

RhoA/ROCK Pathway Is Upregulated in Experimental Autoimmune Myocarditis and Is Inhibited by Simvastatin at the Stage of Myosin Light Chain Phosphorylation
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Induction of Active EAM and Protocol of Experiment
2.3. Sample Collection
2.4. Immunohistochemical Studies
2.5. Western Blot
2.6. Statistical Analysis
3. Results
3.1. RhoA Expression in Heart Tissue Was Not Upregulated and Not Affected by Simvastatin
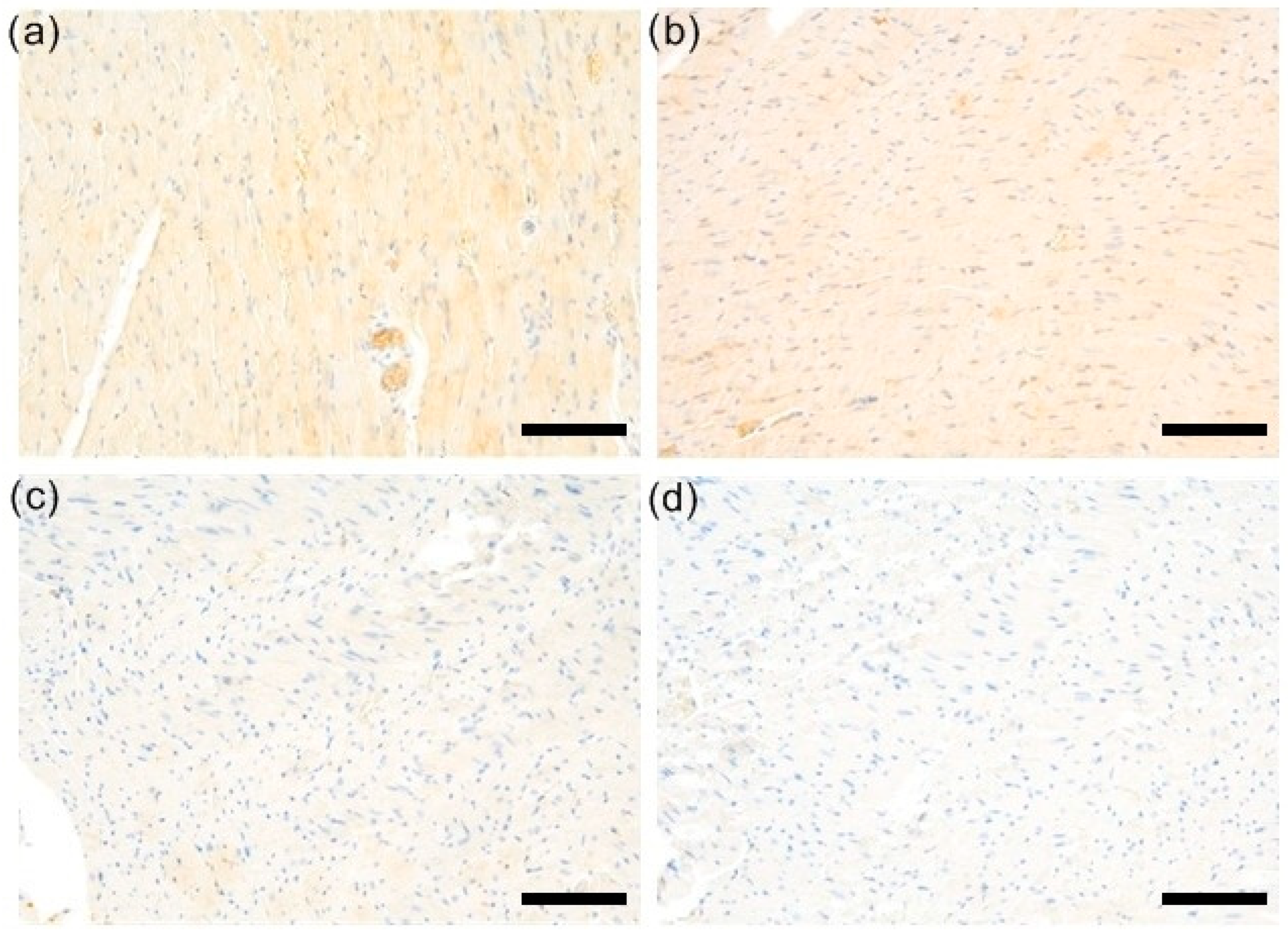
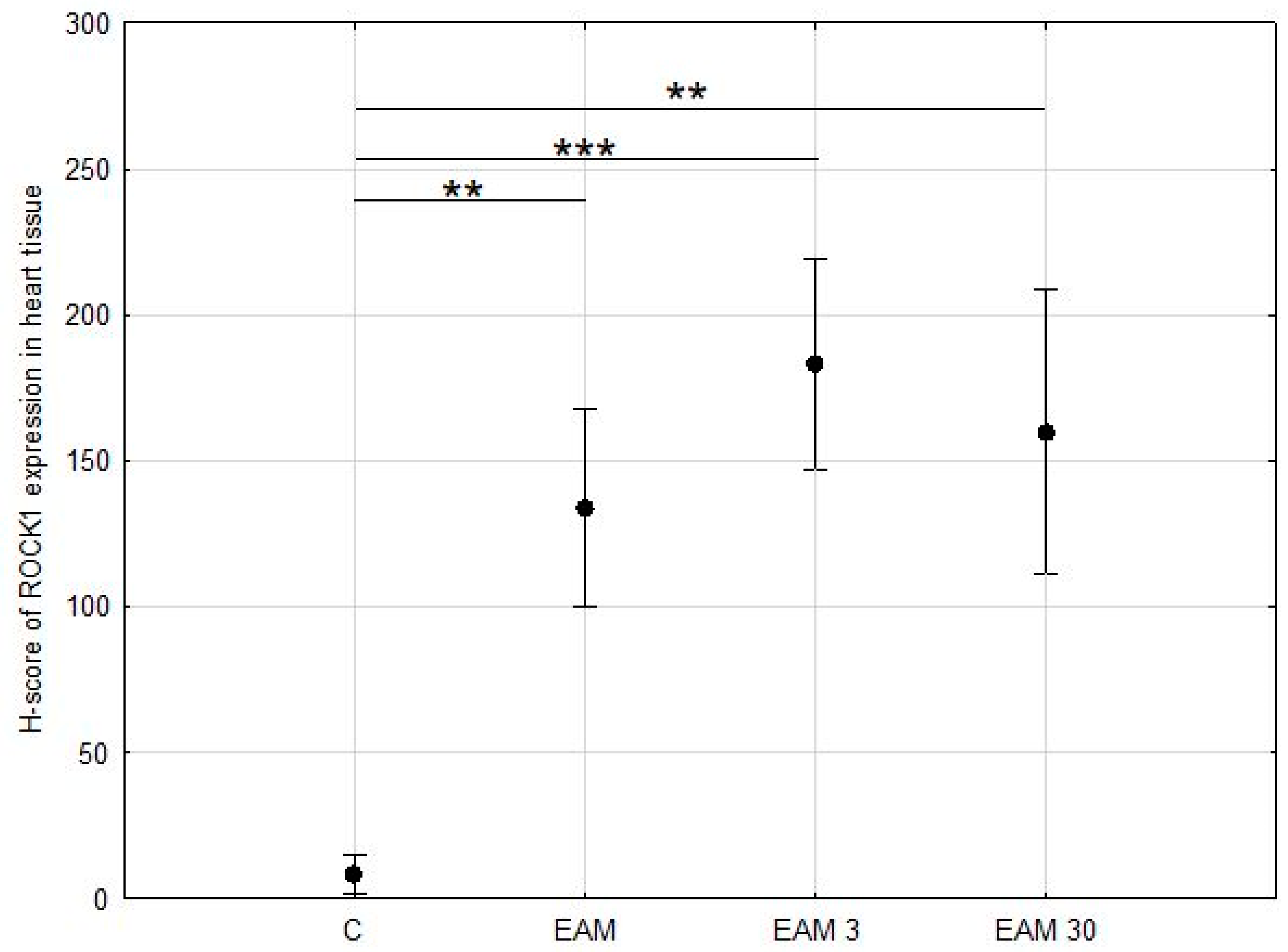
3.2. Expression of ROCK1 Was Upregulated EAM but Not Affected by Simvastatin
3.3. The Western Blotting Analysis for ROCK1 Was Not Reliable
3.4. Phosphorylated Myosin Light Chain Protein Content Was Upregulated in EAM and the Treatment with Simvastatin Prevented the EAM-Induced Increase in Phosphorylation of MLC
3.5. Myosin Light Chain Protein Content Was Not Upregulated in EAM
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Caforio, A.L.; Pankuweit, S.; Arbustini, E.; Basso, C.; Gimeno-Blanes, J.; Felix, S.B.; Fu, M.; Heliö, T.; Heymans, S.; Jahns, R.; et al. Current state of knowledge on aetiology, diagnosis, management, and therapy of myocarditis: A position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2013, 34, 26362648. [Google Scholar] [CrossRef]
- Lerner, A.; Jeremias, P.; Matthias, T. The World Incidence and Prevalence of Autoimmune Diseases is Increasing. Int. J. Celiac Dis. 2015, 3, 151–155. [Google Scholar] [CrossRef]
- Busteed, S.; Sparrow, P.; Molloy, C.; Molloy, M. Myocarditis as a prognostic indicator in systemic lupus erythematosus. Postgrad. Med. J. 2004, 80, 366–367. [Google Scholar] [CrossRef] [PubMed]
- Cooper, L.T., Jr.; Berry, G.J.; Shabetai, R. Idiopathic giant-cell myocarditis—Natural history and treatment. N. Engl. J. Med. 1997, 336, 1860–1866. [Google Scholar] [CrossRef]
- Bracamonte-Baran, W.; Čiháková, D. Cardiac Autoimmunity: Myocarditis. In The Immunology of Cardiovascular Homeostasis and Pathology; Sattler, S., Kennedy-Lydon, T., Eds.; Springer: Berlin/Heidelberg, Germany, 2017; pp. 187–221. [Google Scholar]
- Aggarwal, H.K.; Jain, D.; Kaverappa, V.; Jain, P.; Kumar, A.; Yadav, S. Idiopathic hypereosinophilic syndrome presenting as severe Loeffler’s endocarditis. Arq. Bras. Cardiol. 2013, 100, e43–e46. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Judson, M.A.; Donnino, R.; Gold, M.; Cooper, L.T., Jr.; Prystowsky, E.N.; Prystowsky, S. Cardiac sarcoidosis. Am. Heart J. 2009, 157, 9–21. [Google Scholar] [CrossRef]
