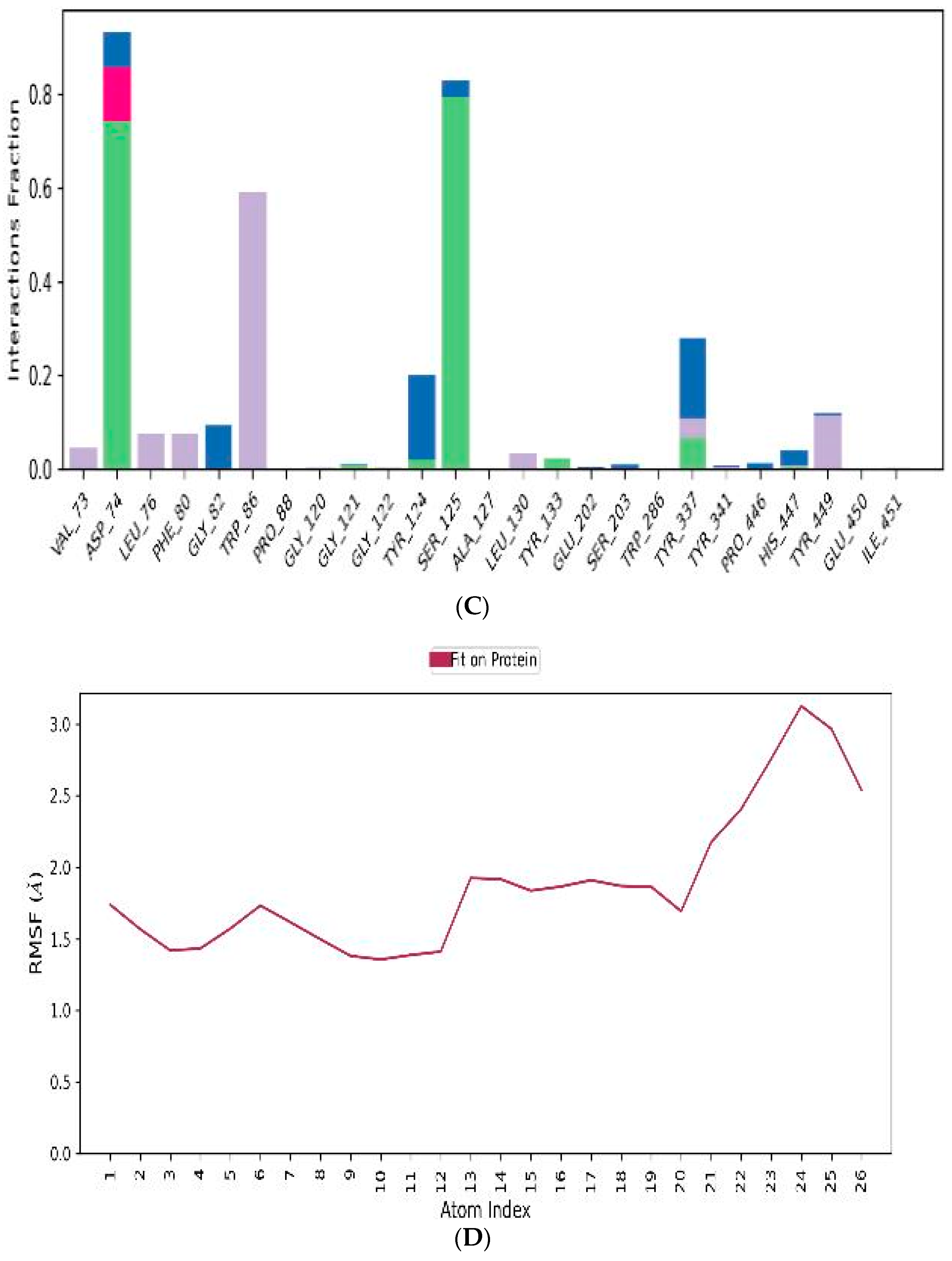
3.4.2. Prime/MM–GBSA Simulation
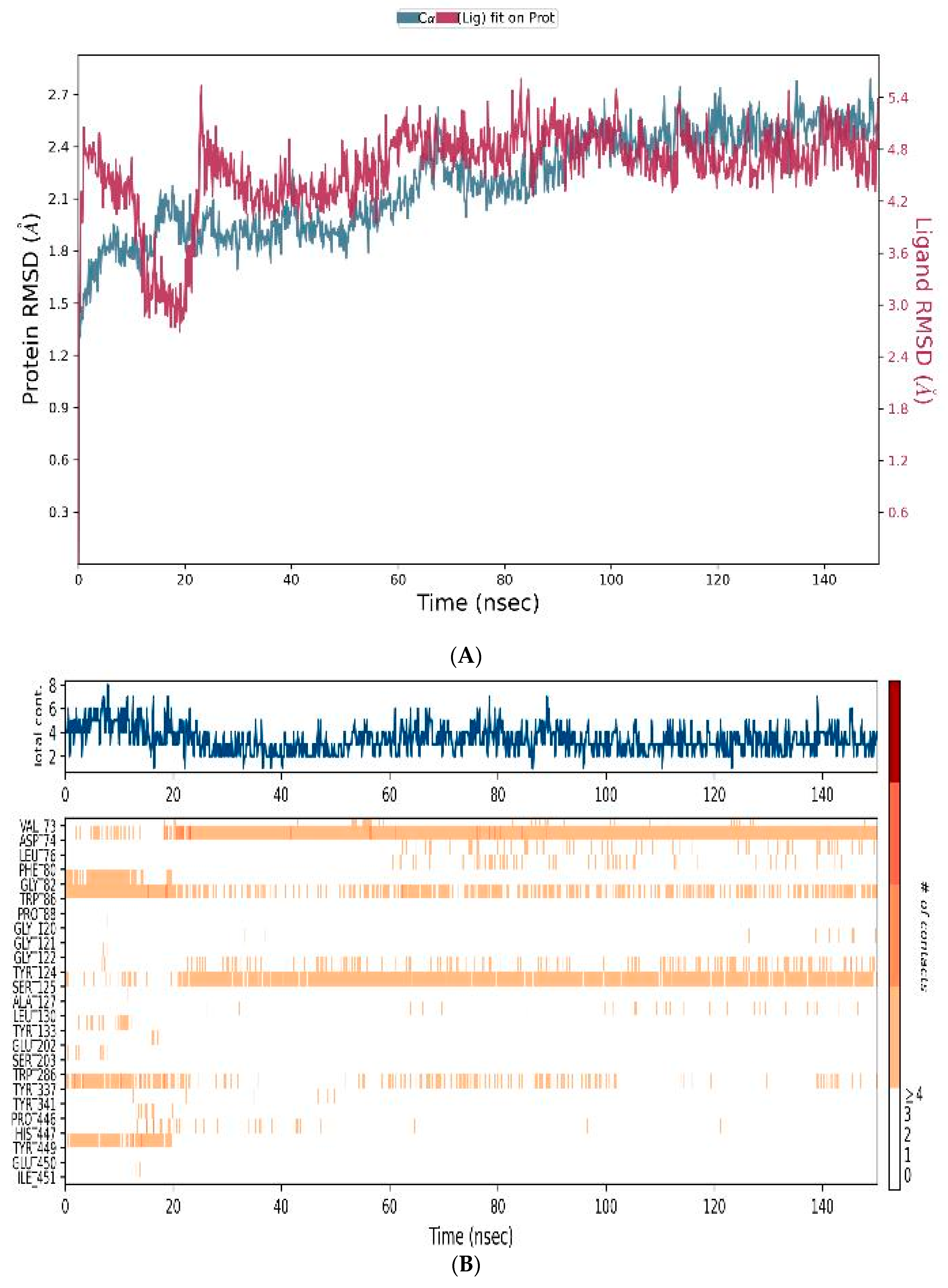
