
Impact of Obesity-Related Endoplasmic Reticulum Stress on Cancer and Associated Molecular Targets

Abstract
:1. Introduction
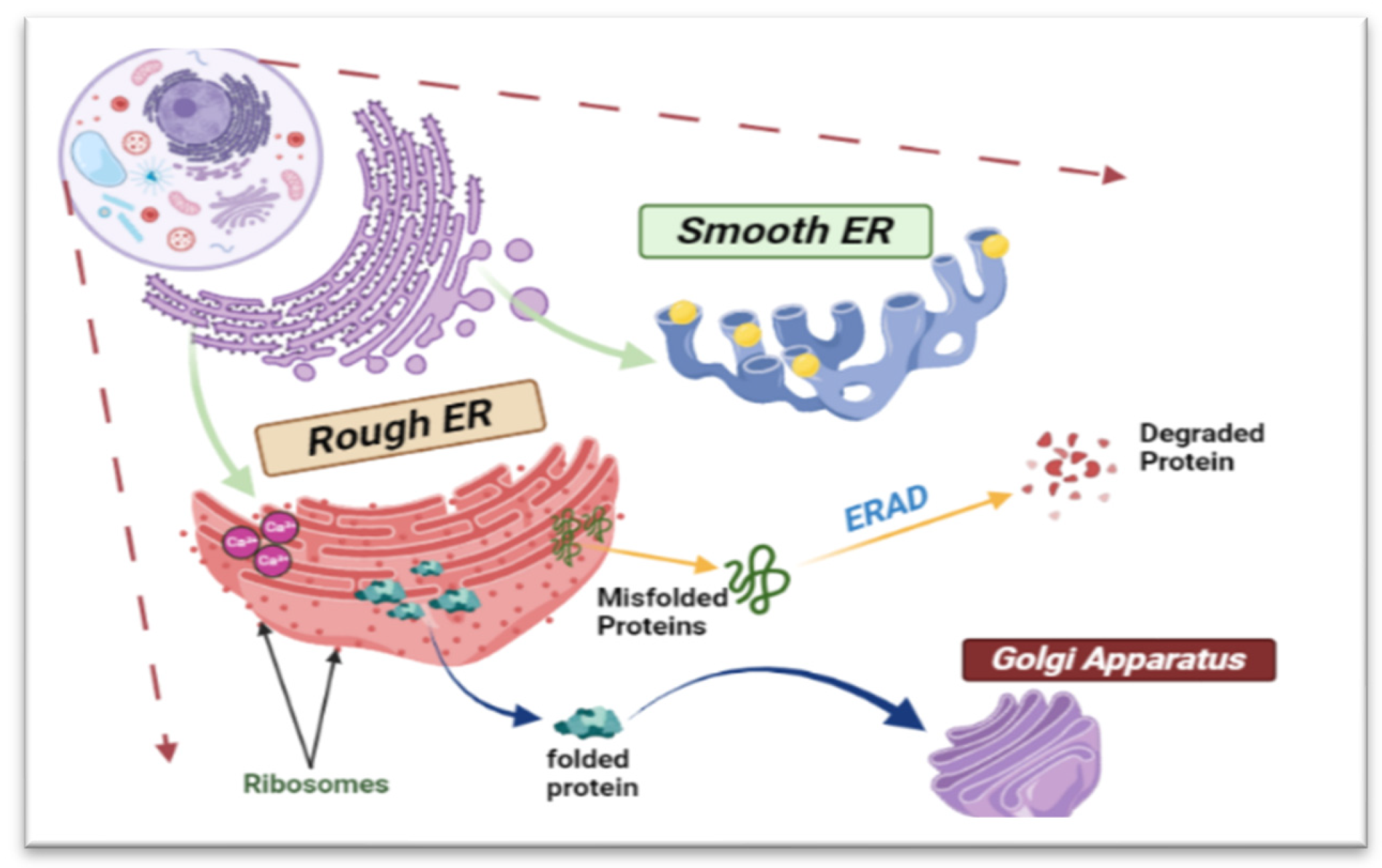
2. Endoplasmic Reticulum Structure and Functions
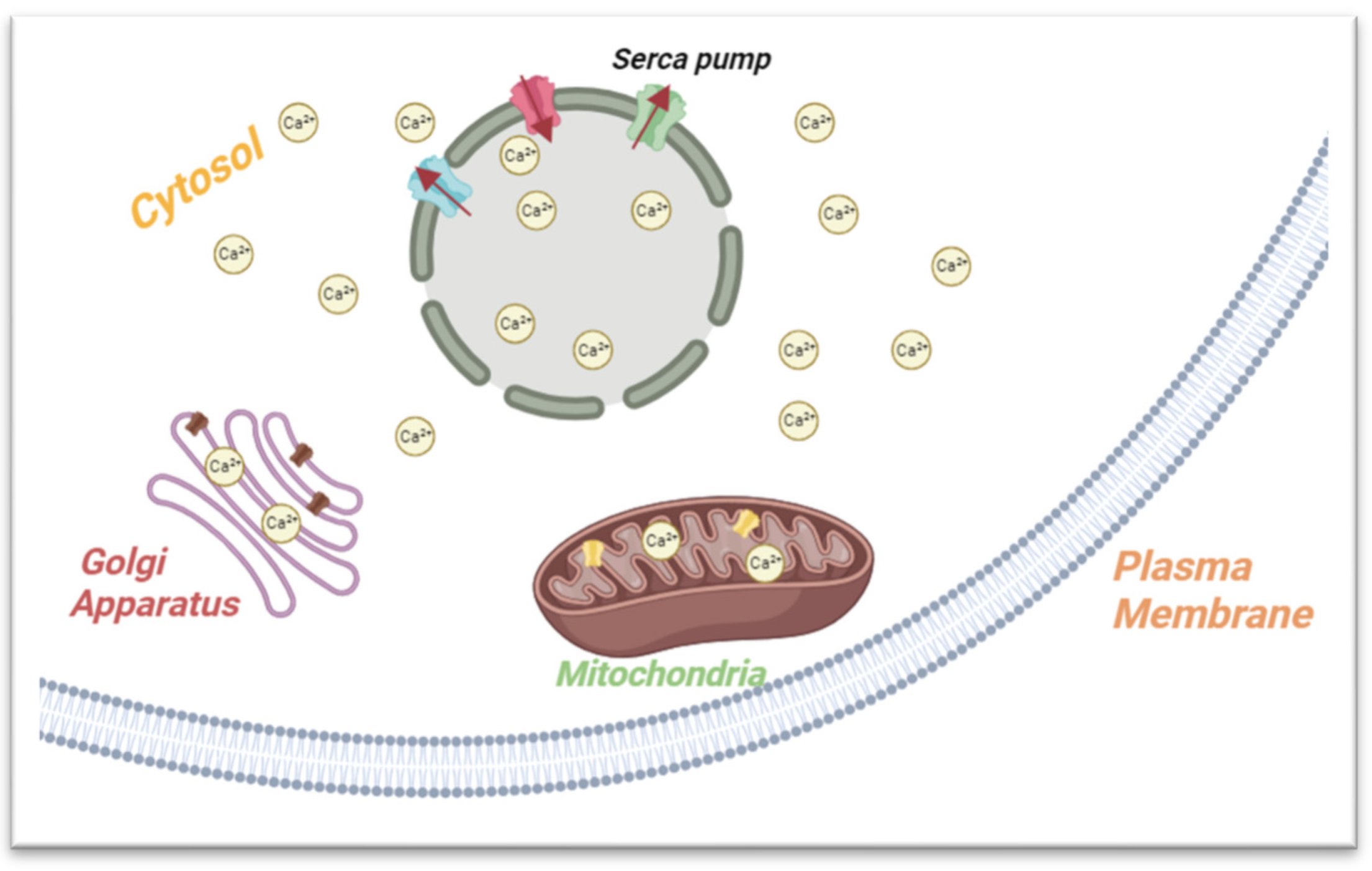
2.1. Calcium Storage and Metabolism
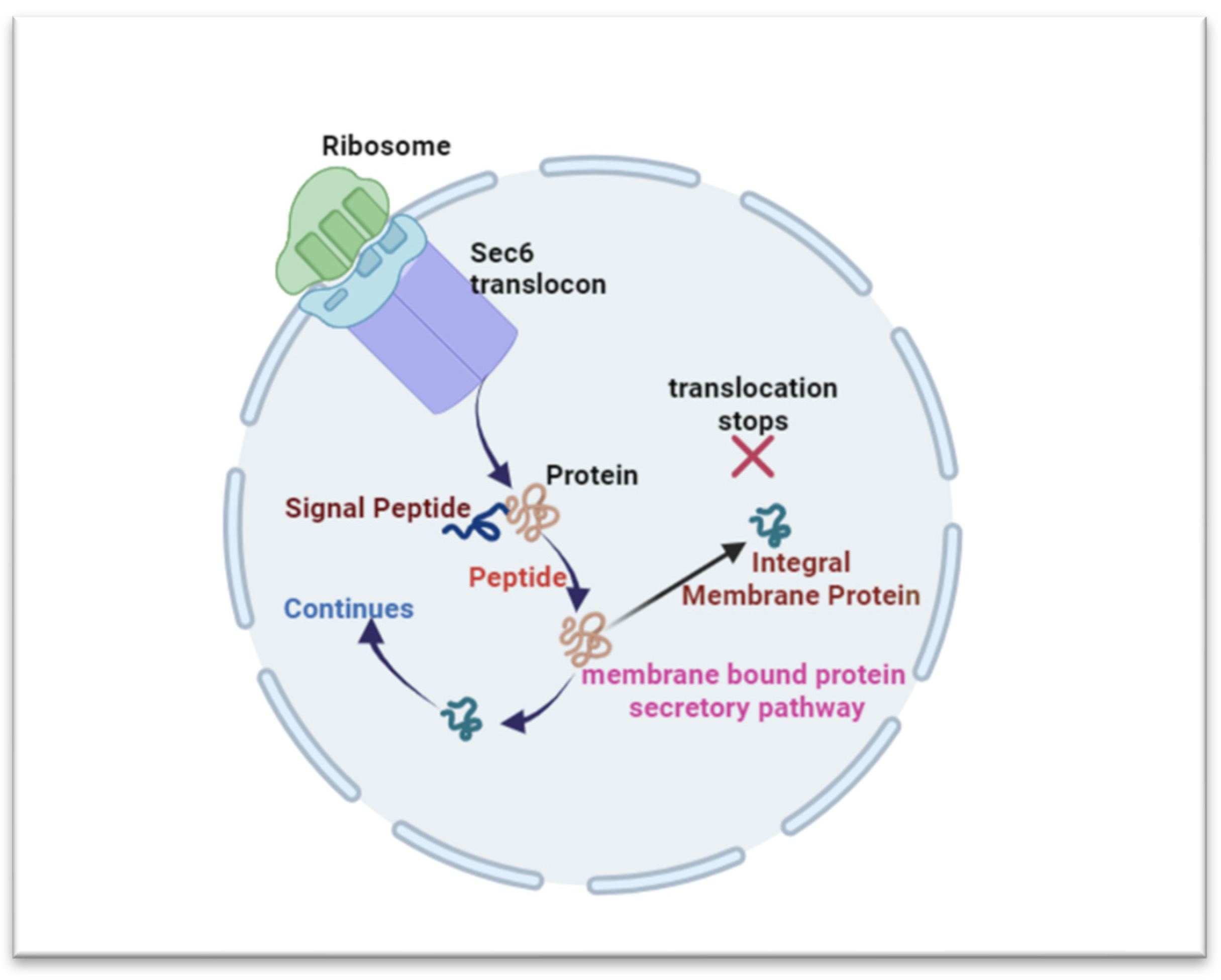
2.2. Protein Synthesis and Folding
2.3. Lipid Metabolism
2.4. Regulation of Lipid Metabolism