- Biggs, R.; Patel, B.; Martinez, M.W.; McCambridge, M.; Kim, S. Cardiac sarcoidosis mimicking arrhythmogenic right ventricular dysplasia in a patient presenting with monomorphic ventricular tachycardia. HeartRhythm Case Rep. 2017, 3, 418–421. [Google Scholar] [CrossRef]
- Blauwet, L.A.; Cooper, L.T. Idiopathic giant cell myocarditis and cardiac sarcoidosis. Heart Fail. Rev. 2013, 18, 733–746. [Google Scholar] [CrossRef]
- Baughman, R.P.; Teirstein, A.S.; Judson, M.A.; Rossman, M.D.; Yeager, H., Jr.; Bresnitz, E.A.; DePalo, L.; Hunninghake, G.; Iannuzzi, M.C.; Johns, C.J. Clinical characteristics of patients in a case control study of sarcoidosis. Am. J. Respir. Crit. Care Med. 2001, 164, 1885–1889. [Google Scholar] [CrossRef]
- Manresa-Arraut, A.; Johansen, F.F.; Brakebusch, C.; Issazadeh-Navikas, S.; Hasseldam, H. RhoA Drives T-Cell Activtion and Encephalitogenic Potential in an Animal Model of Multiple Sclerosis. Front. Immunol. 2018, 9, 1235. [Google Scholar] [CrossRef]
- Sun, X.; Minohara, M.; Kikuchi, H.; Ishizu, T.; Tanaka, M.; Piao, H.; Osoegawa, M.; Ohyagi, Y.; Shimokawa, H.; Kira, J.-I. The selective Rho-kinase inhibitor Fasudil is protective and therapeutic in experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2006, 180, 126–134. [Google Scholar] [CrossRef]
- Stirzaker, R.A.; Biswas, P.S.; Gupta, S.; Song, L.; Bhagat, G.; Pernis, A.B. Administration of fasudil, a ROCK inhibitor, at-tenuates disease in lupus-prone NZB/W F1 female mice. Lupus 2012, 21, 656–661. [Google Scholar] [CrossRef]
- Dai, Y.; Luo, W.; Chang, J. Rho kinase signaling and cardiac physiology. Curr. Opin. Physiol. 2018, 1, 14–20. [Google Scholar] [CrossRef]
- Dai, K.; Wang, Y.; Tai, S.; Ni, H.; Lian, H.; Yu, Y.; Liao, W.; Zheng, C.; Chen, Q.; Kuver, A.; et al. Fasudil exerts a car-dio-protective effect on mice with coxsackievirus B3-induced acute viral myocarditis. Cardiovasc. Ther. 2018, 36, e1247. [Google Scholar] [CrossRef]
- Tkacz, K.; Rolski, F.; Czepiel, M.; Działo, E.; Siedlar, M.; Eriksson, U.; Kania, G.; Błyszczuk, P. Haploinsufficient Rock1+/− and Rock2+/− Mice Are Not Protected from Cardiac Inflammation and Postinflammatory Fibrosis in Experimental Autoimmune Myocarditis. Cells 2020, 9, 700. [Google Scholar] [CrossRef]
