
The Impact of Gut Microbiota Changes on Methotrexate-Induced Neurotoxicity in Developing Young Rats


 ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
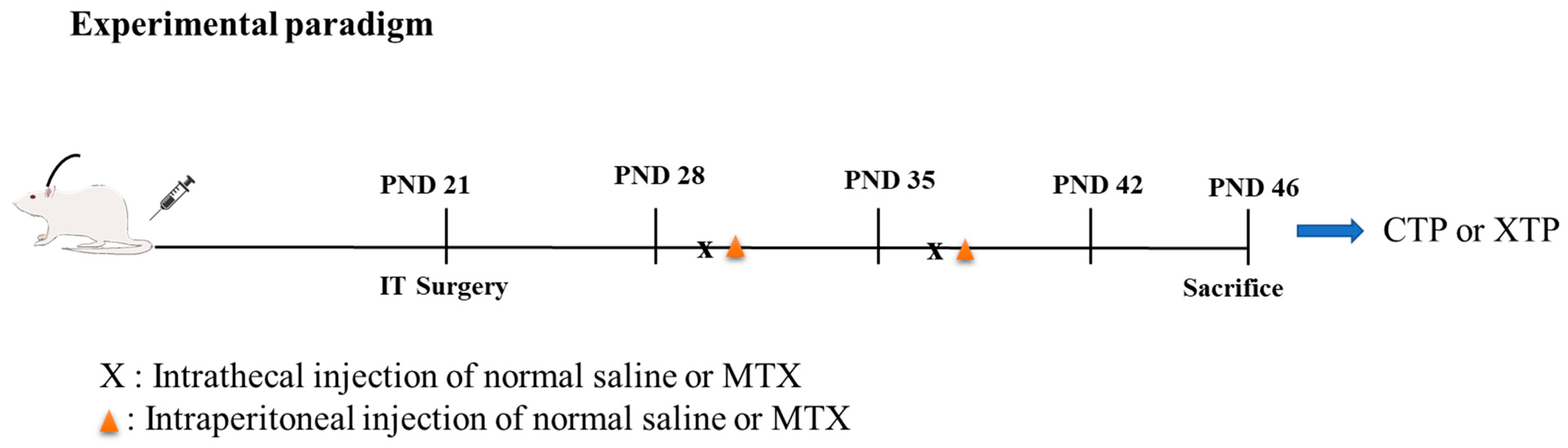
2.1. Experimental Model and Subject Details
- CTP: rats that underwent catheter implantation surgery and received an equal volume of normal saline to the MTX treatment group via IT and intraperitoneal injection (IP).
- XTP: rats that underwent catheter implantation surgery and received 0.5 mg/kg of methotrexate diluted with 0.9% normal saline in 5~10 μL as the final volume via IT once per week for two weeks. Then, 24 h after IT injection, rats received 100 mg/kg of methotrexate via IP injection once per week for two weeks.
2.2. 16S Metagenomics Studies Using 16S rDNA Next Generation Sequencing (NGS) for Gut-Microbiota Measurements
2.3. Brain Tissue Collection
2.4. Quantitative Real-Time Polymerase Chain Reaction (PCR) Analysis
2.5. Short-Chain Fatty Acid Analysis
2.6. Hippocampus Metabolite Analysis
2.7. Prediction of Metabolic Pathway and Metabolite Changes Following MTX Treatment
2.8. Statistical Analysis
3. Results
3.1. MTX Treatment Induced Gut Microbiota Dysbiosis
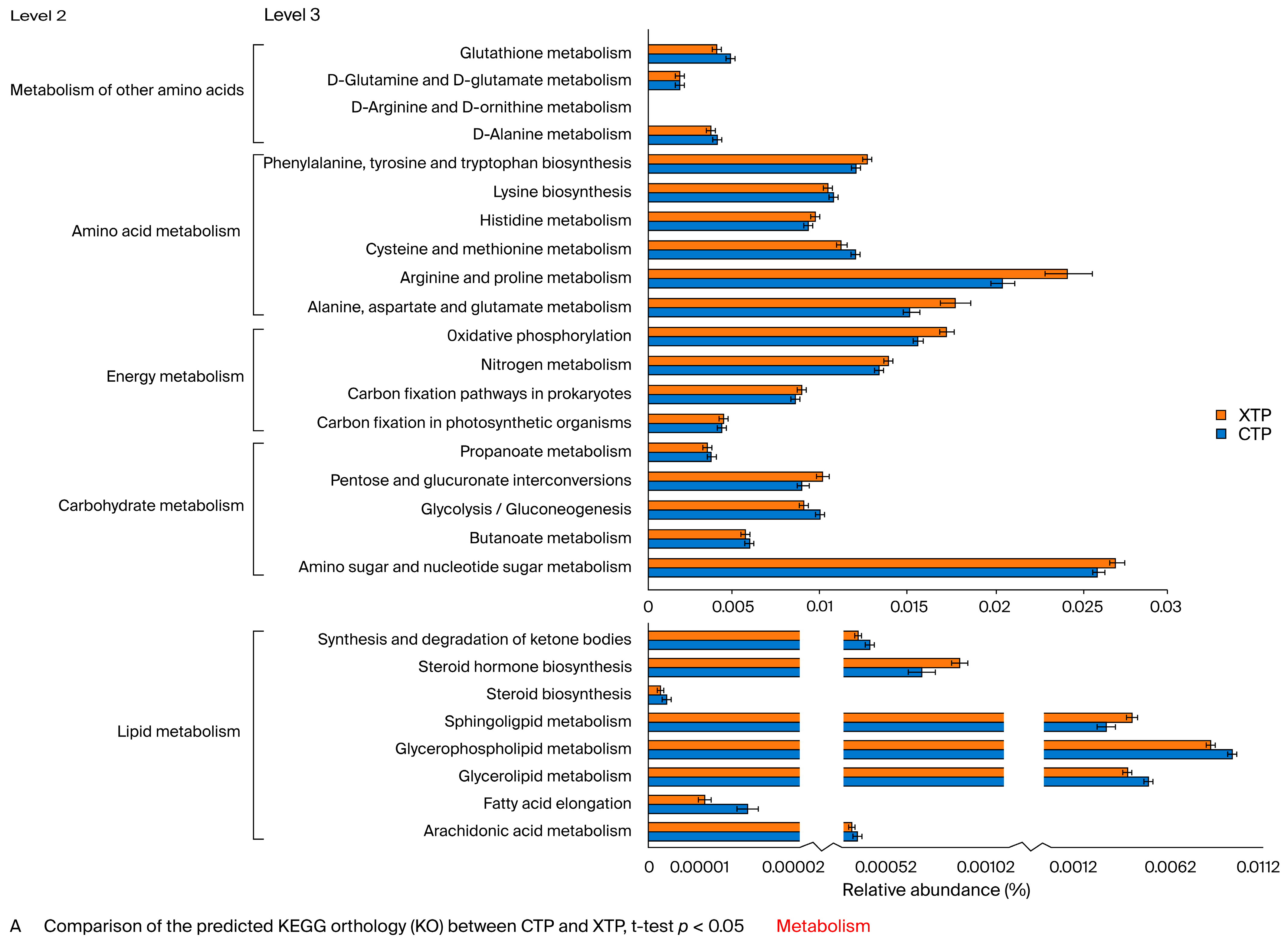
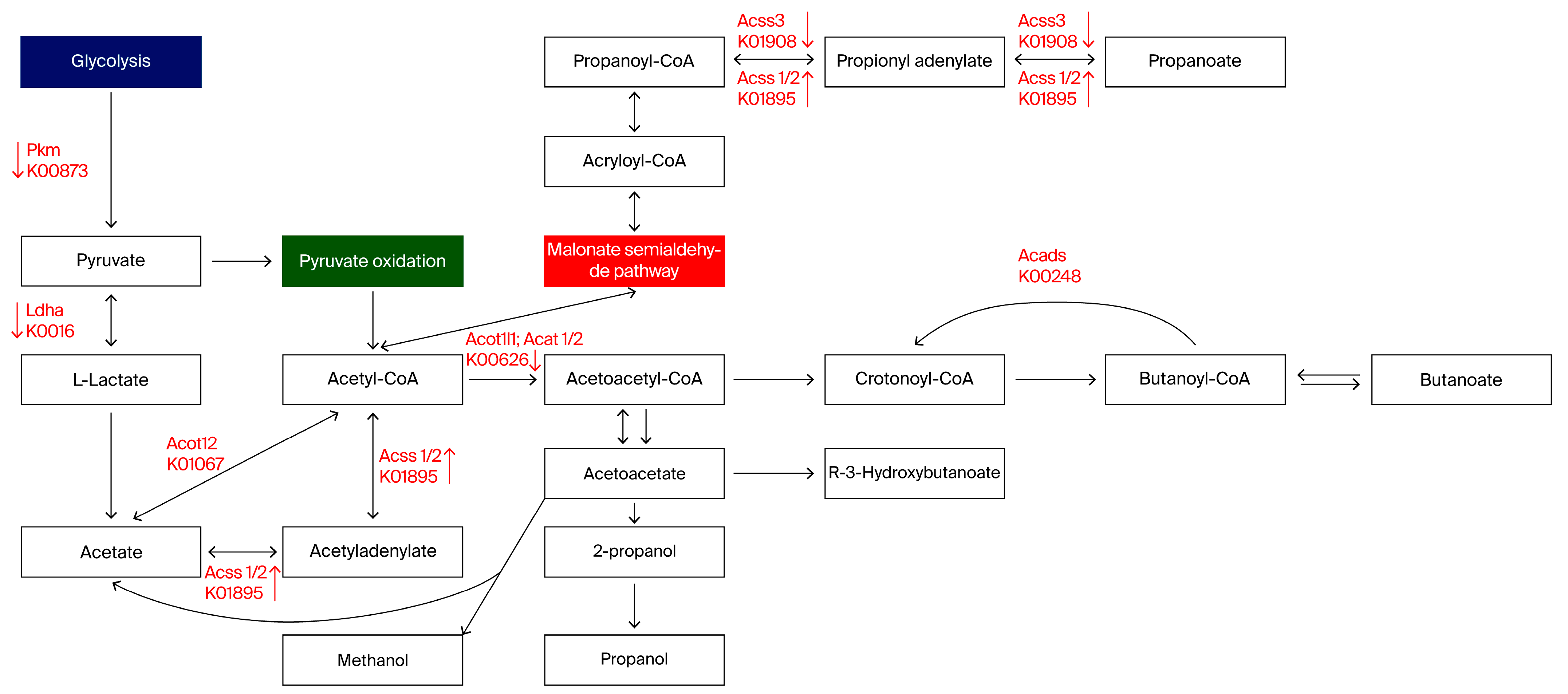
3.2. Metabolic Pathway Alterations Following MTX Treatment
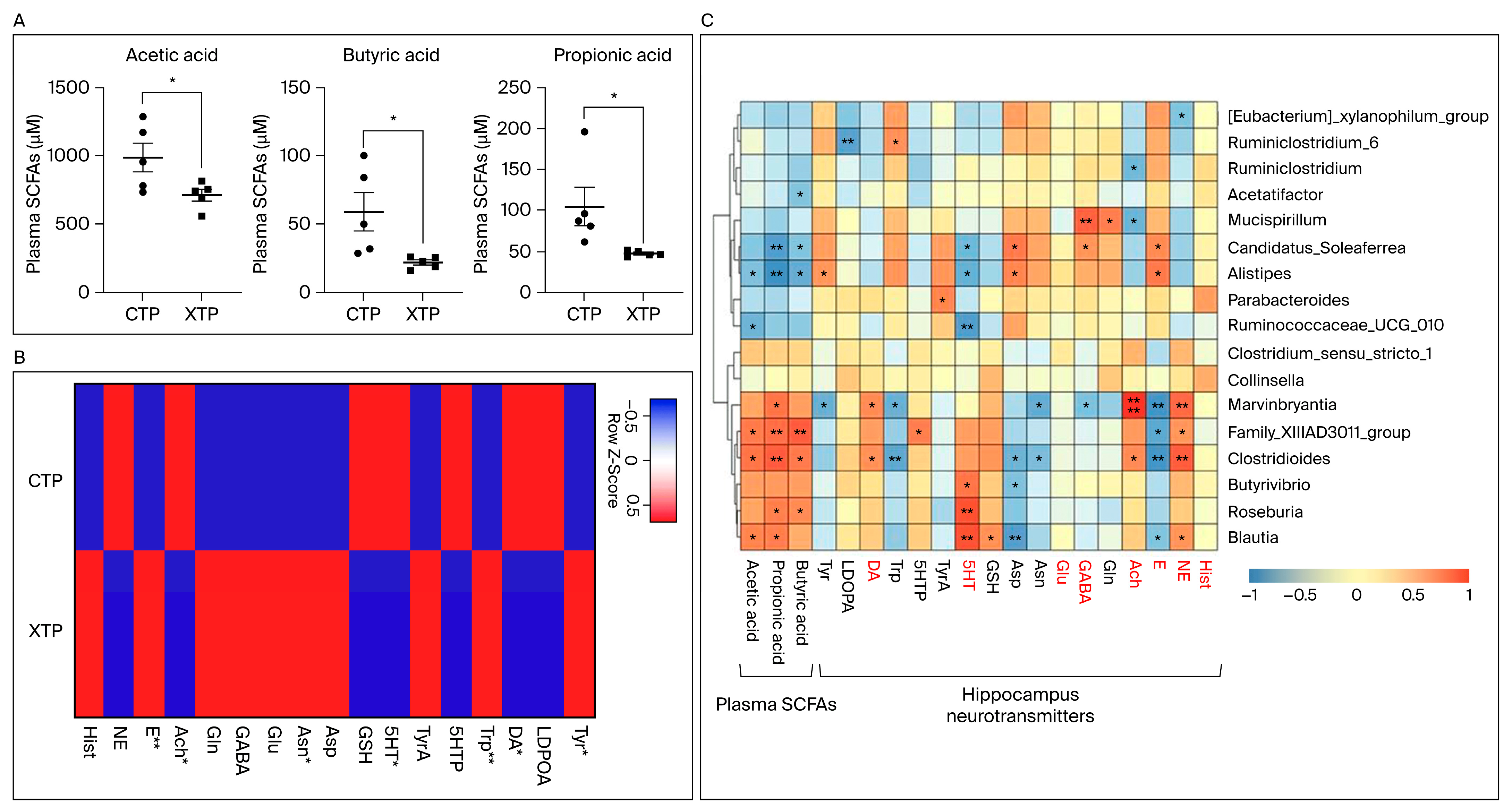
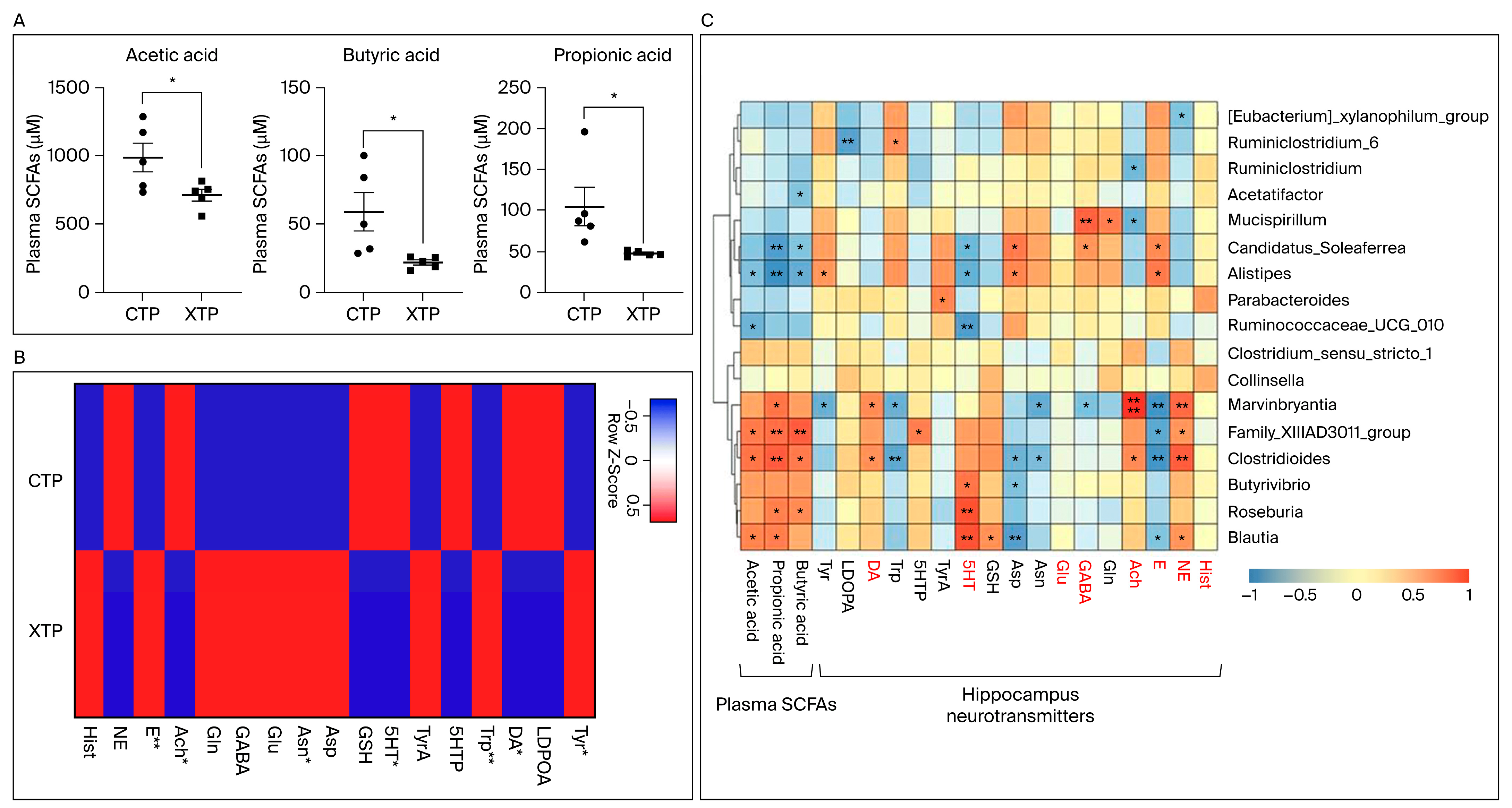
3.3. SCFA and NT Metabolism Were Affected in Response to MTX Treatment
3.4. MTX Results in NT Alteration in Brain Tissue and Dysregulated SCFA Concentration Levels in the Circulating System
3.5. A Strong Relationship between Gut Microbiota Changes and MTX Neurotoxicity
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pui, C.-H.; Campana, D.; Pei, D.; Bowman, W.P.; Sandlund, J.T.; Kaste, S.C.; Ribeiro, R.C.; Rubnitz, J.E.; Raimondi, S.C.; Onciu, M.; et al. Treating childhood acute lymphoblastic leukemia without cranial irradiation. N. Engl. J. Med. 2009, 360, 2730–2741. [Google Scholar] [CrossRef] [PubMed]