2.4.1. IRE1
2.4.2. PERK
2.4.3. ATF6
2.5. ER Homeostasis and ER Stress
2.6. ER Stress and Cancer
2.6.1. IRE1
2.6.2. PERK
2.6.3. ATF6
2.6.4. GRP78
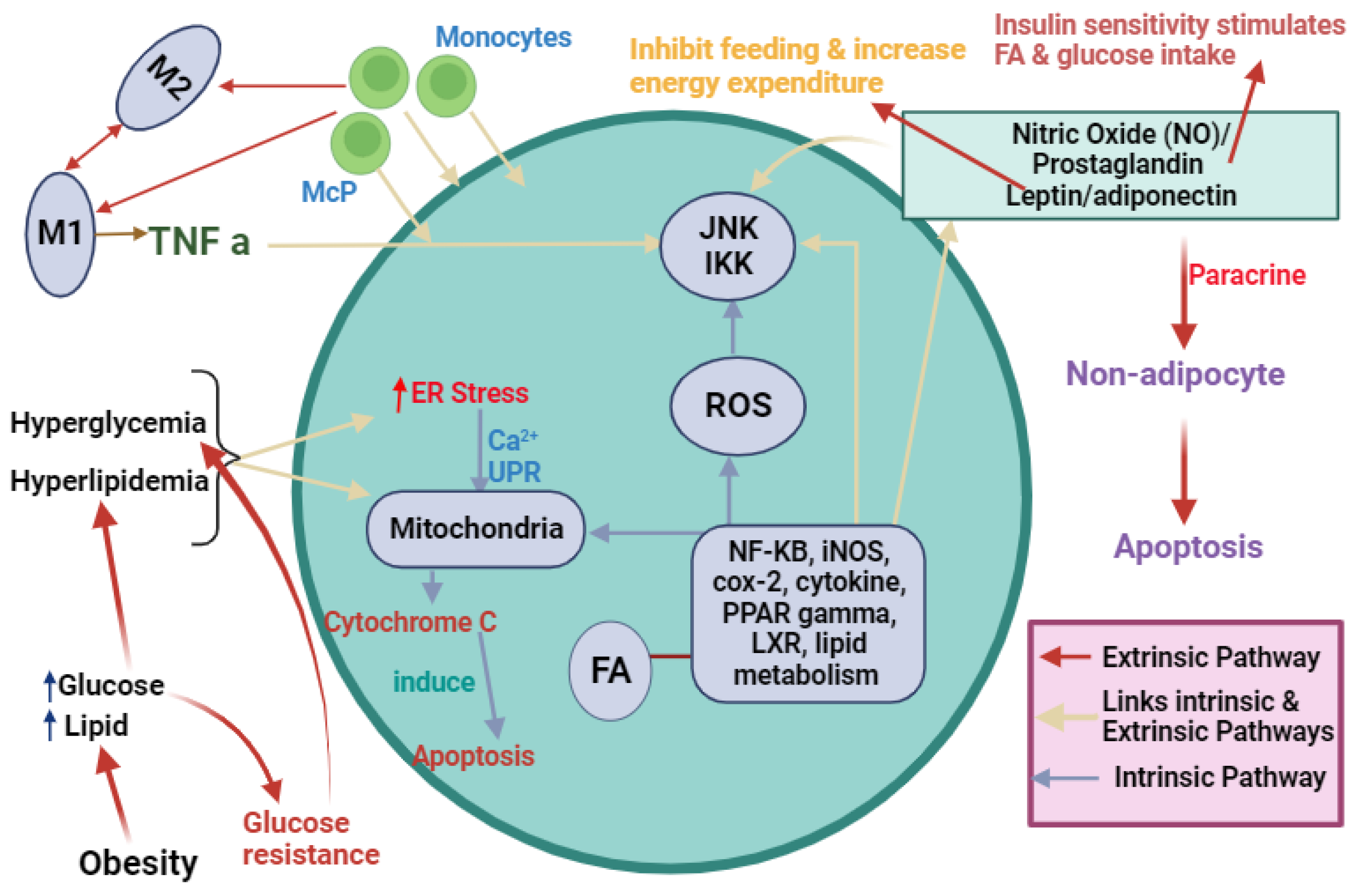
3. Targeting Obesity Induced ER Stress Mediator for Cancer Therapy
Author Contributions
Funding
Conflicts of Interest
References
- World Health Organization. Cancer. 2022. Available online: https://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 3 February 2022).
- Mattiuzzi, C.; Lippi, G. Current cancer epidemiology. J. Epidemiol. Glob. Health 2019, 9, 217. [Google Scholar] [CrossRef]
- Staff AMCaN. Risk of Dying from Cancer Continues to Drop at an Accelerated Pace: American Cancer Society. 2022. Available online: https://www.cancer.org/research/acs-research-news/facts-and-figures-2022.html (accessed on 25 January 2024).
