

Immunological Mechanisms behind Anti-PD-1/PD-L1 Immune Checkpoint Blockade: Intratumoral Reinvigoration or Systemic Induction?

{kind=link}
{kind=link}
Abstract
:1. Introduction
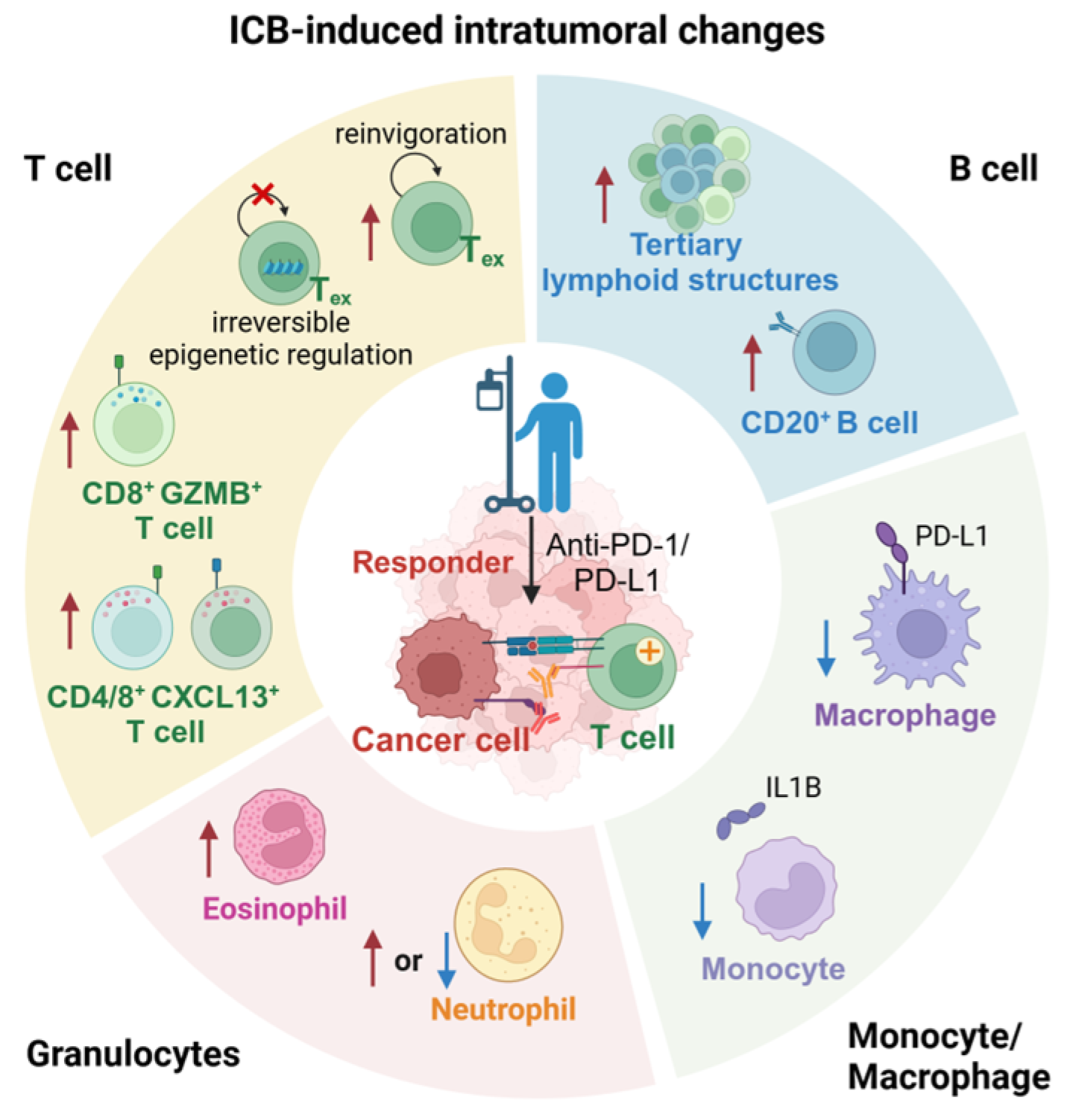
2. ICB-Induced Intratumoral Changes
2.1. Inactivation and Depletion of Regulatory T Cells
2.2. ICB-Induced Reinvigoration of Exhausted T Cells
2.3. Terminally Exhausted T Cells Due to Irreversible Epigenetic Regulation
2.4. ICB-Induced CD8+GZMB+ T Cells Expansion
2.5. ICB-Induced CXCL13+ T Cells Expansion
2.6. ICB-Induced CD20+ B Cells Expansion
2.7. Abundant Tertiary Lymphoid Structures in ICB Responding Patients
2.8. ICB-Induced Tumor-Associated Macrophages Reprograming
2.9. ICB-Induced Neutrophils Expansion in Non-Responders
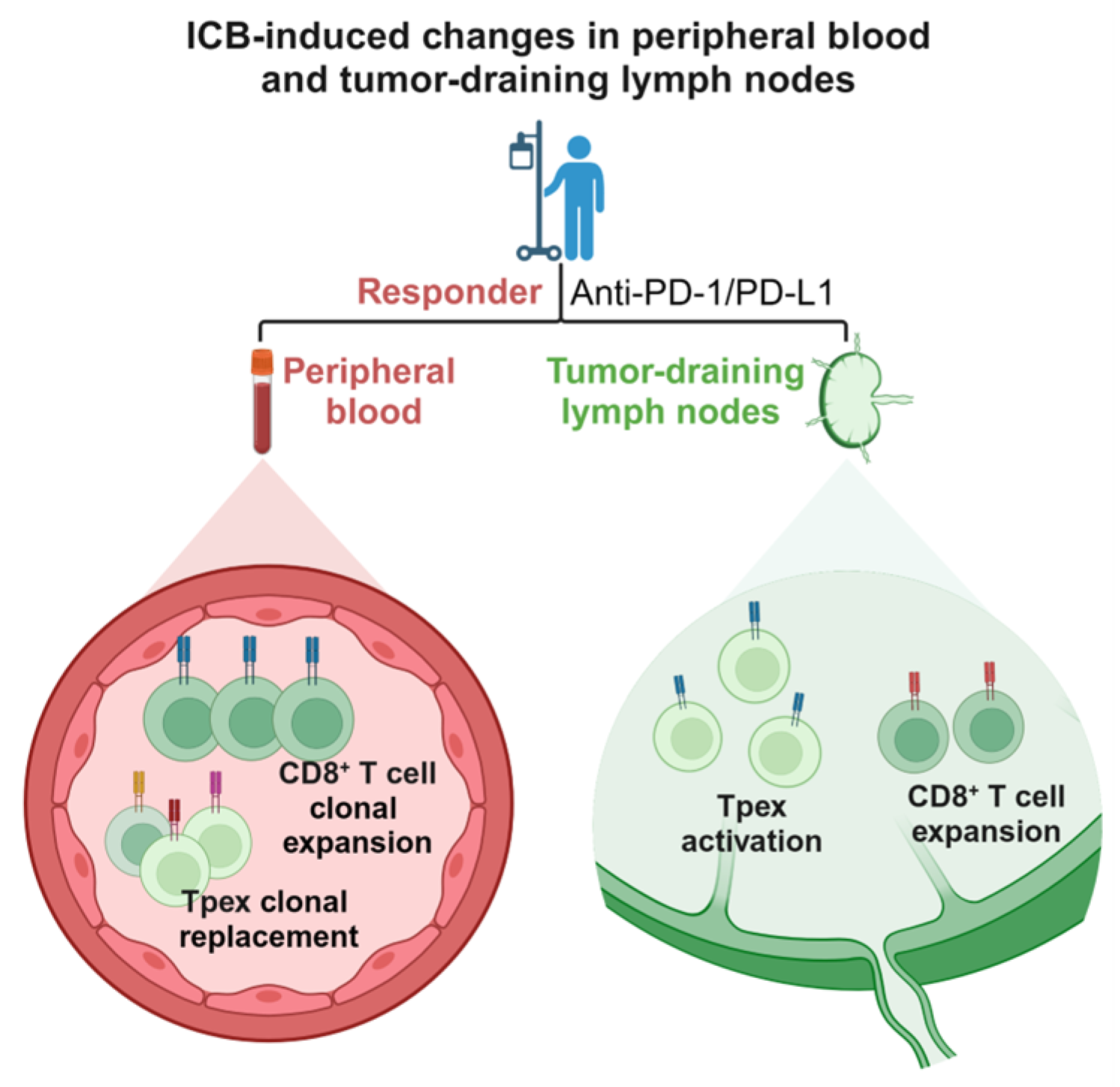
3. ICB-Induced Changes in Peripheral Blood T Cells
3.1. ICB-Induced Clonal Replacement of T Cells in Peripheral Blood
3.2. ICB-Induced Clonal Expansion of T Cells in Peripheral Blood
4. ICB-Induced Changes in Tumor-Draining Lymph Nodes (TDLNs)
4.1. Cancer-Immunity Cycle
4.2. ICB-Induced Tpex Activation in TDLNs
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of Antitumor Immunity by CTLA-4 Blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Sun, Q.; Zhang, X. PD-1 and Its Ligands Are Important Immune Checkpoints in Cancer. Oncotarget 2016, 8, 2171–2186. [Google Scholar] [CrossRef] [PubMed]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the PD-1 Immunoinhibitory Receptor by a Novel B7 Family Member Leads to Negative Regulation of Lymphocyte Activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; Long, G.V.; Robert, C.; Brady, B.; Dutriaux, C.; Di Giacomo, A.M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; et al. Survival Outcomes in Patients With Previously Untreated BRAF Wild-Type Advanced Melanoma Treated With Nivolumab Therapy: Three-Year Follow-up of a Randomized Phase 3 Trial. JAMA Oncol. 2019, 5, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.M.; Hwu, W.-J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and Activity of Anti–PD-L1 Antibody in Patients with Advanced Cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, Activity, and Immune Correlates of Anti–PD-1 Antibody in Cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Balar, A.V.; Kamat, A.M.; Kulkarni, G.S.; Uchio, E.M.; Boormans, J.L.; Bajorin, D.F.; Roumiguié, M.; Singer, E.A.; Krieger, L.E.M.; Grivas, P.; et al. Pembrolizumab (Pembro) for the Treatment of Patients with Bacillus Calmette-Guérin (BCG) Unresponsive, High-Risk (HR) Non–Muscle-Invasive Bladder Cancer (NMIBC): Over Two Years Follow-up of KEYNOTE-057. J. Clin. Oncol. 2020, 38, 5041. [Google Scholar] [CrossRef]