- Pui, C.H.; Mullighan, C.G.; Evans, W.E.; Relling, M.V. Pediatric acute lymphoblastic leukemia: Where are we going and how do we get there? Blood 2012, 120, 1165–1174. [Google Scholar] [CrossRef] [PubMed]
- Pui, C.-H.; Yang, J.J.; Hunger, S.P.; Pieters, R.; Schrappe, M.; Biondi, A.; Vora, A.; Baruchel, A.; Silverman, L.B.; Schmiegelow, K.; et al. Childhood Acute Lymphoblastic Leukemia: Progress Through Collaboration. J. Clin. Oncol. 2015, 33, 2938–2948. [Google Scholar] [CrossRef] [PubMed]
- Cascella, M.; Di Napoli, R.; Carbone, D.; Cuomo, G.F.; Bimonte, S.; Muzio, M.R. Chemotherapy-related cognitive impairment: Mechanisms, clinical features and research perspectives. Recenti. Prog. Med. 2018, 109, 523–530. [Google Scholar]
- Torrente, N.C.; Pastor, J.-B.N.; Chaparro, N.d.l.O. Systematic review of cognitive sequelae of non-central nervous system cancer and cancer therapy. J. Cancer Surviv. 2020, 14, 464–482. [Google Scholar] [CrossRef] [PubMed]
- Iyer, N.S.; Balsamo, L.M.; Bracken, M.B.; Kadan-Lottick, N.S. Chemotherapy-only treatment effects on long-term neurocognitive functioning in childhood ALL survivors: A review and meta-analysis. Blood 2015, 126, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Cheung, Y.T.; Krull, K.R. Neurocognitive outcomes in long-term survivors of childhood acute lymphoblastic leukemia treated on contemporary treatment protocols: A systematic review. Neurosci. Biobehav. Rev. 2015, 53, 108–120. [Google Scholar] [CrossRef] [PubMed]
- Millan, N.C.; Pastrana, A.; Guitter, M.R.; Zubizarreta, P.A.; Monges, M.S.; Felice, M.S. Acute and sub-acute neurological toxicity in children treated for acute lymphoblastic leukemia. Leuk. Res. 2018, 65, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Moe, P.J.; Holen, A. High-dose methotrexate in childhood all. Pediatr. Hematol. Oncol. 2000, 17, 615–622. [Google Scholar] [CrossRef]