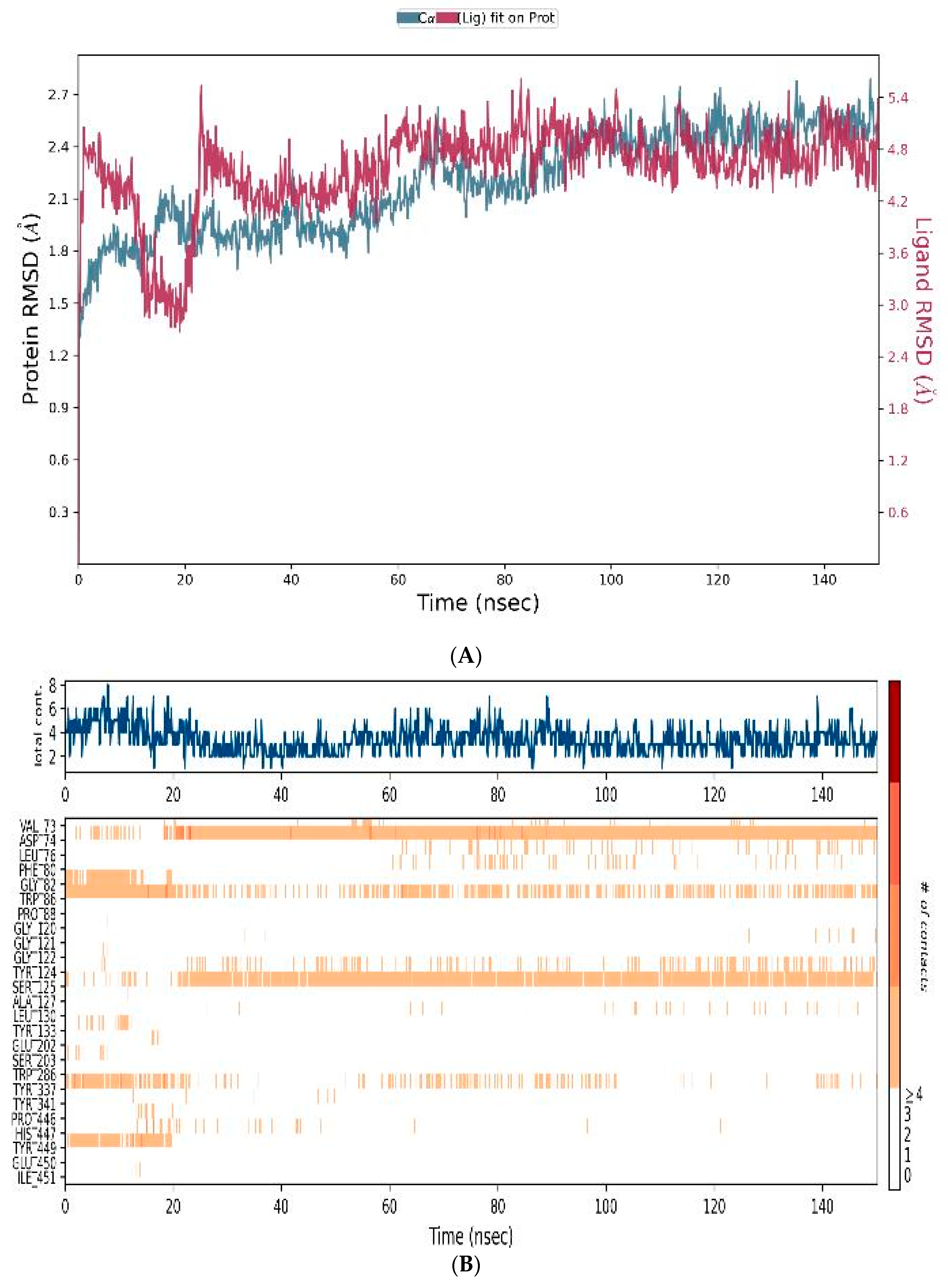
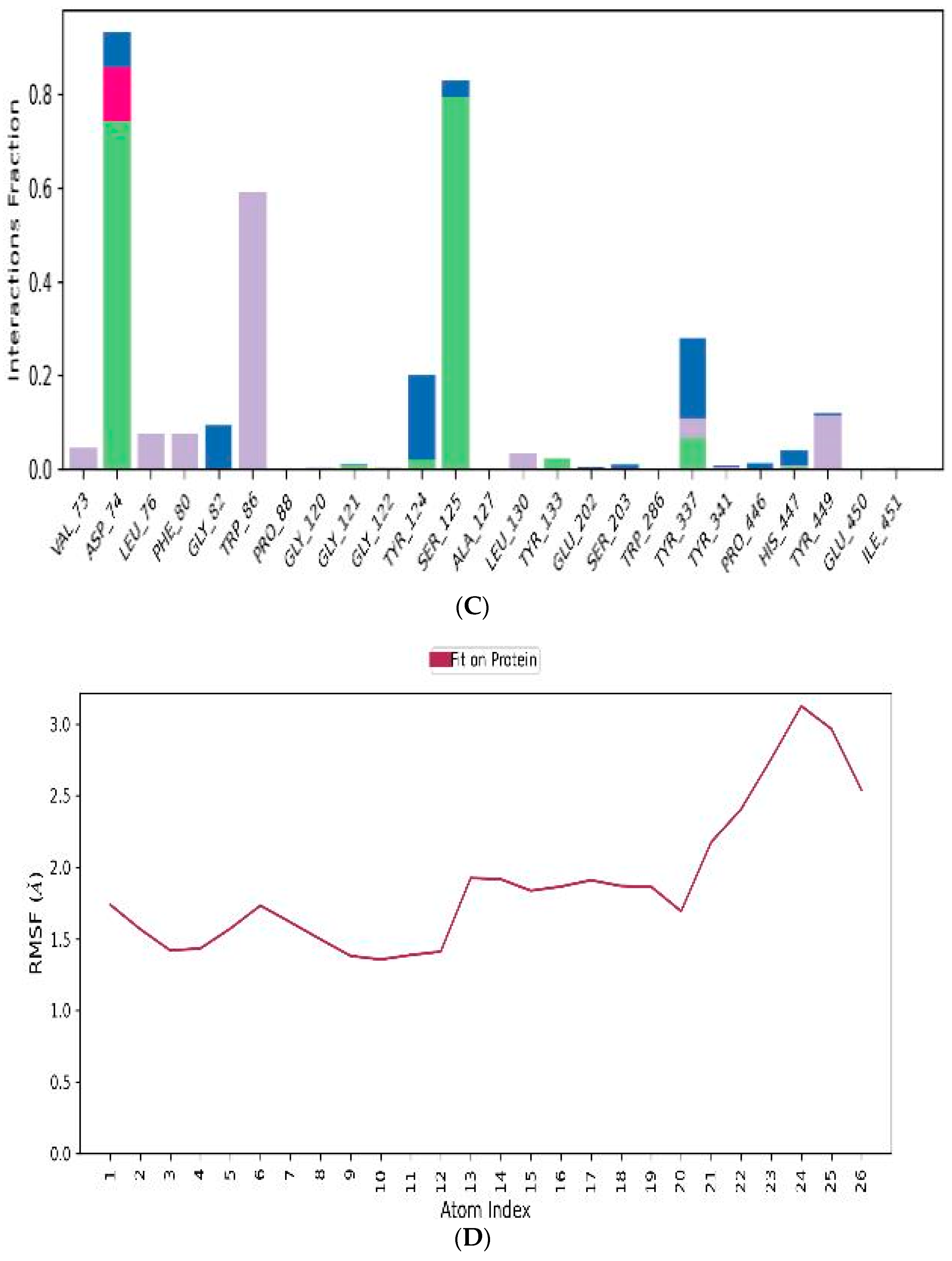
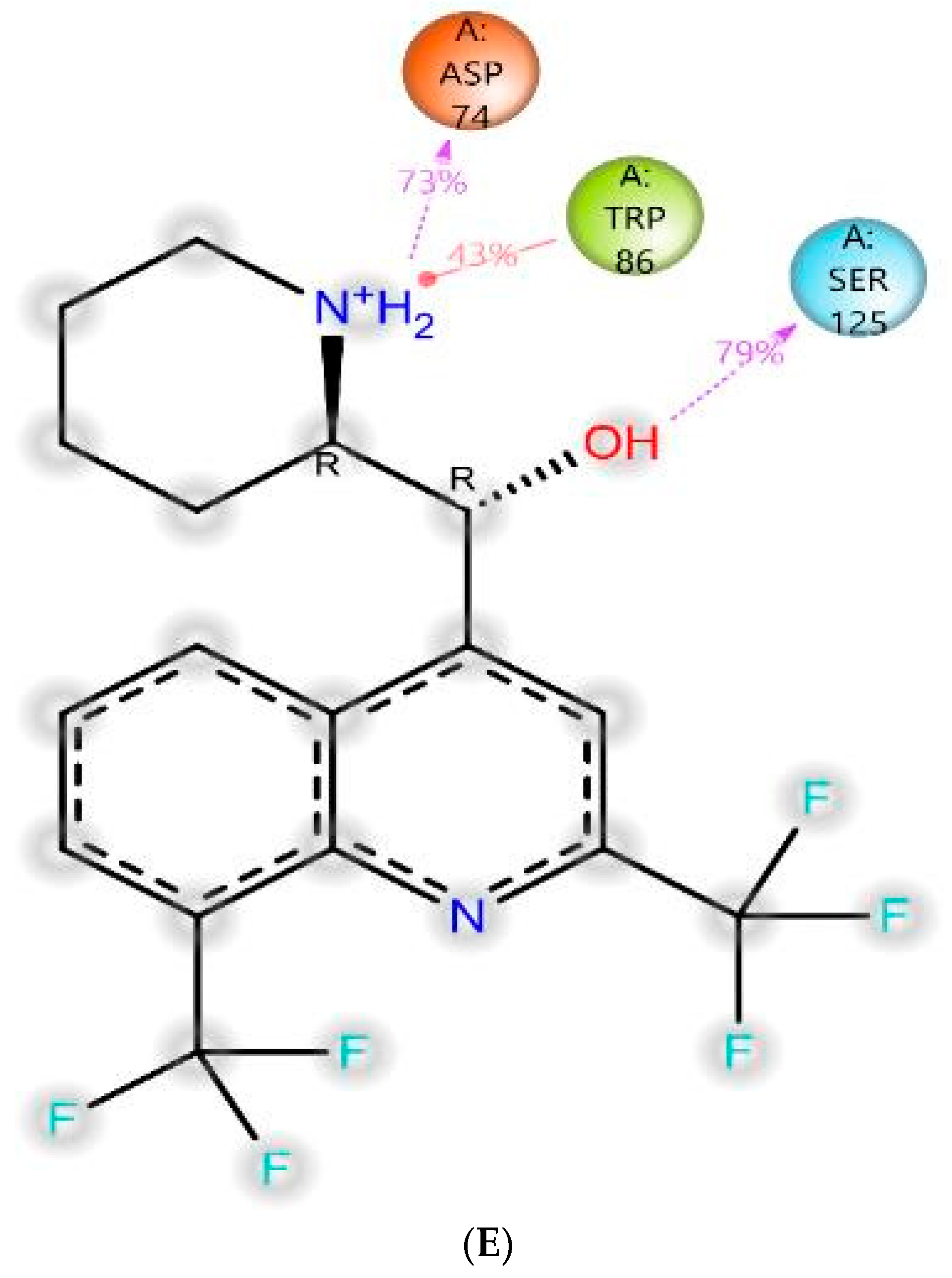
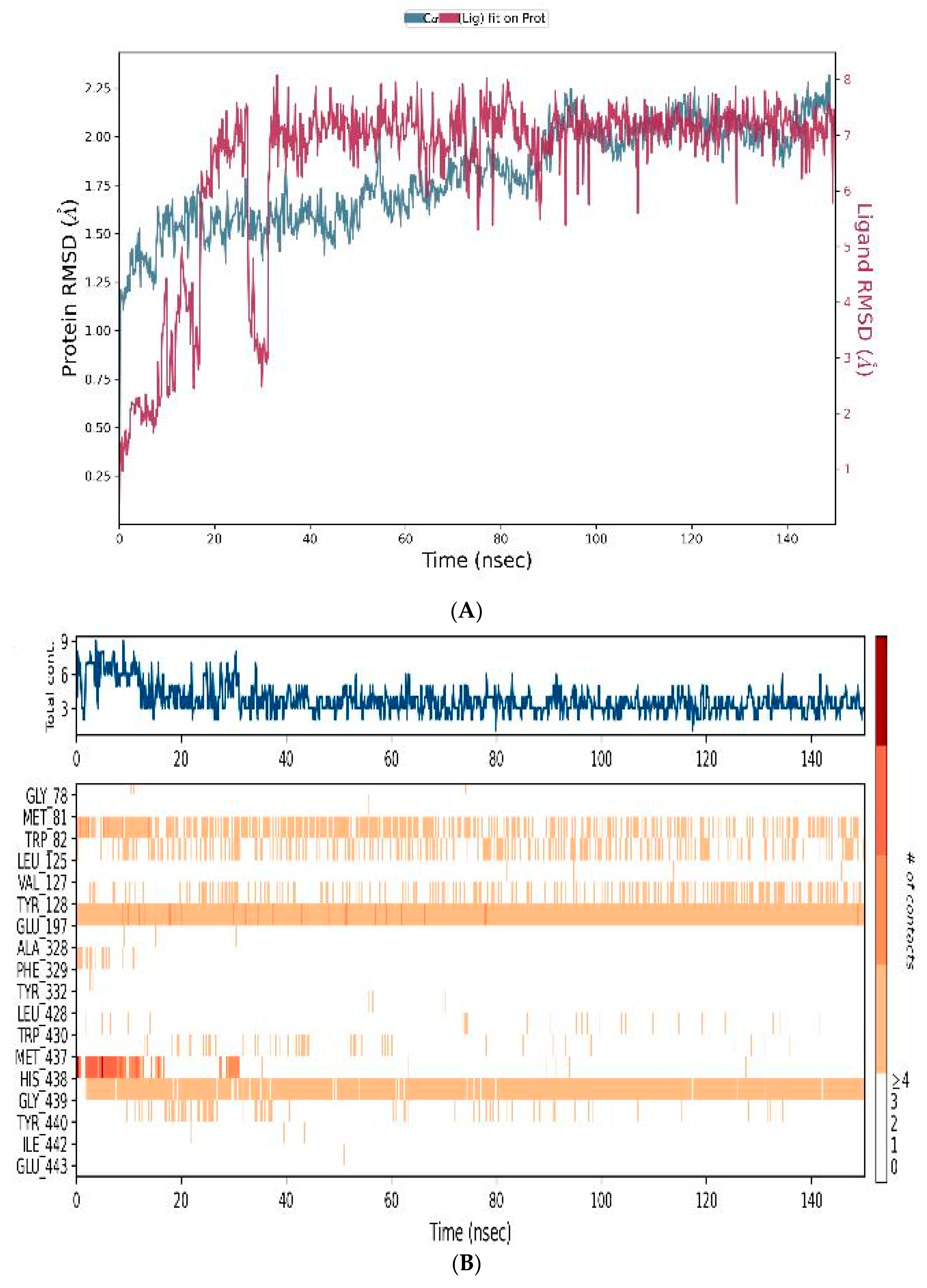