- Morad, G.; Helmink, B.A.; Sharma, P.; Wargo, J.A. Hallmarks of Response, Resistance, and Toxicity to Immune Checkpoint Blockade. Cell 2021, 184, 5309–5337. [Google Scholar] [CrossRef]
- Rodriguez Abreu, D.; Garassino, M.C.; Esteban, E.; Speranza, G.; Felip, E.; Domine, M.; Hochmair, M.J.; Powell, S.; Cheng, S.Y.-S.; Bischoff, H.G.; et al. KEYNOTE-189 Study of Pembrolizumab (Pembro) plus Pemetrexed (Pem) and Platinum vs Placebo plus Pem and Platinum for Untreated, Metastatic, Nonsquamous NSCLC: Does Choice of Platinum Affect Outcomes? Ann. Oncol. 2018, 29, ix164. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef]
- Hamid, O.; Robert, C.; Daud, A.; Hodi, F.S.; Hwu, W.J.; Kefford, R.; Wolchok, J.D.; Hersey, P.; Joseph, R.; Weber, J.S.; et al. Five-Year Survival Outcomes for Patients with Advanced Melanoma Treated with Pembrolizumab in KEYNOTE-001. Ann. Oncol. 2019, 30, 582–588. [Google Scholar] [CrossRef]
- Chen, R.; Zinzani, P.L.; Fanale, M.A.; Armand, P.; Johnson, N.A.; Brice, P.; Radford, J.; Ribrag, V.; Molin, D.; Vassilakopoulos, T.P.; et al. Phase II Study of the Efficacy and Safety of Pembrolizumab for Relapsed/Refractory Classic Hodgkin Lymphoma. J. Clin. Oncol. 2017, 35, 2125–2132. [Google Scholar] [CrossRef]
- Fradet, Y.; Bellmunt, J.; Vaughn, D.J.; Lee, J.L.; Fong, L.; Vogelzang, N.J.; Climent, M.A.; Petrylak, D.P.; Choueiri, T.K.; Necchi, A.; et al. Randomized Phase III KEYNOTE-045 Trial of Pembrolizumab versus Paclitaxel, Docetaxel, or Vinflunine in Recurrent Advanced Urothelial Cancer: Results of >2 Years of Follow-Up. Ann. Oncol. 2019, 30, 970–976. [Google Scholar] [CrossRef]
- Walk, E.E.; Yohe, S.L.; Beckman, A.; Schade, A.; Zutter, M.M.; Pfeifer, J.; Berry, A.B.; on behalf of the College of American Pathologists Personalized Health Care Committee. The Cancer Immunotherapy Biomarker Testing Landscape. Arch. Pathol. Lab. Med. 2019, 144, 706–724. [Google Scholar] [CrossRef]
- Jardim, D.L.; Goodman, A.; de Melo Gagliato, D.; Kurzrock, R. The Challenges of Tumor Mutational Burden as an Immunotherapy Biomarker. Cancer Cell 2021, 39, 154–173. [Google Scholar] [CrossRef]
- Schoenfeld, A.J.; Hellmann, M.D. Acquired Resistance to Immune Checkpoint Inhibitors. Cancer Cell 2020, 37, 443–455. [Google Scholar] [CrossRef]
- Ribas, A.; Hamid, O.; Daud, A.; Hodi, F.S.; Wolchok, J.D.; Kefford, R.; Joshua, A.M.; Patnaik, A.; Hwu, W.-J.; Weber, J.S.; et al. Association of Pembrolizumab With Tumor Response and Survival Among Patients With Advanced Melanoma. JAMA 2016, 315, 1600–1609. [Google Scholar] [CrossRef]
- Rosner, S.; Reuss, J.E.; Zahurak, M.; Zhang, J.; Zeng, Z.; Taube, J.; Anagnostou, V.; Smith, K.N.; Riemer, J.; Illei, P.B.; et al. Five-Year Clinical Outcomes after Neoadjuvant Nivolumab in Resectable Non–Small Cell Lung Cancer. Clin. Cancer Res. 2023, 29, 705–710. [Google Scholar] [CrossRef]