- Vezmar, S.; Schüsseler, P.; Becker, A.; Bode, U.; Jaehde, U. Methotrexate-associated alterations of the folate and methyl-transfer pathway in the CSF of ALL patients with and without symptoms of neurotoxicity. Pediatr. Blood Cancer 2009, 52, 26–32. [Google Scholar] [CrossRef]
- Cohen, I.J. Neurotoxicity after high-dose methotrexate (MTX) is adequately explained by insufficient folinic acid rescue. Cancer Chemother. Pharmacol. 2017, 79, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Maxwell, R.R.; Wolf, A.J.; Spira, M.; Gulinello, M.E.; Cole, P.D. Methotrexate causes persistent deficits in memory and executive function in a juvenile animal model. Neuropharmacology 2018, 139, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-C.; Sheen, J.-M.; Hsu, M.-H.; Hsiao, C.-C.; Wang, S.-C.; Huang, L.-T. Melatonin rescued methotrexate-induced spatial deficit and hyperhomocysteinemia and increased asymmetric dimethylarginine in plasma and dorsal hippocampus in developing rats. Life Sci. 2020, 242, 116931. [Google Scholar] [CrossRef]
- Chen, Y.-C.; Sheen, J.-M.; Wang, S.-C.; Hsu, M.-H.; Hsiao, C.-C.; Chang, K.-A.; Huang, L.-T. Methotrexate Neurotoxicity Is Related to Epigenetic Modification of the Myelination Process. Int. J. Mol. Sci. 2021, 22, 6718. [Google Scholar] [CrossRef]
- Zhou, B.; Xia, X.; Wang, P.; Chen, S.; Yu, C.; Huang, R.; Zhang, R.; Wang, Y.; Lu, L.; Yuan, F.; et al. Induction and Amelioration of Methotrexate-Induced Gastrointestinal Toxicity Are Related to Immune Response and Gut Microbiota. eBioMedicine 2018, 33, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Letertre, M.P.M.; Munjoma, N.; Wolfer, K.; Pechlivanis, A.; McDonald, J.A.K.; Hardwick, R.N.; Cherrington, N.J.; Coen, M.; Nicholson, J.K.; Hoyles, L.; et al. A Two-Way Interaction between Methotrexate and the Gut Microbiota of Male Sprague–Dawley Rats. J. Proteome Res. 2020, 19, 3326–3339. [Google Scholar] [CrossRef]
- Huang, X.; Fang, Q.; Rao, T.; Zhou, L.; Zeng, X.; Tan, Z.; Chen, L.; Ouyang, D. Leucovorin Ameliorated Methotrexate Induced Intestinal Toxicity via Modulation of the Gut Microbiota. Toxicol. Appl. Pharmacol. 2020, 391, 114900. [Google Scholar] [CrossRef]