- Balistreri, C.R.; Caruso, C.; Candore, G. The role of adipose tissue and adipokines in obesity-related inflammatory diseases. Mediat. Inflamm. 2010, 2010, 802078. [Google Scholar] [CrossRef]
- Zhang, A.M.; Wellberg, E.A.; Kopp, J.L.; Johnson, J.D. Hyperinsulinemia in obesity, inflammation, and cancer. Diabetes Metab. J. 2021, 45, 285–311. [Google Scholar] [CrossRef]
- Kawasaki, N.; Asada, R.; Saito, A.; Kanemoto, S.; Imaizumi, K. Obesity-induced endoplasmic reticulum stress causes chronic inflammation in adipose tissue. Sci. Rep. 2012, 2, 799. [Google Scholar] [CrossRef]
- Krzysztof, M.; Katarzyna, K.; Jolanta, S.-K.; Christine, W. Excessive Endoplasmic Reticulum Stress Correlates with Impaired Mitochondrial Dynamics, Mitophagy and Apoptosis. Liver and Adipose Tissue, but Not in Muscles in EMS Horses. Int. J. Mol. Sci. 2018, 19, 165. [Google Scholar]
- Cubillos-Ruiz, J.R.; Bettigole, S.E.; Glimcher, L.H. Tumorigenic and immunosuppressive effects of endoplasmic reticulum stress in cancer. Cell 2017, 168, 692–706. [Google Scholar] [CrossRef]
- You, M.; Xie, Z.; Zhang, N.; Zhang, Y.; Xiao, D.; Liu, S.; Zhuang, W.; Li, L.; Tao, Y. Signaling pathways in cancer metabolism: Mechanisms and therapeutic targets. Signal Transduct. Target. Ther. 2023, 8, 196. [Google Scholar] [CrossRef]
- Bravo, R.; Parra, V.; Gatica, D.; Rodriguez, A.E.; Torrealba, N.; Paredes, F.; Wang, Z.V.; Zorzano, A.; Hill, J.A.; Jaimovich, E.; et al. Endoplasmic reticulum and the unfolded protein response: Dynamics and metabolic integration. Int. Rev. Cell Mol. Biol. 2013, 301, 215–290. [Google Scholar]
- Schwarz, D.S.; Blower, M.D. The endoplasmic reticulum: Structure, function and response to cellular signaling. Cell. Mol. Life Sci. 2016, 73, 79–94. [Google Scholar] [CrossRef]
- Zhao, L.; Shen, Y.; Wang, Y.; Wang, L.; Zhang, L.; Zhao, Z.; Li, S. Lactobacillus plantarum S9 alleviates lipid profile, insulin resistance, and inflammation in high-fat diet-induced metabolic syndrome rats. Sci. Rep. 2022, 12, 15490. [Google Scholar] [CrossRef]
- Zhai, X.; Sterea, A.M.; El Hiani, Y. Lessons from the endoplasmic reticulum Ca2+ Transporters—A Cancer Connection. Cells 2020, 9, 1536. [Google Scholar] [CrossRef]
- Pedriali, G.; Rimessi, A.; Sbano, L.; Giorgi, C.; Wieckowski, M.R.; Previati, M.; Pinton, P. Regulation of endoplasmic reticulum–mitochondria Ca2+ transfer and its importance for anti-cancer therapies. Front. Oncol. 2017, 7, 180. [Google Scholar] [CrossRef]
- Jaud, M.; Philippe, C.; Di Bella, D.; Tang, W.; Pyronnet, S.; Laurell, H.; Mazzolini, L.; Rouault-Pierre, K.; Touriol, C. Translational regulations in response to endoplasmic reticulum stress in cancers. Cells 2020, 9, 540. [Google Scholar] [CrossRef]
- Fu, X.; Cui, J.; Meng, X.; Jiang, P.; Zheng, Q.; Zhao, W.; Chen, X. Endoplasmic reticulum stress, cell death and tumor: Association between endoplasmic reticulum stress and the apoptosis pathway in tumors. Oncol. Rep. 2021, 45, 801–808. [Google Scholar] [CrossRef]
- Bian, X.; Liu, R.; Meng, Y.; Xing, D.; Xu, D.; Lu, Z. Lipid metabolism and cancer. J. Exp. Med. 2021, 218, e20201606. [Google Scholar] [CrossRef]
- Broadfield, L.A.; Pane, A.A.; Talebi, A.; Swinnen, J.V.; Fendt, S.-M. Lipid metabolism in cancer: New perspectives and emerging mechanisms. Dev. Cell 2021, 56, 1363–1393. [Google Scholar] [CrossRef]
- Jin, Y.; Tan, Y.; Wu, J.; Ren, Z. Lipid droplets: A cellular organelle vital in cancer cells. Cell Death Discov. 2023, 9, 254. [Google Scholar] [CrossRef]
- Kadowaki, H.; Nishitoh, H. Signaling pathways from the endoplasmic reticulum and their roles in disease. Genes 2013, 4, 306–333. [Google Scholar] [CrossRef]
- Siwecka, N.; Rozpędek-Kamińska, W.; Wawrzynkiewicz, A.; Pytel, D.; Diehl, J.A.; Majsterek, I. The structure, activation and signaling of IRE1 and its role in determining cell fate. Biomedicines 2021, 9, 156. [Google Scholar] [CrossRef]
- Bell, M.C.; Meier, S.E.; Ingram, A.L.; Abisambra, J.F. PERK-opathies: An endoplasmic reticulum stress mechanism underlying neurodegeneration. Curr. Alzheimer Res. 2016, 13, 150–163. [Google Scholar] [CrossRef]
- Allen, D.; Seo, J. ER Stress activates the TOR pathway through Atf6. J. Mol. Signal. 2018, 13, 1. [Google Scholar] [CrossRef] [PubMed]
- Chuang, J.-C.; Cui, H.; Mason, B.L.; Mahgoub, M.; Bookout, A.L.; Hana, G.Y.; Perello, M.; Elmquist, J.K.; Repa, J.J.; Zigman, J.M.; et al. Chronic social defeat stress disrupts regulation of lipid synthesis. J. Lipid Res. 2010, 51, 1344–1353. [Google Scholar] [CrossRef] [PubMed]