- Prokhnevska, N.; Cardenas, M.A.; Valanparambil, R.M.; Sobierajska, E.; Barwick, B.G.; Jansen, C.; Reyes Moon, A.; Gregorova, P.; delBalzo, L.; Greenwald, R.; et al. CD8+ T Cell Activation in Cancer Comprises an Initial Activation Phase in Lymph Nodes Followed by Effector Differentiation within the Tumor. Immunity 2023, 56, 107–124.e5. [Google Scholar] [CrossRef]
- Sharma, P.; Goswami, S.; Raychaudhuri, D.; Siddiqui, B.A.; Singh, P.; Nagarajan, A.; Liu, J.; Subudhi, S.K.; Poon, C.; Gant, K.L.; et al. Immune Checkpoint Therapy—Current Perspectives and Future Directions. Cell 2023, 186, 1652–1669. [Google Scholar] [CrossRef]
- Anderson, N.M.; Simon, M.C. The Tumor Microenvironment. Curr. Biol. 2020, 30, R921–R925. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Vesely, M.D.; Zhang, T.; Chen, L. Resistance Mechanisms to Anti-PD Cancer Immunotherapy. Annu. Rev. Immunol. 2022, 40, 45–74. [Google Scholar] [CrossRef] [PubMed]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the Tumor Immune Microenvironment (TIME) for Effective Therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Carthon, B.C.; Wolchok, J.D.; Yuan, J.; Kamat, A.; Ng Tang, D.S.; Sun, J.; Ku, G.; Troncoso, P.; Logothetis, C.J.; Allison, J.P.; et al. Preoperative CTLA-4 Blockade: Tolerability and Immune Monitoring in the Setting of a Presurgical Clinical Trial. Clin. Cancer Res. 2010, 16, 2861–2871. [Google Scholar] [CrossRef] [PubMed]
- Carlino, M.S.; Larkin, J.; Long, G.V. Immune Checkpoint Inhibitors in Melanoma. The Lancet 2021, 398, 1002–1014. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.C.; Levine, J.H.; Cogdill, A.P.; Zhao, Y.; Anang, N.-A.A.S.; Andrews, M.C.; Sharma, P.; Wang, J.; Wargo, J.A.; Pe’er, D.; et al. Distinct Cellular Mechanisms Underlie Anti-CTLA-4 and Anti-PD-1 Checkpoint Blockade. Cell 2017, 170, 1120–1133.e17. [Google Scholar] [CrossRef] [PubMed]
- Tay, C.; Tanaka, A.; Sakaguchi, S. Tumor-Infiltrating Regulatory T Cells as Targets of Cancer Immunotherapy. Cancer Cell 2023, 41, 450–465. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.C.; Zappasodi, R. A Decade of Checkpoint Blockade Immunotherapy in Melanoma: Understanding the Molecular Basis for Immune Sensitivity and Resistance. Nat. Immunol. 2022, 23, 660–670. [Google Scholar] [CrossRef]
- Tang, F.; Du, X.; Liu, M.; Zheng, P.; Liu, Y. Anti-CTLA-4 Antibodies in Cancer Immunotherapy: Selective Depletion of Intratumoral Regulatory T Cells or Checkpoint Blockade? Cell Biosci. 2018, 8, 30. [Google Scholar] [CrossRef]
- Hurst, J.H. Cancer Immunotherapy Innovator James Allison Receives the 2015 Lasker~DeBakey Clinical Medical Research Award. J. Clin. Investig. 2015, 125, 3732–3736. [Google Scholar] [CrossRef]