- Tang, D.; Zeng, T.; Wang, Y.; Cui, H.; Wu, J.; Zou, B.; Tao, Z.; Zhang, L.; Garside, G.B.; Tao, S. Dietary restriction increases protective gut bacteria to rescue lethal methotrexate-induced intestinal toxicity. Gut Microbes 2020, 12, 1714401. [Google Scholar] [CrossRef]
- Luo, Y.; Zhou, T. Connecting the dots: Targeting the microbiome in drug toxicity. Med. Res. Rev. 2022, 42, 83–111. [Google Scholar] [CrossRef]
- Sherwin, E.; Dinan, T.G.; Cryan, J.F. Recent developments in understanding the role of the gut microbiota in brain health and disease. Ann. N. Y. Acad. Sci. 2018, 1420, 5–25. [Google Scholar] [CrossRef]
- Mayneris-Perxachs, J.; Castells-Nobau, A.; Arnoriaga-Rodríguez, M.; Martin, M.; de la Vega-Correa, L.; Zapata, C.; Burokas, A.; Blasco, G.; Coll, C.; Escrichs, A.; et al. Microbiota alterations in proline metabolism impact depression. Cell Metab. 2022, 34, 681–701.e10. [Google Scholar] [CrossRef]
- Zhang, Y.; Fan, Q.; Hou, Y.; Zhang, X.; Yin, Z.; Cai, X.; Wei, W.; Wang, J.; He, D.; Wang, G.; et al. Bacteroides species differentially modulate depression-like behavior via gut-brain metabolic signaling. Brain Behav. Immun. 2022, 102, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, C.B.; Bowen, J.M.; Gladman, M.A.; Lustberg, M.B.; Mayo, S.J.; Wardill, H.R. The microbiota-gut-brain axis: An emerging therapeutic target in chemotherapy-induced cognitive impairment. Neurosci. Biobehav. Rev. 2020, 116, 470–479. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.T.; Chen, C.C.; Sheen, J.M.; Chen, Y.J.; Hsieh, C.S.; Tain, Y.L. The interaction between high ammonia diet and bileduct ligation in developing rats: Assessment by spatial memory and asymmetric dimethylarginine. Int. J. Dev. Neurosci. 2010, 28, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Vijayanathan, V.; Gulinello, M.; Cole, P.D. Intrathecal methotrexate induces focal cognitive deficits and increases cerebrospinal fluid homocysteine. Pharmacol. Biochem. Behav. 2010, 95, 428–433. [Google Scholar] [CrossRef] [PubMed]