- Maduka, I.; Neboh, E.; Ufelle, S. The relationship between serum cortisol, adrenaline, blood glucose and lipid profile of undergraduate students under examination stress. Afr. Health Sci. 2015, 15, 131–136. [Google Scholar] [CrossRef]
- Welihinda, A.A.; Tirasophon, W.; Kaufman, R.J. The cellular response to protein misfolding in the endoplasmic reticulum. Gene Expr. 1999, 7, 293–300. [Google Scholar]
- Basaiawmoit, R.V.; Rattan, S.I. Cellular stress and protein misfolding during aging. In Protein Misfolding and Cellular Stress in Disease and Aging: Concepts and Protocols; Humana Press: Totowa, NJ, USA, 2010; Volume 648, pp. 107–117. [Google Scholar]
- Gidalevitz, T.; Prahlad, V.; Morimoto, R.I. The stress of protein misfolding: From single cells to multicellular organisms. Cold Spring Harb. Perspect. Biol. 2011, 3, a009704. [Google Scholar] [CrossRef]
- Kiani, A.K.; Dhuli, K.; Donato, K.; Aquilanti, B.; Velluti, V.; Matera, G.; Iaconelli, A.; Connelly, S.T.; Bellinato, F.; Gisondi, P.; et al. Main nutritional deficiencies. J. Prev. Med. Hyg. 2022, 63 (Suppl. S3), E93. [Google Scholar]
- Mekahli, D.; Bultynck, G.; Parys, J.B.; De Smedt, H.; Missiaen, L. Endoplasmic-reticulum calcium depletion and disease. Cold Spring Harb. Perspect. Biol. 2011, 3, a004317. [Google Scholar] [CrossRef]
- Lemmer, I.L.; Willemsen, N.; Hilal, N.; Bartelt, A. A guide to understanding endoplasmic reticulum stress in metabolic disorders. Mol. Metab. 2021, 47, 101169. [Google Scholar] [CrossRef]
- Haeri, M.; Knox, B.E. Endoplasmic reticulum stress and unfolded protein response pathways: Potential for treating age-related retinal degeneration. J. Ophthalmic Vis. Res. 2012, 7, 45–59. [Google Scholar] [PubMed]
- Haynes, C.M.; Titus, E.A.; Cooper, A.A. Degradation of misfolded proteins prevents ER-derived oxidative stress and cell death. Mol. Cell 2004, 15, 767–776. [Google Scholar] [CrossRef]
- Shacham, T.; Sharma, N.; Lederkremer, G.Z. Protein misfolding and ER stress in Huntington’s disease. Front. Mol. Biosci. 2019, 6, 20. [Google Scholar] [CrossRef]
- Lang, S.; Pfeffer, S.; Lee, P.-H.; Cavalié, A.; Helms, V.; Förster, F.; Zimmermann, R. An update on Sec61 channel functions, mechanisms, and related diseases. Front. Physiol. 2017, 8, 887. [Google Scholar] [CrossRef]
- Balsa, E.; Soustek, M.S.; Thomas, A.; Cogliati, S.; García-Poyatos, C.; Martín-García, E.; Jedrychowski, M.; Gygi, S.P.; Enriquez, J.A.; Puigserver, P. ER and nutrient stress promote assembly of respiratory chain supercomplexes through the PERK-eIF2α axis. Mol. Cell 2019, 74, 877–890.e6. [Google Scholar] [CrossRef] [PubMed]
- Chong, W.C.; Shastri, M.D.; Eri, R. Endoplasmic reticulum stress and oxidative stress: A vicious nexus implicated in bowel disease pathophysiology. Int. J. Mol. Sci. 2017, 18, 771. [Google Scholar] [CrossRef]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative stress: Harms and benefits for human health. Oxidative Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef] [PubMed]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef]
- Yoshida, H. ER stress and diseases. FEBS J. 2007, 274, 630–658. [Google Scholar] [CrossRef]
- Choi, J.-A.; Song, C.-H. Insights into the role of endoplasmic reticulum stress in infectious diseases. Front. Immunol. 2020, 10, 3147. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Chen, J.; Hua, X.; Sun, Y.; Cui, R.; Sha, J.; Zhu, X. The emerging role of XBP1 in cancer. Biomed. Pharmacother. 2020, 127, 110069. [Google Scholar] [CrossRef]
- Palade, G.E. The endoplasmic reticulum. J. Biophys. Biochem. Cytol. 1956, 2, 85. [Google Scholar] [CrossRef]
- Basseri, S.; Austin, R.C. Endoplasmic reticulum stress and lipid metabolism: Mechanisms and therapeutic potential. Biochem. Res. Int. 2012, 2012, 841362. [Google Scholar] [CrossRef] [PubMed]
- Sheng, X.; Nenseth, H.Z.; Qu, S.; Kuzu, O.F.; Frahnow, T.; Simon, L.; Greene, S.; Zeng, Q.; Fazli, L.; Rennie, P.S.; et al. IRE1α-XBP1s pathway promotes prostate cancer by activating c-MYC signaling. Nat. Commun. 2019, 10, 32. [Google Scholar] [CrossRef] [PubMed]
- Pike, L.R.G.; Singleton, D.C.; Buffa, F.; Abramczyk, O.; Phadwal, K.; Li, J.-L.; Simon, A.K.; Murray, J.T.; Harris, A.L. Transcriptional up-regulation of ULK1 by ATF4 contributes to cancer cell survival. Biochem. J. 2013, 449, 389–400. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Liu, Y.; Yang, Y.; Qiu, Y.; Wang, Z.; Li, X.; Zhang, W. Emerging roles of activating transcription factor (ATF) family members in tumourigenesis and immunity: Implications in cancer immunotherapy. Genes Dis. 2022, 9, 981–999. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wey, S.; Zhang, Y.; Ye, R.; Lee, A.S. Role of the unfolded protein response regulator GRP78/BiP in development, cancer, and neurological disorders. Antioxid. Redox Signal. 2009, 11, 2307–2316. [Google Scholar] [CrossRef]