- Liu, Y.; Zheng, P. Preserving the CTLA-4 Checkpoint for Safer and More Effective Cancer Immunotherapy. Trends Pharmacol. Sci. 2020, 41, 4–12. [Google Scholar] [CrossRef]
- Hashimoto, M.; Kamphorst, A.O.; Im, S.J.; Kissick, H.T.; Pillai, R.N.; Ramalingam, S.S.; Araki, K.; Ahmed, R. CD8 T Cell Exhaustion in Chronic Infection and Cancer: Opportunities for Interventions. Annu. Rev. Med. 2018, 69, 301–318. [Google Scholar] [CrossRef]
- Wang, X.Q.; Danenberg, E.; Huang, C.-S.; Egle, D.; Callari, M.; Bermejo, B.; Dugo, M.; Zamagni, C.; Thill, M.; Anton, A.; et al. Spatial Predictors of Immunotherapy Response in Triple-Negative Breast Cancer. Nature 2023, 621, 868–876. [Google Scholar] [CrossRef]
- Yost, K.E.; Satpathy, A.T.; Wells, D.K.; Qi, Y.; Wang, C.; Kageyama, R.; McNamara, K.L.; Granja, J.M.; Sarin, K.Y.; Brown, R.A.; et al. Clonal Replacement of Tumor-Specific T Cells Following PD-1 Blockade. Nat. Med. 2019, 25, 1251–1259. [Google Scholar] [CrossRef]
- Oliveira, G.; Egloff, A.M.; Afeyan, A.B.; Wolff, J.O.; Zeng, Z.; Chernock, R.D.; Zhou, L.; Messier, C.; Lizotte, P.; Pfaff, K.L.; et al. Preexisting Tumor-Resident T Cells with Cytotoxic Potential Associate with Response to Neoadjuvant Anti–PD-1 in Head and Neck Cancer. Sci. Immunol. 2023, 8, eadf4968. [Google Scholar] [CrossRef]
- Bassez, A.; Vos, H.; Van Dyck, L.; Floris, G.; Arijs, I.; Desmedt, C.; Boeckx, B.; Vanden Bempt, M.; Nevelsteen, I.; Lambein, K.; et al. A Single-Cell Map of Intratumoral Changes during Anti-PD1 Treatment of Patients with Breast Cancer. Nat. Med. 2021, 27, 820–832. [Google Scholar] [CrossRef]
- Sen, D.R.; Kaminski, J.; Barnitz, R.A.; Kurachi, M.; Gerdemann, U.; Yates, K.B.; Tsao, H.-W.; Godec, J.; LaFleur, M.W.; Brown, F.D.; et al. The Epigenetic Landscape of T Cell Exhaustion. Science 2016, 354, 1165–1169. [Google Scholar] [CrossRef] [PubMed]
- Franco, F.; Jaccard, A.; Romero, P.; Yu, Y.-R.; Ho, P.-C. Metabolic and Epigenetic Regulation of T-Cell Exhaustion. Nat. Metab. 2020, 2, 1001–1012. [Google Scholar] [CrossRef] [PubMed]
- Mellman, I.; Chen, D.S.; Powles, T.; Turley, S.J. The Cancer-Immunity Cycle: Indication, Genotype, and Immunotype. Immunity 2023, 56, 2188–2205. [Google Scholar] [CrossRef]
- Liu, B.; Zhang, Y.; Wang, D.; Hu, X.; Zhang, Z. Single-Cell Meta-Analyses Reveal Responses of Tumor-Reactive CXCL13+ T Cells to Immune-Checkpoint Blockade. Nat. Cancer 2022, 3, 1123–1136. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wu, C.; Hu, H.; Qin, G.; Wu, X.; Bai, F.; Zhang, J.; Cai, Y.; Huang, Y.; Wang, C.; et al. Remodeling of the Immune and Stromal Cell Compartment by PD-1 Blockade in Mismatch Repair-Deficient Colorectal Cancer. Cancer Cell 2023, 41, 1152–1169.e7. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.; Giladi, A.; Barboy, O.; Hamon, P.; Li, B.; Zada, M.; Gurevich-Shapiro, A.; Beccaria, C.G.; David, E.; Maier, B.B.; et al. The Interaction of CD4+ Helper T Cells with Dendritic Cells Shapes the Tumor Microenvironment and Immune Checkpoint Blockade Response. Nat. Cancer 2022, 3, 303–317. [Google Scholar] [CrossRef] [PubMed]