- Cole, P.D.; Vijayanathan, V.; Ali, N.F.; Wagshul, M.E.; Tanenbaum, E.J.; Price, J.; Dalal, V.; Gulinello, M.E. Memantine protects rats treated with intrathecal methotrexate from developing spatial memory deficits. Clin. Cancer Res. 2013, 19, 4446–4454. [Google Scholar] [CrossRef] [PubMed]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Gonzalez Peña, A.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Bokulich, N.A.; Subramanian, S.; Faith, J.J.; Gevers, D.; Gordon, J.I.; Knight, R.; Mills, D.A.; Caporaso, J.G. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 2013, 10, 57–59. [Google Scholar] [CrossRef]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef]
- Haas, B.J.; Gevers, D.; Earl, A.M.; Feldgarden, M.; Ward, D.V.; Giannoukos, G.; Ciulla, D.; Tabbaa, D.; Highlander, S.K.; Sodergren, E.; et al. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 2011, 21, 494–504. [Google Scholar] [CrossRef]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Yilmaz, P.; Parfrey, L.W.; Yarza, P.; Gerken, J.; Pruesse, E.; Quast, C.; Schweer, T.; Peplies, J.; Ludwig, W.; Glöckner, F.O. The SILVA and “All-species Living Tree Project (LTP)” taxonomic frameworks. Nucleic Acids Res. 2014, 42, D643–D648. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree: Computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 2009, 26, 1641–1650. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Whittaker, R.H. Evolution and measurement of species diversity. Taxon 1972, 21, 213–251. [Google Scholar] [CrossRef]
- Jiang, X.-T.; Peng, X.; Deng, G.-H.; Sheng, H.-F.; Wang, Y.; Zhou, H.-W.; Tam, N.F.-Y. Illumina sequencing of 16S rRNA tag revealed spatial variations of bacterial communities in a mangrove wetland. Microb. Ecol. 2013, 66, 96–104. [Google Scholar] [CrossRef]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Aßhauer, K.P.; Wemheuer, B.; Daniel, R.; Meinicke, P. Tax4Fun: Predicting functional profiles from metagenomic 16S rRNA data. Bioinformatics 2015, 31, 2882–2884. [Google Scholar] [CrossRef]