- Skrzypiec-Spring, M.; Sapa-Wojciechowska, A.; Haczkiewicz-Leśniak, K.; Piasecki, T.; Kwiatkowska, J.; Podhorska-Okołów, M.; Szeląg, A. HMG-CoA Reductase Inhibitor, Simvastatin Is Effective in Decreasing Degree of Myocarditis by Inhibiting Metalloproteinases Activation. Biomolecules 2021, 11, 1415. [Google Scholar] [CrossRef]
- Skrzypiec-Spring, M.; Sapa-Wojciechowska, A.; Rak-Pasikowska, A.; Kaczorowski, M.; Bil Lula, I.; Hałoń, A.; Szeląg, A. The Protective Effect of Simvastatin on the Systolic Function of the Heart in the Model of Acute Ischemia and Reperfusion Is Due to Inhibition of the RhoA Pathway and Independent of Reduction of MMP-2 Activity. Biomolecules 2022, 12, 1291. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.L.; Sung, K.R.; Kwon, J.; Shin, J.A. Statins Suppress TGF-beta 2-Mediated MMP-2 and MMP-9 Expression and Acti-vation Through RhoA/ROCK Inhibition in Astrocytes of the Human Optic Nerve Head. Investig. Ophthalmol. Vis. Sci. 2020, 61, 29. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, U.; Frantz, S. Role of lymphocytes in myocardial injury, healing, and remodeling after myocardial infarction. Circ. Res. 2015, 16, 354–367. [Google Scholar] [CrossRef]
- Bruestle, K.; Hackner, K.; Kreye, G.; Heidecker, B. Autoimmunity in Acute Myocarditis: How Immunopathogenesis Steers New Directions for Diagnosis and Treatment. Curr. Cardiol. Rep. 2020, 20, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Aronoff, L.; Epelman, S.; Clemente-Casares, X. Isolation and Identification of Extravascular Immune Cells of the Heart. J. Vis. Exp. 2018, 23, 58114. [Google Scholar] [CrossRef]
- Kilian, L.S.; Frank, D.; Rangrez, A.Y. RhoA Signaling in Immune Cell Response and Cardiac Disease. Cells 2021, 10, 1681. [Google Scholar] [CrossRef] [PubMed]
- Bros, M.; Haas, K.; Moll, L.; Grabbe, S. RhoA as a Key Regulator of Innate and Adaptive Immunity. Cells 2019, 17, 733. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.L.; Brown, M.S. Regulation of the mevalonate pathway. Nature 1990, 343, 425–430. [Google Scholar] [CrossRef]
- Liao, J.K.; Laufs, U. Pleiotropic effects of statins. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 89–118. [Google Scholar] [CrossRef] [PubMed]
- Wassmann, S.; Laufs, U.; Bäumer, A.T.; Müller, K.; Ahlbory, K.; Linz, W.; Itter, G.; Rösen, R.; Böhm, M.; Nickenig, G. HMG-CoA reductase inhibitors improve endothelial dysfunction in normocholesterolemic hypertension via reduced production of reactive oxygen species. Hypertension 2001, 37, 1450–1457. [Google Scholar] [CrossRef] [PubMed]