- Mahoney, D.J.; Lefebvre, C.; Allan, K.; Brun, J.; Sanaei, C.A.; Baird, S.; Pearce, N.; Grönberg, S.; Wilson, B.; Prakesh, M.; et al. Virus-tumor interactome screen reveals ER stress response can reprogram resistant cancers for oncolytic virus-triggered caspase-2 cell death. Cancer Cell 2011, 20, 443–456. [Google Scholar] [CrossRef] [PubMed]
- Robinson, C.M.; Talty, A.; Logue, S.E.; Mnich, K.; Gorman, A.M.; Samali, A. An emerging role for the unfolded protein response in pancreatic cancer. Cancers 2021, 13, 261. [Google Scholar] [CrossRef]
- Raymundo, D.P.; Doultsinos, D.; Guillory, X.; Carlesso, A.; Eriksson, L.A.; Chevet, E. Pharmacological targeting of IRE1 in cancer. Trends Cancer 2020, 6, 1018–1030. [Google Scholar] [CrossRef]
- Auf, G.; Jabouille, A.; Guérit, S.; Pineau, R.; Delugin, M.; Bouchecareilh, M.; Magnin, N.; Favereaux, A.; Maitre, M.; Gaiser, T.; et al. Inositol-requiring enzyme 1α is a key regulator of angiogenesis and invasion in malignant glioma. Proc. Natl. Acad. Sci. USA 2010, 107, 15553–15558. [Google Scholar] [CrossRef]
- Tripathi, Y.B.; Pandey, V. Obesity and endoplasmic reticulum (ER) stresses. Front. Immunol. 2012, 3, 240. [Google Scholar] [CrossRef]
- Yadav, R.K.; Chae, S.-W.; Kim, H.-R.; Chae, H.J. Endoplasmic reticulum stress and cancer. J. Cancer Prev. 2014, 19, 75. [Google Scholar] [CrossRef] [PubMed]
- Ajoolabady, A.; Liu, S.; Klionsky, D.J.; Lip, G.Y.; Tuomilehto, J.; Kavalakatt, S.; Pereira, D.M.; Samali, A.; Ren, J. ER stress in obesity pathogenesis and management. Trends Pharmacol. Sci. 2021, 43, 97–109. [Google Scholar] [CrossRef]
- Iyengar, N.M.; Gucalp, A.; Dannenberg, A.J.; Hudis, C.A. Obesity and cancer mechanisms: Tumor microenvironment and inflammation. J. Clin. Oncol. 2016, 34, 4270–4276. [Google Scholar] [CrossRef]
- Pan, D.; Yang, Y.; Nong, A.; Tang, Z.; Li, Q.X. GRP78 activity moderation as a therapeutic treatment against obesity. Int. J. Environ. Res. Public Health 2022, 19, 15965. [Google Scholar] [CrossRef] [PubMed]
- Luo, D.; Fan, N.; Zhang, X.; Ngo, F.Y.; Zhao, J.; Zhao, W.; Huang, M.; Li, D.; Wang, Y.; Rong, J. Covalent inhibition of endoplasmic reticulum chaperone GRP78 disconnects the transduction of ER stress signals to inflammation and lipid accumulation in diet-induced obese mice. Elife 2022, 11, e72182. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F. Targeting endoplasmic reticulum signaling pathways in cancer. Acta Oncol. 2012, 51, 822–830. [Google Scholar] [CrossRef]
- Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. The Yin-Yang of tumor-associated macrophages in neoplastic progression and immune surveillance. Immunol. Rev. 2008, 222, 155–161. [Google Scholar] [CrossRef]
- Gupta, S.; McGrath, B.; Cavener, D.R. PERK regulates the proliferation and development of insulin-secreting beta-cell tumors in the endocrine pancreas of mice. PLoS ONE 2009, 4, e8008. [Google Scholar] [CrossRef]
- Feng, Y.-X.; Jin, D.X.; Sokol, E.S.; Reinhardt, F.; Miller, D.H.; Gupta, P.B. Cancer-specific PERK signaling drives invasion and metastasis through CREB3L1. Nat. Commun. 2017, 8, 1079. [Google Scholar] [CrossRef]
- Delie, F.; Petignat, P.; Cohen, M. GRP78 protein expression in ovarian cancer patients and perspectives for a drug-targeting approach. J. Oncol. 2012, 2012, 468615. [Google Scholar] [CrossRef]
- Casas, C. GRP78 at the Centre of the Stage in Cancer and Neuroprotection. Front. Neurosci. 2017, 11, 177. [Google Scholar] [CrossRef]
- Overley-Adamson, B.; Artlett, C.M.; Stephens, C.; Sassi-Gaha, S.; Weis, R.D.; Thacker, J.D. Targeting the unfolded protein response, XBP1, and the NLRP3 inflammasome in fibrosis and cancer. Cancer Biol. Ther. 2014, 15, 452–462. [Google Scholar] [CrossRef]
- Daneshmand, S.; Quek, M.L.; Lin, E.; Lee, C.; Cote, R.J.; Hawes, D.; Cai, J.; Groshen, S.; Lieskovsky, G.; Skinner, D.G.; et al. Glucose-regulated protein GRP78 is up-regulated in prostate cancer and correlates with recurrence and survival. Hum. Pathol. 2007, 38, 1547–1552. [Google Scholar] [CrossRef]
- Déry, M.-A.; Jodoin, J.; Ursini-Siegel, J.; Aleynikova, O.; Ferrario, C.; Hassan, S.; Basik, M.; LeBlanc, A.C. Endoplasmic reticulum stress induces PRNP prion protein gene expression in breast cancer. Breast Cancer Res. 2013, 15, R22. [Google Scholar] [CrossRef]