- Sloan, H.L.; Good, M.; Dunnett, S.B. Double dissociation between hippocampal and prefrontal lesions on an operant delayed matching task and a water maze reference memory task. Behav. Brain Res. 2006, 171, 116–126. [Google Scholar] [CrossRef]
- Hsu, C.-N.; Hou, C.-Y.; Chan, J.Y.H.; Lee, C.-T.; Tain, Y.-L. Hypertension Programmed by Perinatal High-Fat Diet: Effect of Maternal Gut Microbiota-Targeted Therapy. Nutrients 2019, 11, 2908. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.-M.T.; Malec, P.A.; Mabrouk, O.S.; Ro, J.; Dus, M.; Kennedy, R.T. Benzoyl chloride derivatization with liquid chromatography–mass spectrometry for targeted metabolomics of neurochemicals in biological samples. J. Chromatogr. A 2016, 1446, 78–90. [Google Scholar] [CrossRef] [PubMed]
- Addona, T.A.; Abbatiello, S.E.; Schilling, B.; Skates, S.J.; Mani, D.R.; Bunk, D.M.; Spiegelman, C.H.; Zimmerman, L.J.; Ham, A.-J.L.; Keshishian, H.; et al. Multi-site assessment of the precision and reproducibility of multiple reaction monitoring–based measurements of proteins in plasma. Nat. Biotechnol. 2009, 27, 633–641. [Google Scholar] [CrossRef]
- Mani, D.R.; Abbatiello, S.E.; Carr, S.A. Statistical characterization of multiple-reaction monitoring mass spectrometry (MRM-MS) assays for quantitative proteomics. BMC Bioinform. 2012, 13 (Suppl. 16), S9. [Google Scholar] [CrossRef]
- Paulson, J.N.; Stine, O.C.; Bravo, H.C.; Pop, M. Differential abundance analysis for microbial marker-gene surveys. Nat. Methods 2013, 10, 1200–1202. [Google Scholar] [CrossRef]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef]
- Angelucci, F.; Cechova, K.; Amlerova, J.; Hort, J. Antibiotics, gut microbiota, and Alzheimer’s disease. J. Neuroinflamm. 2019, 16, 108. [Google Scholar] [CrossRef]