- Laumont, C.M.; Banville, A.C.; Gilardi, M.; Hollern, D.P.; Nelson, B.H. Tumour-Infiltrating B Cells: Immunological Mechanisms, Clinical Impact and Therapeutic Opportunities. Nat. Rev. Cancer 2022, 22, 414–430. [Google Scholar] [CrossRef] [PubMed]
- Griss, J.; Bauer, W.; Wagner, C.; Simon, M.; Chen, M.; Grabmeier-Pfistershammer, K.; Maurer-Granofszky, M.; Roka, F.; Penz, T.; Bock, C.; et al. B Cells Sustain Inflammation and Predict Response to Immune Checkpoint Blockade in Human Melanoma. Nat. Commun. 2019, 10, 4186. [Google Scholar] [CrossRef] [PubMed]
- Hardy, R.R.; Hayakawa, K. B Cell Development Pathways. Annu. Rev. Immunol. 2001, 19, 595–621. [Google Scholar] [CrossRef] [PubMed]
- Stewart, A.; Ng, J.C.-F.; Wallis, G.; Tsioligka, V.; Fraternali, F.; Dunn-Walters, D.K. Single-Cell Transcriptomic Analyses Define Distinct Peripheral B Cell Subsets and Discrete Development Pathways. Front. Immunol. 2021, 12, 602539. [Google Scholar] [CrossRef] [PubMed]
- Glass, D.R.; Tsai, A.G.; Oliveria, J.P.; Hartmann, F.J.; Kimmey, S.C.; Calderon, A.A.; Borges, L.; Glass, M.C.; Wagar, L.E.; Davis, M.M.; et al. An Integrated Multi-Omic Single-Cell Atlas of Human B Cell Identity. Immunity 2020, 53, 217–232.e5. [Google Scholar] [CrossRef]
- Holmes, A.B.; Corinaldesi, C.; Shen, Q.; Kumar, R.; Compagno, N.; Wang, Z.; Nitzan, M.; Grunstein, E.; Pasqualucci, L.; Dalla-Favera, R.; et al. Single-Cell Analysis of Germinal-Center B Cells Informs on Lymphoma Cell of Origin and Outcome. J. Exp. Med. 2020, 217, e20200483. [Google Scholar] [CrossRef]
- He, S.; Wang, L.-H.; Liu, Y.; Li, Y.-Q.; Chen, H.-T.; Xu, J.-H.; Peng, W.; Lin, G.-W.; Wei, P.-P.; Li, B.; et al. Single-Cell Transcriptome Profiling of an Adult Human Cell Atlas of 15 Major Organs. Genome Biol. 2020, 21, 294. [Google Scholar] [CrossRef] [PubMed]
- Fridman, W.H.; Petitprez, F.; Meylan, M.; Chen, T.W.-W.; Sun, C.-M.; Roumenina, L.T.; Sautès-Fridman, C. B Cells and Cancer: To B or Not to B? J. Exp. Med. 2021, 218, e20200851. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.; Kwong, D.L.-W.; Dai, W.; Wu, P.; Li, S.; Yan, Q.; Zhang, Y.; Zhang, B.; Fang, X.; Liu, L.; et al. Comprehensive Single-Cell Sequencing Reveals the Stromal Dynamics and Tumor-Specific Characteristics in the Microenvironment of Nasopharyngeal Carcinoma. Nat. Commun. 2021, 12, 1540. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhong, Y.; Zhuang, Z.; Xie, J.; Lu, Y.; Huang, C.; Sun, Y.; Wu, L.; Yin, J.; Yu, H.; et al. Multiregion Single-Cell Sequencing Reveals the Transcriptional Landscape of the Immune Microenvironment of Colorectal Cancer. Clin. Transl. Med. 2021, 11, e253. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Mandal, G.; Payne, K.K.; Anadon, C.M.; Gatenbee, C.D.; Chaurio, R.A.; Costich, T.L.; Moran, C.; Harro, C.M.; Rigolizzo, K.E.; et al. IgA Transcytosis and Antigen Recognition Govern Ovarian Cancer Immunity. Nature 2021, 591, 464–470. [Google Scholar] [CrossRef] [PubMed]
- Mazor, R.D.; Nathan, N.; Gilboa, A.; Stoler-Barak, L.; Moss, L.; Solomonov, I.; Hanuna, A.; Divinsky, Y.; Shmueli, M.D.; Hezroni, H.; et al. Tumor-Reactive Antibodies Evolve from Non-Binding and Autoreactive Precursors. Cell 2022, 185, 1208–1222.e21. [Google Scholar] [CrossRef] [PubMed]
- Ng, K.W.; Boumelha, J.; Enfield, K.S.S.; Almagro, J.; Cha, H.; Pich, O.; Karasaki, T.; Moore, D.A.; Salgado, R.; Sivakumar, M.; et al. Antibodies against Endogenous Retroviruses Promote Lung Cancer Immunotherapy. Nature 2023, 616, 563–573. [Google Scholar] [CrossRef] [PubMed]
- Fridman, W.H.; Meylan, M.; Petitprez, F.; Sun, C.-M.; Italiano, A.; Sautès-Fridman, C. B Cells and Tertiary Lymphoid Structures as Determinants of Tumour Immune Contexture and Clinical Outcome. Nat. Rev. Clin. Oncol. 2022, 19, 441–457. [Google Scholar] [CrossRef] [PubMed]
- Sautès-Fridman, C.; Petitprez, F.; Calderaro, J.; Fridman, W.H. Tertiary Lymphoid Structures in the Era of Cancer Immunotherapy. Nat. Rev. Cancer 2019, 19, 307–325. [Google Scholar] [CrossRef]
- Helmink, B.A.; Reddy, S.M.; Gao, J.; Zhang, S.; Basar, R.; Thakur, R.; Yizhak, K.; Sade-Feldman, M.; Blando, J.; Han, G.; et al. B Cells and Tertiary Lymphoid Structures Promote Immunotherapy Response. Nature 2020, 577, 549–555. [Google Scholar] [CrossRef]
- Vanhersecke, L.; Brunet, M.; Guégan, J.-P.; Rey, C.; Bougouin, A.; Cousin, S.; Le Moulec, S.; Besse, B.; Loriot, Y.; Larroquette, M.; et al. Mature Tertiary Lymphoid Structures Predict Immune Checkpoint Inhibitor Efficacy in Solid Tumors Independently of PD-L1 Expression. Nat. Cancer 2021, 2, 794–802. [Google Scholar] [CrossRef]
- Li, M.O.; Wolf, N.; Raulet, D.H.; Akkari, L.; Pittet, M.J.; Rodriguez, P.C.; Kaplan, R.N.; Munitz, A.; Zhang, Z.; Cheng, S.; et al. Innate Immune Cells in the Tumor Microenvironment. Cancer Cell 2021, 39, 725–729. [Google Scholar] [CrossRef]
- Engblom, C.; Pfirschke, C.; Pittet, M.J. The Role of Myeloid Cells in Cancer Therapies. Nat. Rev. Cancer 2016, 16, 447–462. [Google Scholar] [CrossRef]
- Colligan, S.H.; Amitrano, A.M.; Zollo, R.A.; Peresie, J.; Kramer, E.D.; Morreale, B.; Barbi, J.; Singh, P.K.; Yu, H.; Wang, J.; et al. Inhibiting the Biogenesis of Myeloid-Derived Suppressor Cells Enhances Immunotherapy Efficacy against Mammary Tumor Progression. J. Clin. Investig. 2022, 132, e158661. [Google Scholar] [CrossRef]