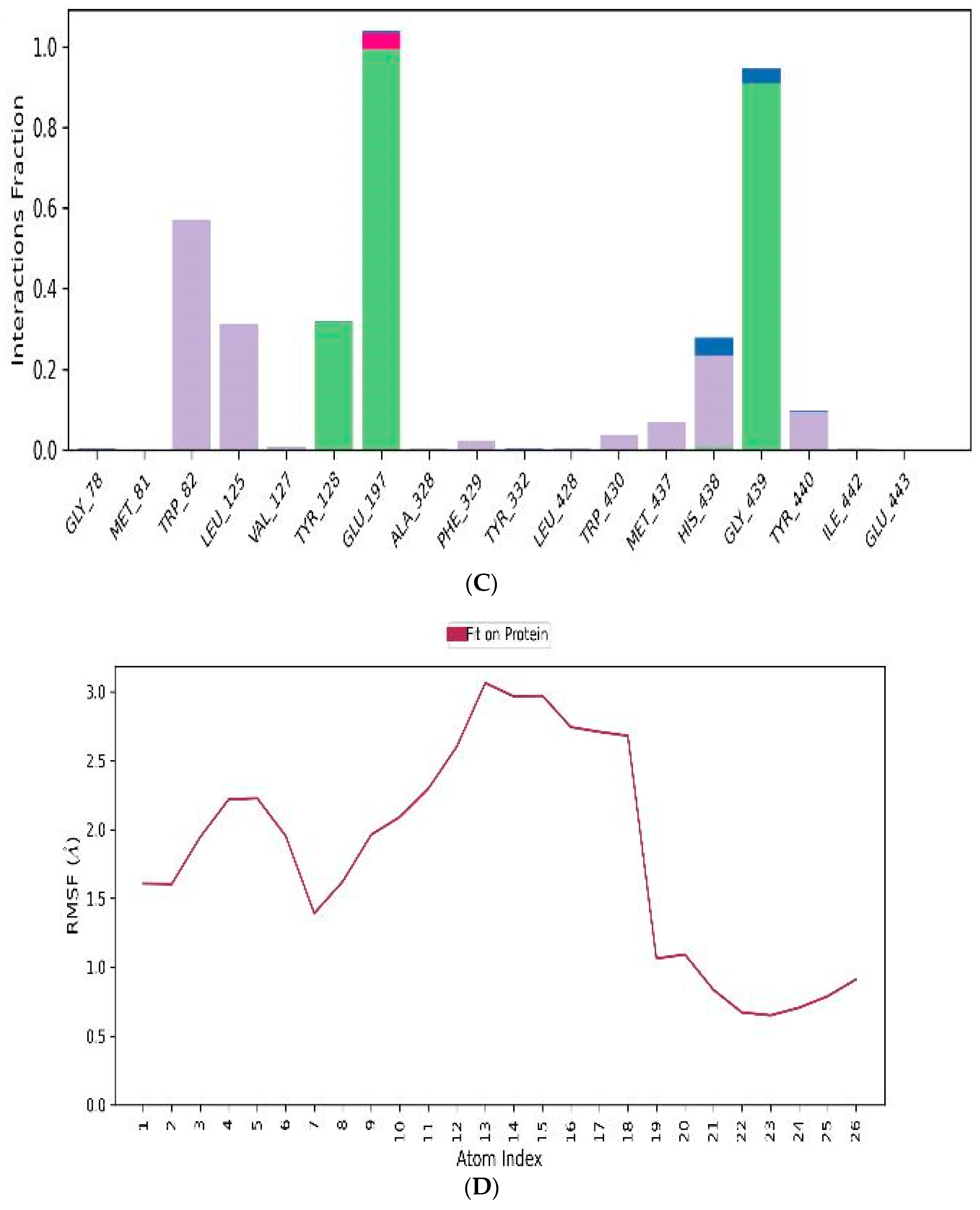
3.4.3. MD-Simulations
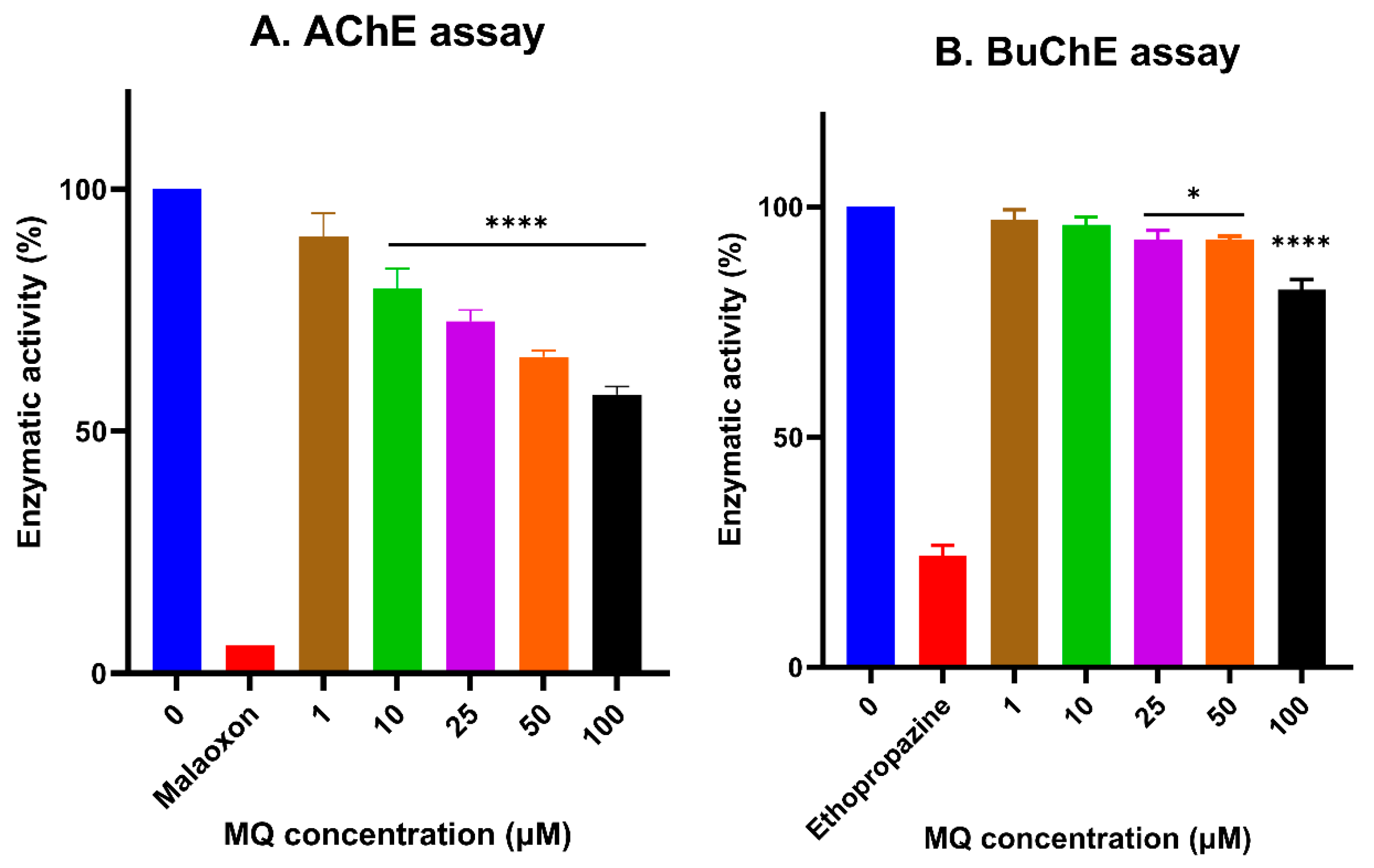