- Megur, A.; Baltriukienė, D.; Bukelskienė, V.; Burokas, A. The Microbiota-Gut-Brain Axis and Alzheimer’s Disease: Neuroinflammation Is to Blame? Nutrients 2020, 13, 37. [Google Scholar] [CrossRef] [PubMed]
- Raben, D.M.; Barber, C.N. Phosphatidic acid and neurotransmission. Adv. Biol. Regul. 2017, 63, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Barber, C.N.; Goldschmidt, H.L.; Lilley, B.; Bygrave, A.M.; Johnson, R.C.; Huganir, R.L.; Zack, D.J.; Raben, D.M. Differential expression patterns of phospholipase D isoforms 1 and 2 in the mammalian brain and retina. J. Lipid Res. 2022, 63, 100247. [Google Scholar] [CrossRef] [PubMed]
- Roza, C.; Campos-Sandoval, J.A.; Gómez-García, M.C.; Peñalver, A.; Márquez, J. Lysophosphatidic Acid and Glutamatergic Transmission. Front. Mol. Neurosci. 2019, 12, 138. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Huang, Y.; Zhao, L.; Li, Y.; Zheng, J. Glutaminase 1 is essential for the differentiation, proliferation, and survival of human neural progenitor cells. Stem Cells Dev. 2014, 23, 2782–2790. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.-C.; Shieh, Y.-D.; Chien, Y.-H.; Tzen, K.-Y.; Yu, I.-S.; Chen, P.-W.; Hu, M.-H.; Hu, M.-K.; Muramatsu, S.-I.; Ichinose, H.; et al. Regulation of the dopaminergic system in a murine model of aromatic L-amino acid decarboxylase deficiency. Neurobiol. Dis. 2013, 52, 177–190. [Google Scholar] [CrossRef] [PubMed]
- Shih, D.-F.; Hsiao, C.-D.; Min, M.-Y.; Lai, W.-S.; Yang, C.-W.; Lee, W.-T.; Lee, S.-J. Aromatic L-amino acid decarboxylase (AADC) is crucial for brain development and motor functions. PLoS ONE 2013, 8, e71741. [Google Scholar] [CrossRef]
- Trifonov, S.; Yamashita, Y.; Kase, M.; Maruyama, M.; Sugimoto, T. Glutamic acid decarboxylase 1 alternative splicing isoforms: Characterization, expression and quantification in the mouse brain. BMC Neurosci. 2014, 15, 114. [Google Scholar] [CrossRef] [PubMed]