- Takemoto, M.; Node, K.; Nakagami, H.; Liao, Y.; Grimm, M.; Takemoto, Y.; Kitakaze, M.; Liao, J.K. Statins as antioxidant therapy for preventing cardiac myocyte hypertrophy. J. Clin. Investig. 2001, 108, 1429–1437. [Google Scholar] [CrossRef]
- Van Aelst, L.; D’Souza-Schorey, C. Rho GTPases and signaling networks. Genes Dev. 1997, 11, 2295–2322. [Google Scholar] [CrossRef] [PubMed]
- Laufs, U.; Endres, M.; Stagliano, N.; Amin-Hanjani, S.; Chui, D.S.; Yang, S.X.; Simoncini, T.; Yamada, M.; Rabkin, E.; Allen, P.G.; et al. Neuroprotection mediated by changes in the endothelial actin cytoskeleton. J. Clin. Investig. 2000, 106, 15–24. [Google Scholar] [CrossRef]
- Laufs, U.; Endres, M.; Custodis, F.; Gertz, K.; Nickenig, G.; Liao, J.K.; Böhm, M. Suppression of endothelial nitric oxide production after withdrawal of statin treatment is mediated by negative feedback regulation of rho GTPase gene transcription. Circulation 2000, 102, 3104–3110. [Google Scholar] [CrossRef]
- Kwak, B.; Mulhaupt, F.; Myit, S.; Mach, F. Statins as a newly recognized type of immunomodulator. Nat. Med. 2000, 6, 1399–1402. [Google Scholar] [CrossRef] [PubMed]
- Mulhaupt, F.; Matter, C.M.; Kwak, B.R.; Pelli, G.; Veillard, N.R.; Burger, F.; Graber, P.; Lüscher, T.F.; Mach, F. Statins (HMG-CoA reductase inhibitors) reduce CD40 expression in human vascular cells. Cardiovasc. Res. 2003, 59, 755–766. [Google Scholar] [CrossRef]
- Weitz-Schmidt, G.; Welzenbach, K.; Brinkmann, V.; Kamata, T.; Kallen, J.; Bruns, C.; Cottens, S.; Takada, Y.; Hommel, U. Statins selectively inhibit leukocyte function antigen-1 by binding to a novel regulatory integrin site. Nat. Med. 2001, 7, 687–692. [Google Scholar] [CrossRef]
- Ma, Z.; Shen, Z.; Gong, Y.; Zhou, J.; Chen, X.; Lv, Q.; Wang, M.; Chen, J.; Yu, M.; Fu, G.; et al. Weighted gene co-expression network analysis identified underlying hub genes and mechanisms in the occurrence and development of viral myocarditis. Ann. Transl. Med. 2020, 8, 1348. [Google Scholar] [CrossRef] [PubMed]
- Dubey, P.K.; Patil, M.; Singh, S.; Dubey, S.; Ahuja, P.; Verma, S.K.; Krishnamurthy, P. Increased m6A-RNA methylation and FTO suppression is associated with myocardial inflammation and dysfunction during endotoxemia in mice. Mol. Cell. Biochem. 2022, 477, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Preau, S.; Delguste, F.; Yu, Y.; Remy-Jouet, I.; Richard, V.; Saulnier, F.; Boulanger, E.; Neviere, R. Endotoxemia Engages the RhoA Kinase Pathway to Impair Cardiac Function By Altering Cytoskeleton, Mitochondrial Fission, and Autophagy. Antioxid. Redox Signal. 2016, 24, 529–542. [Google Scholar] [CrossRef]