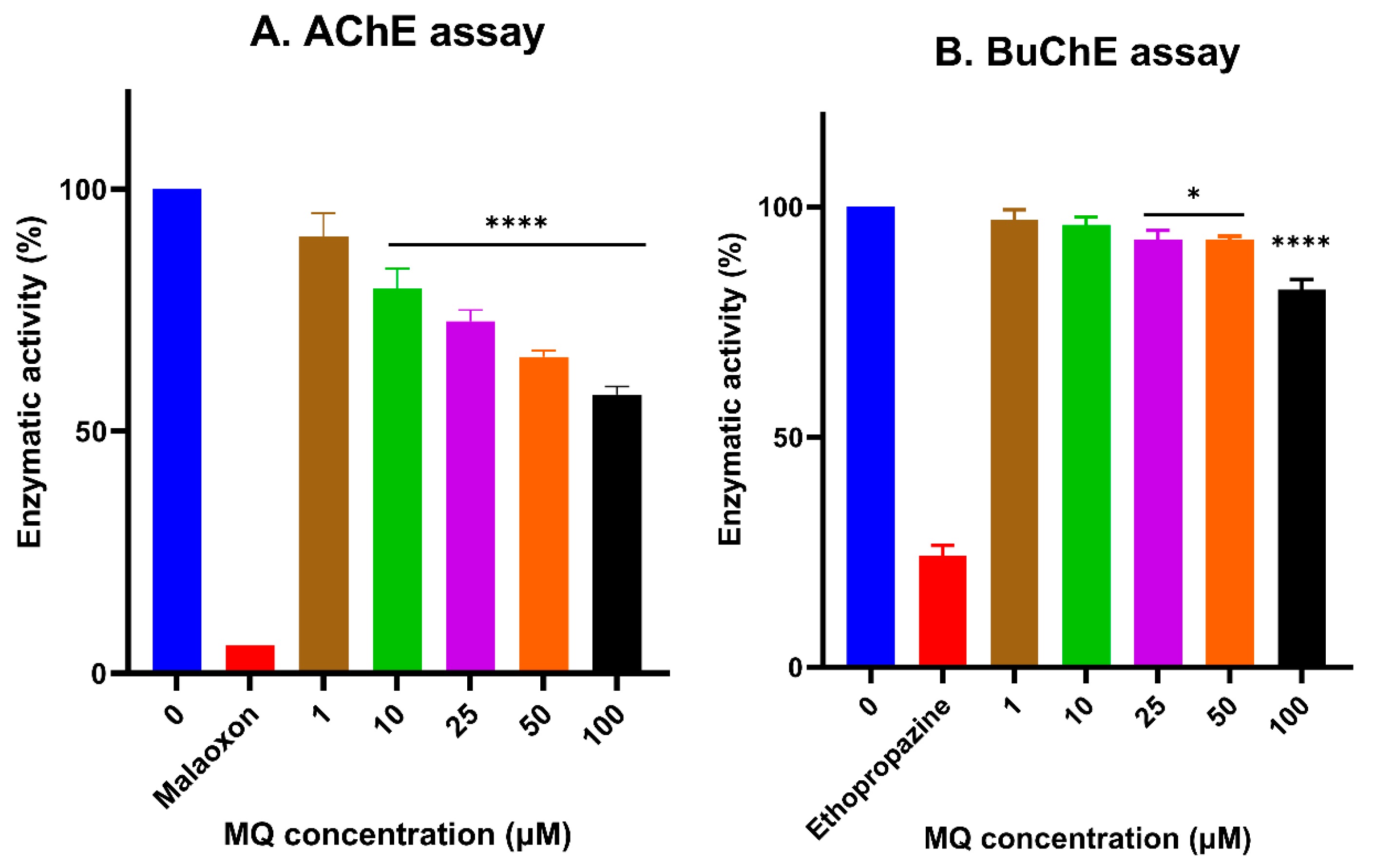
3.5. In Vitro Assessment of Cholinesterase Inhibitor Activity
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Malaria Report 2020. Geneva. Available online: https://www.who.int/publications/i/item/9789240015791 (accessed on 19 January 2024).