- Nagelkerke, A.; Bussink, J.; Mujcic, H.; Wouters, B.G.; Lehmann, S.; Sweep, F.C.; Span, P.N. Hypoxia stimulates migration of breast cancer cells via the PERK/ATF4/LAMP3-arm of the unfolded protein response. Breast Cancer Res. 2013, 15, R2. [Google Scholar] [CrossRef]
- Davies, M.P.; Barraclough, D.L.; Stewart, C.; Joyce, K.A.; Eccles, R.M.; Barraclough, R.; Rudland, P.S.; Sibson, D.R. Expression and splicing of the unfolded protein response gene XBP-1 are significantly associated with clinical outcome of endocrine-treated breast cancer. Int. J. Cancer 2008, 123, 85–88. [Google Scholar] [CrossRef]
- Zhang, Y.; Tseng, C.-C.; Tsai, Y.-L.; Fu, X.; Schiff, R.; Lee, A.S. Cancer cells resistant to therapy promote cell surface relocalization of GRP78 which complexes with PI3K and enhances PI(3,4,5)P3 production. PLoS ONE 2013, 8, e80071. [Google Scholar] [CrossRef] [PubMed]
- Pfaffenbach, K.T.; Lee, A.S. The critical role of GRP78 in physiologic and pathologic stress. Curr. Opin. Cell Biol. 2011, 23, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-T.; Zhu, G.; Pfaffenbach, K.; Kanel, G.; Stiles, B.; Lee, A.S. GRP78 as a regulator of liver steatosis and cancer progression mediated by loss of the tumor suppressor PTEN. Oncogene 2014, 33, 4997–5005. [Google Scholar] [CrossRef] [PubMed]
- Su, R.; Li, Z.; Li, H.; Song, H.; Bao, C.; Wei, J.; Cheng, L. Grp78 promotes the invasion of hepatocellular carcinoma. BMC Cancer 2010, 10, 20. [Google Scholar] [CrossRef] [PubMed]
- Akinyemi, A.O.; Simpson, K.E.; Oyelere, S.F.; Nur, M.; Ngule, C.M.; Owoyemi, B.C.D.; Ayarick, V.A.; Oyelami, F.F.; Obaleye, O.; Esoe, D.-P.; et al. Unveiling the dark side of glucose-regulated protein 78 (GRP78) in cancers and other human pathology: A systematic review. Mol. Med. 2023, 29, 122. [Google Scholar] [CrossRef]
- Lee, H.K.; Xiang, C.; Cazacu, S.; Finniss, S.; Kazimirsky, G.; Lemke, N.; Lehman, N.L.; Rempel, S.A.; Mikkelsen, T.; Brodie, C. GRP78 is overexpressed in glioblastomas and regulates glioma cell growth and apoptosis. Neuro-Oncol. 2008, 10, 236–243. [Google Scholar] [CrossRef]
- Koong, A.C.; Chauhan, V.; Romero-Ramirez, L. Targeting XBP-1 as a novel anti-cancer strategy. Cancer Biol. Ther. 2006, 5, 756–759. [Google Scholar] [CrossRef] [PubMed]
- Koumenis, C.; Naczki, C.; Koritzinsky, M.; Rastani, S.; Diehl, A.; Sonenberg, N.; Koromilas, A.; Wouters, B.G. Regulation of protein synthesis by hypoxia via activation of the endoplasmic reticulum kinase PERK and phosphorylation of the translation initiation factor eIF2α. Mol. Cell. Biol. 2002, 22, 7405–7416. [Google Scholar] [CrossRef] [PubMed]
- Oakes, S.A. Endoplasmic reticulum stress signaling in cancer cells. Am. J. Pathol. 2020, 190, 934–946. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wen, W.; Luo, J. Targeting endoplasmic reticulum stress as an effective treatment for alcoholic pancreatitis. Biomedicines 2022, 10, 108. [Google Scholar] [CrossRef] [PubMed]
- Bonsignore, G.; Martinotti, S.; Ranzato, E. Endoplasmic reticulum stress and cancer: Could unfolded protein response be a druggable target for cancer therapy? Int. J. Mol. Sci. 2023, 24, 1566. [Google Scholar] [CrossRef] [PubMed]
- Hill, D.S.; Lovat, P.E.; Haass, N.K. Induction of endoplasmic reticulum stress as a strategy for melanoma therapy: Is there a future? Melanoma Manag. 2014, 1, 127–137. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Normal Function of ER | Produced Molecule | ER Stress Impact on Normal Cellular Functions | Reference |
|---|---|---|---|
| Lipid biosynthesis | Phosphatidylcholine (PtdCho), phosphatidylethanolamine (PtdEtn), phosphatidylinositol (PtdIns), and basic sphingolipid structures | Overproduction of lipids will often lead to lipotoxicity and cause diseases such as insulin resistance and dyslipidemia | [24,25,43,44] |
| Protein synthesis and folding | Glycoproteins, soluble proteins, and membrane bound proteins | ER no longer synthesizes nor folds protein properly, leading to diseases such as ovarian cancer | [26,27,28,29,43] |
| Calcium metabolism | Calcium | Anti-apoptotic activity | [30,31,43] |
| ER Stress Inducers | ER Stress Stimulation Mechanism | Resulting Diseases | References |
|---|---|---|---|
| Accumulation of misfolded protein | Misfolded protein buildup and aggregation in the ER hinder normal cellular function and can be toxic, resulting in cell death. | Diabetes, Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease | [32,33,34] |