- Shimokawa, H.; Sunamura, S.; Satoh, K. RhoA/Rho-Kinase in the Cardiovascular System. Circ. Res. 2016, 118, 352–366. [Google Scholar] [CrossRef]
- Yang, Y.; Lu, N.; Zhou, J.; Chen, Z.N.; Zhu, P. Cyclophilin A up-regulates MMP-9 expression and adhesion of monocytes/macrophages via CD147 signalling pathway in rheumatoid arthritis. Rheumatology 2008, 47, 1299–1310. [Google Scholar] [CrossRef]
- Luo, D.; Chen, H.; Li, X.; Lu, P.; Long, M.; Peng, X.; Lin, S.; Tan, L.; Zhu, Y.; Ouyang, N.; et al. Activation of the ROCK1/MMP-9 pathway is associated with the invasion and poor prognosis in papillary thyroid carcinoma. Int. J. Oncol. 2017, 51, 1209–1218. [Google Scholar] [CrossRef]
- Cao, M.; Yuan, W.; Peng, M.; Mao, Z.; Zhao, Q.; Sun, X.; Yan, J. Role of CyPA in cardiac hypertrophy and remodeling. Biosci. Rep. 2019, 39, BSR20193190. [Google Scholar] [CrossRef]
- Björkhem-Bergman, L.; Lindh, J.D.; Bergman, P. What is the appropriate concentration of statin in the experiments on cells confirms pleiotropic effect? Br. J. Clin. Pharmacol. 2011, 72, 164–165. [Google Scholar] [CrossRef] [PubMed]
- Westwood, F.R.; Bigley, A.; Randall, K.; Marsden, A.M.; Scott, R.C. Statin-induced myonecrosis in rats: Distribution, development and fiber selectivity. Toxicol. Pathol. 2005, 33, 246–257. [Google Scholar] [CrossRef]
- Garip, S.; Bayari, S.H.; Severcan, M.; Abbas, S.; Lednev, I.K.; Severcan, F. Structural effects of simvastatin on rat liver tissue [revised]: Fourier transform infrared and Raman microscopic studies. J. Biomed. Opt. 2016, 21, 25008. [Google Scholar] [CrossRef]
- Root-Bernstein, R.; Fairweather, D. Unresolved issues in theories of autoimmune disease using myocarditis as a framework. J. Theor. Biol. 2015, 375, 101–123. [Google Scholar] [CrossRef] [PubMed]
- Afanasyeva, M.; Georgakopoulos, D.; Belardi, D.F.; Ramsundar, A.C.; Barin, J.G.; Kass, D.A.; Rose, N.R. Quantitative analysis of myocardial inflammation by flow cytometry in murine autoimmune myocarditis: Correlation with cardiac function. Am. J. Pathol. 2004, 164, 807–815. [Google Scholar] [CrossRef]