- National Center for Biotechnology Information. “PubChem Compound Summary for CID 40692, Mefloquine Hydrochloride” PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Mefloquine-hydrochloride (accessed on 19 January 2024).
- World Health Organization. WHO Model List of Essential Medicines—23rd List, 2023. Available online: https://www.who.int/publications/i/item/WHO-MHP-HPS-EML-2023.02 (accessed on 19 January 2024).
- National Insititute for Health and Care Excellence. “British National Formulary, Drugs, Mefloquine”. Available online: https://bnf.nice.org.uk/drugs/mefloquine/ (accessed on 19 January 2024).
- Lobel, H.O.; Miani, M.; Eng, T.; Bernard, K.W.; Hightower, A.W.; Campbell, C.C. Long-term malaria prophylaxis with weekly mefloquine. Lancet 1993, 341, 8481. [Google Scholar] [CrossRef] [PubMed]
- Bukirwa, H.; Orton, L. Artesunate plus mefloquine versus mefloquine for treating uncomplicated malaria. Cochrane Database Syst. Rev. 2005, 2005, CD004531. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.; Bai, X.C.; Sleebs, B.E.; Triglia, T.; Brown, A.; Thompson, J.K.; Jackson, K.E.; Hanssen, E.; Marapana, D.S.; Fernandez, I.S.; et al. Mefloquine targets the Plasmodium falciparum 80S ribosome to inhibit protein synthesis. Nat. Microbiol. 2017, 2, 17031. [Google Scholar] [CrossRef] [PubMed]
- de Villiers, K.A.; Egan, T.J. Heme Detoxification in the Malaria Parasite: A Target for Antimalarial Drug Development. Acc. Chem. Res. 2021, 54, 2649–2659. [Google Scholar] [CrossRef] [PubMed]
- Hoppe, H.C.; van Schalkwyk, D.A.; Wiehart, U.I.; Meredith, S.A.; Egan, J.; Weber, B.W. Antimalarial quinolines and artemisinin inhibit endocytosis in P. falciparum. Antimicrob. Agents Chemother. 2004, 48, 2370–2378. [Google Scholar] [CrossRef]
- Gunjan, S.; Singh, S.K.; Sharma, T.; Dwivedi, H.; Chauhan, B.S.; Imran Siddiqi, M.; Tripathi, R. Mefloquine induces ROS mediated programmed cell death in malaria parasite: Plasmodium. Apoptosis 2016, 21, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Chevli, R.; Fitch, C.D. The antimalarial drug mefloquine binds to membrane phospholipids. Antimicrob. Agents Chemother. 1982, 21, 581–586. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Ghosh, D.K.; Ali, J.; Ranjan, A. Characterization of Lipid Binding Properties of Plasmodium falciparum Acyl-Coenzyme A Binding Proteins and Their Competitive Inhibition by Mefloquine. ACS Chem. Biol. 2019, 14, 901–915. [Google Scholar] [CrossRef]
- Lee, S.J.; Ter Kuile, F.O.; Price, R.N.; Luxemburger, C.; Nosten, F. Adverse effects of mefloquine for the treatment of uncomplicated malaria in Thailand: A pooled analysis of 19, 850 individual patients. PLoS ONE 2017, 12, e0168780. [Google Scholar] [CrossRef]
- Tran, T.M.; Browning, J.; Dell, M.L. Psychosis with paranoid delusions after a therapeutic dose of mefloquine: A case report. Malar. J. 2006, 5, 74. [Google Scholar] [CrossRef]
- Ghosh, D.K.; Kumar, A.; Ranjan, A. Cellular targets of mefloquine. Toxicology 2021, 464, 152995. [Google Scholar] [CrossRef] [PubMed]
- McArdle, J.J.; Sellin, L.C.; Coakley, K.M.; Potian, J.G.; Hognason, K. Mefloquine selectively increases asynchronous acetylcholine release from motor nerve terminals. Neuropharmacology 2006, 50, 345–353. [Google Scholar] [CrossRef]
- Lim, L.Y.; Go, M.L. The anticholinesterase activity of mefloquine. Clin. Exp. Pharmacol. Physiol. 1985, 12, 527–531. [Google Scholar] [CrossRef]
- Zhou, C.; Xiao, C.; McArdle, J.J.; Ye, J.H. Mefloquine enhances nigral gamma-aminobutyric acid release via inhibition of cholinesterase. J. Pharmacol. Exp. Ther. 2006, 317, 1155–1160. [Google Scholar] [CrossRef] [PubMed]
- Toovey, S. Mefloquine neurotoxicity: A literature review. Travel Med. Infect. Dis. 2009, 7, 2–6. [Google Scholar] [CrossRef] [PubMed]
- Martins, A.C.; Paoliello, M.M.B.; Docea, A.O.; Santamaria, A.; Tinkov, A.A.; Skalny, A.V.; Aschner, M. Review of the mechanism underlying mefloquine-induced neurotoxicity. Crit. Rev. Toxicol. 2021, 51, 209–216. [Google Scholar] [CrossRef]
- Cruikshank, S.J.; Hopperstad, M.; Younger, M.; Connors, B.W.; Spray, D.C.; Srinivas, M. Potent block of Cx36 and Cx50 gap junction channels by mefloquine. Proc. Natl. Acad. Sci. USA 2004, 101, 12364–12369. [Google Scholar] [CrossRef]
- Dow, G.S.; Hudson, T.H.; Vahey, M.; Koenig, M.L. The acute neurotoxicity of mefloquine may be mediated through a disruption of calcium homeostasis and ER function in vitro. Malar. J. 2003, 2, 14. [Google Scholar] [CrossRef]
- Gribble, F.M.; Davis, T.M.; Higham, C.E.; Clark, A.; Ashcroft, F.M. The antimalarial agent mefloquine inhibits ATP-sensitive K-channels. Br. J. Pharmacol. 2000, 131, 756–760. [Google Scholar] [CrossRef]
- Hood, J.E.; Jenkins, J.W.; Milatovic, D.; Rongzhu, L.; Aschner, M. Mefloquine induces oxidative stress and neurodegeneration in primary rat cortical neurons. Neurotoxicology 2010, 31, 518–523. [Google Scholar] [CrossRef]
- Denizot, F.; Lang, R. Rapid colorimetric assay for cell growth and survival. Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J. Immunol. Methods 1986, 89, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Elmorsy, E.; Al-Ghafari, A.; Al Doghaither, H.; Hashish, S.; Salama, M.; Mudyanselage, A.W.; James, L.; Carter, W.G. Differential Effects of Paraquat, Rotenone, and MPTP on Cellular Bioenergetics of Undifferentiated and Differentiated Human Neuroblastoma Cells. Brain Sci. 2023, 13, 1717. [Google Scholar] [CrossRef] [PubMed]