- Jaillon, S.; Ponzetta, A.; Di Mitri, D.; Santoni, A.; Bonecchi, R.; Mantovani, A. Neutrophil Diversity and Plasticity in Tumour Progression and Therapy. Nat. Rev. Cancer 2020, 20, 485–503. [Google Scholar] [CrossRef]
- Ren, X.; Zhang, L.; Zhang, Y.; Li, Z.; Siemers, N.; Zhang, Z. Insights Gained from Single-Cell Analysis of Immune Cells in the Tumor Microenvironment. Annu. Rev. Immunol. 2021, 39, 583–609. [Google Scholar] [CrossRef]
- McAllister, S.S.; Gifford, A.M.; Greiner, A.L.; Kelleher, S.P.; Saelzler, M.P.; Ince, T.A.; Reinhardt, F.; Harris, L.N.; Hylander, B.L.; Repasky, E.A.; et al. Systemic Endocrine Instigation of Indolent Tumor Growth Requires Osteopontin. Cell 2008, 133, 994–1005. [Google Scholar] [CrossRef]
- Blomberg, O.S.; Spagnuolo, L.; Garner, H.; Voorwerk, L.; Isaeva, O.I.; van Dyk, E.; Bakker, N.; Chalabi, M.; Klaver, C.; Duijst, M.; et al. IL-5-Producing CD4+ T Cells and Eosinophils Cooperate to Enhance Response to Immune Checkpoint Blockade in Breast Cancer. Cancer Cell 2023, 41, 106–123.e10. [Google Scholar] [CrossRef]
- Hirschhorn, D.; Budhu, S.; Kraehenbuehl, L.; Gigoux, M.; Schröder, D.; Chow, A.; Ricca, J.M.; Gasmi, B.; De Henau, O.; Mangarin, L.M.B.; et al. T Cell Immunotherapies Engage Neutrophils to Eliminate Tumor Antigen Escape Variants. Cell 2023, 186, 1432–1447.e17. [Google Scholar] [CrossRef]
- Sui, Q.; Zhang, X.; Chen, C.; Tang, J.; Yu, J.; Li, W.; Han, K.; Jiang, W.; Liao, L.; Kong, L.; et al. Inflammation Promotes Resistance to Immune Checkpoint Inhibitors in High Microsatellite Instability Colorectal Cancer. Nat. Commun. 2022, 13, 7316. [Google Scholar] [CrossRef]
- Sidiropoulos, D.N.; Ho, W.J.; Jaffee, E.M.; Kagohara, L.T.; Fertig, E.J. Systems Immunology Spanning Tumors, Lymph Nodes, and Periphery. Cell Rep. Methods 2023, 3, 100670. [Google Scholar] [CrossRef]
- Yi, M.; Jiao, D.; Xu, H.; Liu, Q.; Zhao, W.; Han, X.; Wu, K. Biomarkers for Predicting Efficacy of PD-1/PD-L1 Inhibitors. Mol. Cancer 2018, 17, 129. [Google Scholar] [CrossRef]
- Liu, B.; Hu, X.; Feng, K.; Gao, R.; Xue, Z.; Zhang, S.; Zhang, Y.; Corse, E.; Hu, Y.; Han, W.; et al. Temporal Single-Cell Tracing Reveals Clonal Revival and Expansion of Precursor Exhausted T Cells during Anti-PD-1 Therapy in Lung Cancer. Nat. Cancer 2022, 3, 108–121. [Google Scholar] [CrossRef]
- Fairfax, B.P.; Taylor, C.A.; Watson, R.A.; Nassiri, I.; Danielli, S.; Fang, H.; Mahé, E.A.; Cooper, R.; Woodcock, V.; Traill, Z.; et al. Peripheral CD8+ T Cell Characteristics Associated with Durable Responses to Immune Checkpoint Blockade in Patients with Metastatic Melanoma. Nat. Med. 2020, 26, 193–199. [Google Scholar] [CrossRef]