| Changes in calcium homeostasis | Unfolded proteins build up due to changes in ER Ca2+ homeostasis, which, in turn, trigger ER stress and the unfolded protein response. | Diabetes mellitus, neurologic disorders, cancer, kidney disease, obesity | [30,34,35] |
| Nutrient deprivation | The development of respiratory super complexes (SCs) and mitochondrial bioenergetics are stimulated by ER stress and glucose restriction. | Protein energy malnutrition, scurvy, rickets, beriberi, hypocalcemia, osteomalacia, vitamin K deficiency, pellagra, xerophthalmia, and iron deficiency | [29,36] |
| Oxidative stress | Because the process of protein folding depends on redox equilibrium, oxidative stress can interfere with this process and increase the generation of misfolded proteins, which, in turn, increases ER stress. | Parkinson’s disease, asthma, rheumatoid arthritis, amyotrophic lateral sclerosis (ALS), multiple sclerosis, depression | [37,38,39] |
| Protein overexpression | Human cells trigger ER stress as a defense mechanism when the amount of unfolded protein exceeds the ER’s capacity to fold it (combine with protein accumulation). | Diabetes and neurodegenerative disorders | [40] |
| Viral infections | Viral infection triggers ER stress, which helps cells survive by preventing apoptosis. By encouraging the synthesis of C/EBP homologous protein (CHOP), some viral infections cause ER-stress-mediated apoptosis. | Encephalopathy characterized by spongiform changes, neurodegeneration, and gliosis, along with the pathogenic effects of viruses like hepatitis C virus, influenza virus, and herpes simplex virus, leading to associated diseases | [41] |
| Cancer | ER Stress Markers Expressed | Oncogenic Process | Reference |
|---|---|---|---|
| Pancreatic | PERK encourages angiogenesis and boosts beta-cell insulinoma growth | Promote metastasis through the transcription factor CREB3L1 | [61,62] |
| Ovarian | Overexpression of Grp78 | Grp78 induces tumor progression through the activation of JNKs and FAK1 pathways; it also regulates the tumor microenvironment, angiogenesis, and cell proliferation | [63,64] |
| Lymphoma | Under hypoxic conditions, XBP1 splicing encourages tumor growth | XBP1 can induce epithelial–mesenchymal transition (EMT) and cell invasion and metastasis, activating the c-MYC signaling pathway | [42,45,65] |
| Colorectal | ATF4 levels rise in extreme hypoxia | ATF4 promotes tumor progression by activating the c-MYC signaling pathway, inducing epithelial–mesenchymal transition (EMT) and cell invasion | [46,47,48] |
| Breast | Elevated levels of protein and mRNA and Bip/Grp78 protein in severe hypoxia; ATF4-elevated levels of spliced mRNA XBP1, which favors the survival of the tumor | Grp78 initiates the activation of JNKs and FAK1 pathways; ATF4 promotes EMT and angiogenesis | [48,64,66,67,68,69] |
| Prostate | Increased signaling of the Grp78 to the cell surface | Grp78 initiates activation of Akt and the PI3K/AKT oncogenic pathway | [70,71,72] |
| Liver | High levels of Grp78, which leads to the in vivo and ex vivo invasion of hepatocellular carcinoma | Grp78 regulates PTEN-loss-mediated liver injury and cancer progression | [72,73,74] |
| Brain | Overexpression of Grp78 decrease in XBP-1 will stimulate the viral oncolysis of the U373 glioblastoma cells | XBP1 is a transcription factor that promotes the expression of genes implicated in cell metabolism, nutrient uptake, and anti-oxidation | [49,74,75] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
AlBashtawi, J.; Al-Jaber, H.; Ahmed, S.; Al-Mansoori, L. Impact of Obesity-Related Endoplasmic Reticulum Stress on Cancer and Associated Molecular Targets. Biomedicines 2024, 12, 793. https://doi.org/10.3390/biomedicines12040793
AlBashtawi J, Al-Jaber H, Ahmed S, Al-Mansoori L. Impact of Obesity-Related Endoplasmic Reticulum Stress on Cancer and Associated Molecular Targets. Biomedicines. 2024; 12(4):793. https://doi.org/10.3390/biomedicines12040793
Chicago/Turabian StyleAlBashtawi, Joud, Hend Al-Jaber, Sara Ahmed, and Layla Al-Mansoori. 2024. "Impact of Obesity-Related Endoplasmic Reticulum Stress on Cancer and Associated Molecular Targets" Biomedicines 12, no. 4: 793. https://doi.org/10.3390/biomedicines12040793
APA StyleAlBashtawi, J., Al-Jaber, H., Ahmed, S., & Al-Mansoori, L. (2024). Impact of Obesity-Related Endoplasmic Reticulum Stress on Cancer and Associated Molecular Targets. Biomedicines, 12(4), 793. https://doi.org/10.3390/biomedicines12040793