- ALNasser, M.N.; AlSaadi, A.M.; Whitby, A.; Kim, D.H.; Mellor, I.R.; Carter, W.G. Acai Berry (Euterpe sp.) Extracts Are Neuroprotective against L-Glutamate-Induced Toxicity by Limiting Mitochondrial Dysfunction and Cellular Redox Stress. Life 2023, 13, 1019. [Google Scholar] [CrossRef] [PubMed]
- Naseem, A.; Rasool, F.; Ahmed, A.; Carter, W.G. The Potential of Stilbene Compounds to Inhibit Mpro Protease as a Natural Treatment Strategy for Coronavirus Disease-2019. Curr. Issues Mol. Biol. 2022, 45, 12–32. [Google Scholar] [CrossRef] [PubMed]
- Ellman, G.L.; Courtney, K.D.; Andres, V., Jr.; Feather-stone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Carter, W.G.; Tarhoni, M.; Rathbone, A.J.; Ray, D.E. Differential protein adduction by seven organophosphorus pesticides in both brain and thymus. Hum. Exp. Toxicol. 2007, 26, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Dorling, J.L.; Clayton, D.J.; Jones, J.; Carter, W.G.; Thackray, A.E.; King, J.A.; Pucci, A.; Batterham, R.L.; Stensel, D.J. A randomized crossover trial assessing the effects of acute exercise on appetite, circulating ghrelin concentrations, and butyrylcholinesterase activity in normal-weight males with variants of the obesity-linked FTO rs9939609 polymorphism. Am. J. Clin. Nutr. 2019, 110, 1055–1066. [Google Scholar] [CrossRef]
- Mudyanselage, A.W.; Wijamunige, B.C.; Kocon, A.; Carter, W.G. Differentiated Neurons Are More Vulnerable to Organophosphate and Carbamate Neurotoxicity than Undifferentiated Neurons Due to the Induction of Redox Stress and Accumulate Oxidatively-Damaged Proteins. Brain Sci. 2023, 13, 728. [Google Scholar] [CrossRef]
- Nevin, R.L.; Byrd, A.M. Neuropsychiatric Adverse Reactions to Mefloquine: A Systematic Comparison of Prescribing and Patient Safety Guidance in the US, UK, Ireland, Australia, New Zealand, and Canada. Neurol. Ther. 2016, 5, 69–83. [Google Scholar] [CrossRef]
- Kovalevich, J.; Langford, D. Considerations for the use of SH-SY5Y neuroblastoma cells in neurobiology. Methods Mol. Biol. 2013, 1078, 9–21. [Google Scholar]
- Lopez-Suarez, L.; Awabdh, S.A.; Coumoul, X.; Chauvet, C. The SH-SY5Y human neuroblastoma cell line, a relevant in vitro cell model for investigating neurotoxicology in human: Focus on organic pollutants. Neurotoxicology 2022, 92, 131–155. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.H.; Park, S.J.; Jo, Y.K.; Kim, E.S.; Kang, H.; Park, J.H.; Lee, E.H.; Cho, D.H. Suppression of autophagy exacerbates Mefloquine-mediated cell death. Neurosci. Lett. 2012, 515, 162–167. [Google Scholar] [CrossRef] [PubMed]
- Karbwang, J.; White, N.J. Clinical Pharmacokinetics of Mefloquine. Clin. Pharmacokinet. 1990, 19, 264–279. [Google Scholar] [CrossRef] [PubMed]
- Simpson, J.A.; Price, R.; ter Kuile, F.; Teja-Isavatharm, P.; Nosten, F.; Chongsuphajaisiddhi, T.; Looareesuwan, S.; Aarons, L.; White, N.J. Population pharmacokinetics of mefloquine in patients with acute falciparum malaria. Clin. Pharmacol. Ther. 1999, 66, 472–484. [Google Scholar] [CrossRef] [PubMed]
- Kaja, S.; Payne, A.J.; Naumchuk, Y.; Koulen, P. Quantification of Lactate Dehydrogenase for Cell Viability Testing Using Cell Lines and Primary Cultured Astrocytes. Curr. Protoc. Toxicol. 2017, 72, 2.26.1–2.26.10. [Google Scholar] [CrossRef] [PubMed]
- Reuter, S.E.; Upton, R.N.; Evans, A.M.; Navaratnam, V.; Olliaro, P.L. Population pharmacokinetics of orally administered mefloquine in healthy volunteers and patients with uncomplicated Plasmodium falciparum malaria. J. Antimicrob. Chemother. 2015, 70, 868–876. [Google Scholar] [CrossRef]
- Gutman, J.; Green, M.; Durand, S.; Rojas, O.V.; Ganguly, B.; Quezada, W.M.; Utz, G.C.; Slutsker, L.; Ruebush, T.K., 2nd; Bacon, D.J. Mefloquine pharmacokinetics and mefloquine-artesunate effectiveness in Peruvian patients with uncomplicated Plasmodium falciparum malaria. Malar. J. 2009, 8, 58. [Google Scholar] [CrossRef] [PubMed]
- Watson, J.A.; Tarning, J.; Hoglund, R.M.; Baud, F.J.; Megarbane, B.; Clemessy, J.L.; White, N.J. Concentration-dependent mortality of chloroquine in overdose. eLife 2020, 9, e58631. [Google Scholar] [CrossRef]
- Lee, M.-S.; Park, W.-S.; Kim, Y.; Ahn, W.; Kwon, S.-H.; Her, S. Intracellular ATP assay of live cells using PTD-conjugated luciferase. Sensors 2012, 12, 15628–15637. [Google Scholar] [CrossRef]
- Kamiloglu, S.; Sari, G.; Ozdal, T.; Capanoglu, E. Guidelines for cell viability assays. Food Front. 2020, 1, 332–349. [Google Scholar] [CrossRef]
- Xie, Y.; Zhang, J.; Lu, B.; Bao, Z.; Zhao, J.; Lu, X.; Wei, Y.; Yao, K.; Jiang, Y.; Yuan, Q.; et al. Mefloquine Inhibits Esophageal Squamous Cell Carcinoma Tumor Growth by Inducing Mitochondrial Autophagy. Front. Oncol. 2020, 10, 1217. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta 2016, 1863, 2977–2992. [Google Scholar] [CrossRef] [PubMed]
- Elmehy, D.A.; Ismail, H.I.H.; Soliman, N.A.; Amer, B.S.; Elkaliny, H.H.; El-Ebiary, A.A.; Gamea, G.A. Oxidative stress mediated apoptotic potential of mefloquine on experimental trichinellosis. Acta Trop. 2021, 213, 105760. [Google Scholar] [CrossRef] [PubMed]
- Kandiah, N.; Pai, M.C.; Senanarong, V.; Looi, I.; Ampil, E.; Park, K.W.; Karanam, A.K.; Christopher, S. Rivastigmine: The advantages of dual inhibition of acetylcholinesterase and butyrylcholinesterase and its role in subcortical vascular dementia and Parkinson’s disease dementia. Clin. Interv. Aging 2017, 12, 697–707. [Google Scholar] [CrossRef] [PubMed]
- National Insititute for Health and Care Excellence. “British National Formulary, Drugs, Rivastigmine”. Available online: https://bnf.nice.org.uk/drugs/rivastigmine/ (accessed on 15 February 2024).