- Wu, T.D.; Madireddi, S.; de Almeida, P.E.; Banchereau, R.; Chen, Y.-J.J.; Chitre, A.S.; Chiang, E.Y.; Iftikhar, H.; O’Gorman, W.E.; Au-Yeung, A.; et al. Peripheral T Cell Expansion Predicts Tumour Infiltration and Clinical Response. Nature 2020, 579, 274–278. [Google Scholar] [CrossRef]
- Li, Y.-L.; Hung, W.-C. Reprogramming of Sentinel Lymph Node Microenvironment during Tumor Metastasis. J. Biomed. Sci. 2022, 29, 84. [Google Scholar] [CrossRef]
- du Bois, H.; Heim, T.A.; Lund, A.W. Tumor-Draining Lymph Nodes: At the Crossroads of Metastasis and Immunity. Sci. Immunol. 2021, 6, eabg3551. [Google Scholar] [CrossRef]
- Kallies, A.; Zehn, D.; Utzschneider, D.T. Precursor Exhausted T Cells: Key to Successful Immunotherapy? Nat. Rev. Immunol. 2020, 20, 128–136. [Google Scholar] [CrossRef]
- Huang, Q.; Wu, X.; Wang, Z.; Chen, X.; Wang, L.; Lu, Y.; Xiong, D.; Liu, Q.; Tian, Y.; Lin, H.; et al. The Primordial Differentiation of Tumor-Specific Memory CD8+ T Cells as Bona Fide Responders to PD-1/PD-L1 Blockade in Draining Lymph Nodes. Cell 2022, 185, 4049–4066.e25. [Google Scholar] [CrossRef]
- Rahim, M.K.; Okholm, T.L.H.; Jones, K.B.; McCarthy, E.E.; Liu, C.C.; Yee, J.L.; Tamaki, S.J.; Marquez, D.M.; Tenvooren, I.; Wai, K.; et al. Dynamic CD8+ T Cell Responses to Cancer Immunotherapy in Human Regional Lymph Nodes Are Disrupted in Metastatic Lymph Nodes. Cell 2023, 186, 1127–1143.e18. [Google Scholar] [CrossRef]
- Sugiura, D.; Maruhashi, T.; Okazaki, I.; Shimizu, K.; Maeda, T.K.; Takemoto, T.; Okazaki, T. Restriction of PD-1 Function by Cis-PD-L1/CD80 Interactions Is Required for Optimal T Cell Responses. Science 2019, 364, 558–566. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, Z.; Yu, J.; Chen, Z.; Chen, S.; Wang, L. Immunological Mechanisms behind Anti-PD-1/PD-L1 Immune Checkpoint Blockade: Intratumoral Reinvigoration or Systemic Induction? Biomedicines 2024, 12, 764. https://doi.org/10.3390/biomedicines12040764
Guo Z, Yu J, Chen Z, Chen S, Wang L. Immunological Mechanisms behind Anti-PD-1/PD-L1 Immune Checkpoint Blockade: Intratumoral Reinvigoration or Systemic Induction? Biomedicines. 2024; 12(4):764. https://doi.org/10.3390/biomedicines12040764
Chicago/Turabian StyleGuo, Zhikun, Jiangnan Yu, Zihan Chen, Shuxian Chen, and Lei Wang. 2024. "Immunological Mechanisms behind Anti-PD-1/PD-L1 Immune Checkpoint Blockade: Intratumoral Reinvigoration or Systemic Induction?" Biomedicines 12, no. 4: 764. https://doi.org/10.3390/biomedicines12040764
APA StyleGuo, Z., Yu, J., Chen, Z., Chen, S., & Wang, L. (2024). Immunological Mechanisms behind Anti-PD-1/PD-L1 Immune Checkpoint Blockade: Intratumoral Reinvigoration or Systemic Induction? Biomedicines, 12(4), 764. https://doi.org/10.3390/biomedicines12040764