- Frisancho-Kiss, S.; Davis, S.E.; Nyland, J.F.; Frisancho, J.A.; Cihakova, D.; Rose, N.R.; Fairweather, D. Cutting edge: Cross-regulation by TLR4 and T cell Ig mucin-3 determines sex differences in inflammatory heart disease. J. Immunol. 2007, 178, 6710–6714. [Google Scholar] [CrossRef] [PubMed]
- Fairweather, D.; Coronado, M.J.; Garton, A.E.; Dziedzic, J.L.; Bucek, A.; Cooper, L.T., Jr.; Brandt, J.E.; Alikhan, F.S.; Wang, H.; Endres, C.J.; et al. Sex differences in translocator protein 18 kDa (TSPO) in the heart: Implications for imaging myocardial inflammation. J. Cardiovasc. Trans. Res. 2014, 7, 192–202. [Google Scholar] [CrossRef]
- Fairweather, D.; Kaya, Z.; Shellam, G.R.; Lawson, C.M.; Rose, N.R. From infection to autoimmunity. J. Autoimm. 2001, 16, 175–186. [Google Scholar] [CrossRef]
- Mascaro-Blanco, A.; Alvarez, K.; Yu, X.; Lindenfeld, J.; Olansky, L.; Lyons, T.; Duvall, D.; Heuser, J.S.; Gosmanova, A.; Rubenstein, C.J.; et al. Consequences of unlocking the cardiac myosin molecule in human myocarditis and cardiomyopathies. Autoimmunity 2008, 41, 442–453. [Google Scholar] [CrossRef]
- Ponzoni, M.; Coles, J.G.; Maynes, J.T. Rodent Models of Dilated Cardiomyopathy and Heart Failure for Translational Investigations and Therapeutic Discovery. Int. J. Mol. Sci. 2023, 24, 3162. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skrzypiec-Spring, M.; Kaczorowski, M.; Rak-Pasikowska, A.; Sapa-Wojciechowska, A.; Kujawa, K.; Żuryń, A.; Bil-Lula, I.; Hałoń, A.; Szeląg, A. RhoA/ROCK Pathway Is Upregulated in Experimental Autoimmune Myocarditis and Is Inhibited by Simvastatin at the Stage of Myosin Light Chain Phosphorylation. Biomedicines 2024, 12, 596. https://doi.org/10.3390/biomedicines12030596
Skrzypiec-Spring M, Kaczorowski M, Rak-Pasikowska A, Sapa-Wojciechowska A, Kujawa K, Żuryń A, Bil-Lula I, Hałoń A, Szeląg A. RhoA/ROCK Pathway Is Upregulated in Experimental Autoimmune Myocarditis and Is Inhibited by Simvastatin at the Stage of Myosin Light Chain Phosphorylation. Biomedicines. 2024; 12(3):596. https://doi.org/10.3390/biomedicines12030596
Chicago/Turabian StyleSkrzypiec-Spring, Monika, Maciej Kaczorowski, Alina Rak-Pasikowska, Agnieszka Sapa-Wojciechowska, Krzysztof Kujawa, Agnieszka Żuryń, Iwona Bil-Lula, Agnieszka Hałoń, and Adam Szeląg. 2024. "RhoA/ROCK Pathway Is Upregulated in Experimental Autoimmune Myocarditis and Is Inhibited by Simvastatin at the Stage of Myosin Light Chain Phosphorylation" Biomedicines 12, no. 3: 596. https://doi.org/10.3390/biomedicines12030596