- Ngiam, T.L.; Go, M.L. Stereospecific inhibition of cholinesterases by mefloquine enantiomers. Chem. Pharm. Bull. 1987, 35, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Bester, S.M.; Guelta, M.A.; Cheung, J.; Winemiller, M.D.; Bae, S.Y.; Myslinski, J.; Pegan, S.D.; Height, J.J. Structural Insights of Stereospecific Inhibition of Human Acetylcholinesterase by VX and Subsequent Reactivation by HI-6. Chem. Res. Toxicol. 2018, 31, 1405–1417. [Google Scholar] [CrossRef]
- Barraud de Lagerie, S.; Comets, E.; Gautrand, C.; Fernandez, C.; Auchere, D.; Singlas, E.; Mentre, F.; Gimenez, F. Cerebral uptake of mefloquine enantiomers with and without the P-gp inhibitor elacridar (GF1210918) in mice. Br. J. Pharmcol. 2004, 141, 1214–1222. [Google Scholar] [CrossRef]
- Tansley, R.; Lotharius, J.; Priestley, A.; Bull, F.; Duparc, S.; Möhrle, J. A randomized, double-blind, placebo-controlled study to investigate the safety, tolerability, and pharmacokinetics of single enantiomer (+)-mefloquine compared with racemic mefloquine in healthy persons. Am. J. Trop. Med. Hyg. 2010, 83, 1195–1201. [Google Scholar] [CrossRef]
- Thompson, A.J.; Lochner, M.; Lummis, S.C. The antimalarial drugs quinine, chloroquine and mefloquine are antagonists at 5-HT3 receptors. Br. J. Pharmacol. 2007, 151, 666–677. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment Concentration (µM) | 6 h vs. 24 h | 6 h vs. 48 h | 24 h vs. 48 h |
|---|---|---|---|
| 1 | 0.0006 | <0.0001 | NS |
| 10 | <0.0001 | <0.0001 | NS |
| 25 | 0.0003 | <0.0001 | <0.0001 |
| 50 | <0.0001 | <0.0001 | NS |
| 100 | <0.0001 | <0.0001 | NS |
| Treatment Concentration (µM) | 6 h vs. 24 h | 6 h vs. 48 h | 24 h vs. 48 h |
|---|---|---|---|
| 0.1 | NS | NS | NS |
| 1 | NS | NS | 0.0234 |
| 10 | NS | 0.0092 | 0.0045 |
| 25 | <0.0001 | <0.0001 | <0.0001 |
| 50 | NS | <0.0001 | <0.0001 |
| 100 | 0.0307 | <0.0001 | <0.0001 |
| Treatment Concentration (µM) | 6 h vs. 24 h | 6 h vs. 48 h | 24 h vs. 48 h |
|---|---|---|---|
| 0.1 | NS | 0.0025 | NS |
| 1 | 0.0398 | 0.0071 | NS |
| 10 | NS | NS | NS |
| 15 | 0.0019 | <0.0001 | NS |
| 20 | NS | NS | NS |
| 25 | NS | NS | NS |
| 50 | NS | NS | NS |
| 100 | NS | NS | NS |
| Duration of Treatment | MTT Assay | LDH Assay | ATP Assay | |||
|---|---|---|---|---|---|---|
| IC50 | R2 | IC50 | R2 | IC50 | R2 | |
| 6 h | 67.09 | 0.6291 | 26.16 | 0.9079 | 10.24 | 0.9224 |
| 24 h | 23.85 | 0.9329 | 16.20 | 0.8934 | 4.73 | 0.8368 |
| 48 h | 14.23 | 0.9659 | 11.68 | 0.7594 | 3.19 | 0.8499 |
| Ligand | Docking Score | Binding Affinity (XP-Score) | Glide Energy | Glide-Ligand Efficacy | XP-Hbond | Hydrogen Bonding | Hydrogen Bond Distance (Å) |
|---|---|---|---|---|---|---|---|
| MQ-4EY5 | −8.111 | −8.114 | −7.018 | −0.312 | 0.000 | Tyr-337 O–ligand H | 2.1 |
| Gly-120 O–ligand H | 2.4 | ||||||
| MQ-6I0C | −9.755 | −8.759 | −36.623 | −0.337 | −0.700 | Glu-197 O–ligand H | 2.2 |
| His-438 O–ligand H | 2.1 |
| Interaction Type | Residues | |
|---|---|---|
| MQ-6I0C | Polar | Gly-120, Tyr-337 |
| Non-polar | Gln-71, Tyr-72, Val-73, Asp-74, Gly-82, Thr-83, Trp-86, Asn-87, Pro-88, Tyr-119, Gly-121, Gly-122, Tyr-124, Ser-125, Gly-126, Ala-127, Leu-130, Tyr-133, Gln-202, Ser-203, Ala-204, Phe-297, Phe-338, Tyr-341, Trp-439, Gly-440, His-447, Tyr-449, Ile-451 | |
| MQ-4EY5 | Polar | Glu-197, His-438 |
| Non-polar | Asp-70, Gly-78, Ser-79, Trp-82, Trp-112, Tyr-114, Gly-115, Gly-116, Gly-117, Thr-120, Gly-121, Thr-122, Tyr-128, Ser-198, Ala-199, Pro-285, Leu-286, Ala-328, Phe-329, Tyr-332, Trp-430, Met-434, Met-437, Gly-439, Tyr-440, Ile-442 |
| Ligand | Prime Energy | Ligand Efficiency | Ligand Efficiency Ln | ΔG Bind | ΔG Bind Coulomb | ΔG Bind Solv.GB | ΔG Bind (NS) | ΔG Bind (NS) Coulomb | ΔG Bind (NS) Solv. GB |
|---|---|---|---|---|---|---|---|---|---|
| MQ-6I0C | −22,715.70 | −0.337 | −2.056 | −49.43 | −18.42 | 32.93 | −57.93 | −18.67 | 32.99 |
| MQ-4EY5 | −22,881.89 | −0.312 | −1.905 | 1.37 | −48.36 | 74.21 | −25.63 | −47.45 | 74.00 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El Sharazly, B.M.; Ahmed, A.; Elsheikha, H.M.; Carter, W.G. An In Silico and In Vitro Assessment of the Neurotoxicity of Mefloquine. Biomedicines 2024, 12, 505. https://doi.org/10.3390/biomedicines12030505
El Sharazly BM, Ahmed A, Elsheikha HM, Carter WG. An In Silico and In Vitro Assessment of the Neurotoxicity of Mefloquine. Biomedicines. 2024; 12(3):505. https://doi.org/10.3390/biomedicines12030505
Chicago/Turabian StyleEl Sharazly, Basma M., Abrar Ahmed, Hany M. Elsheikha, and Wayne G. Carter. 2024. "An In Silico and In Vitro Assessment of the Neurotoxicity of Mefloquine" Biomedicines 12, no. 3: 505. https://doi.org/10.3390/biomedicines12030505
APA StyleEl Sharazly, B. M., Ahmed, A., Elsheikha, H. M., & Carter, W. G. (2024). An In Silico and In Vitro Assessment of the Neurotoxicity of Mefloquine. Biomedicines, 12(3), 505. https://doi.org/10.3390/biomedicines12030505