- Tao, R.; Davis, K.N.; Li, C.; Shin, J.H.; Gao, Y.; Jaffe, A.E.; Gondré-Lewis, M.C.; Weinberger, D.R.; Kleinman, J.E.; Hyde, T.M. GAD1 alternative transcripts and DNA methylation in human prefrontal cortex and hippocampus in brain development, schizophrenia. Mol. Psychiatry 2018, 23, 1496–1505. [Google Scholar] [CrossRef] [PubMed]
- O’Riordan, K.J.; Collins, M.K.; Moloney, G.M.; Knox, E.G.; Aburto, M.R.; Fülling, C.; Morley, S.J.; Clarke, G.; Schellekens, H.; Cryan, J.F. Short chain fatty acids: Microbial metabolites for gut-brain axis signalling. Mol. Cell. Endocrinol. 2022, 546, 111572. [Google Scholar] [CrossRef]
- Rekha, K.; Venkidasamy, B.; Samynathan, R.; Nagella, P.; Rebezov, M.; Khayrullin, M.; Ponomarev, E.; Bouyahya, A.; Sarkar, T.; Shariati, M.A.; et al. Short-chain fatty acid: An updated review on signaling, metabolism, and therapeutic effects. Crit. Rev. Food Sci. Nutr. 2022, 64, 2461–2489. [Google Scholar] [CrossRef] [PubMed]
- Borre, Y.E.; O’keeffe, G.W.; Clarke, G.; Stanton, C.; Dinan, T.G.; Cryan, J.F. Microbiota and neurodevelopmental windows: Implications for brain disorders. Trends Mol. Med. 2014, 20, 509–518. [Google Scholar] [CrossRef]
- O’Mahony, S.M.; Clarke, G.; Borre, Y.E.; Dinan, T.G.; Cryan, J.F. Serotonin, tryptophan metabolism and the brain-gut-microbiome axis. Behav. Brain Res. 2015, 277, 32–48. [Google Scholar] [CrossRef] [PubMed]
- Fung, T.C.; Olson, C.A.; Hsiao, E.Y. Interactions between the microbiota, immune and nervous systems in health and disease. Nat. Neurosci. 2017, 20, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Dalile, B.; Van Oudenhove, L.; Vervliet, B.; Verbeke, K. The role of short-chain fatty acids in microbiota–gut–brain communication. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 461–478. [Google Scholar] [CrossRef] [PubMed]
- Dinan, T.G.; Cryan, J.F. Microbes, Immunity, and Behavior: Psychoneuroimmunology Meets the Microbiome. Neuropsychopharmacology 2017, 42, 178–192. [Google Scholar] [CrossRef]
- Higuchi, T.; Yoshimura, M.; Oka, S.; Tanaka, K.; Naito, T.; Yuhara, S.; Warabi, E.; Mizuno, S.; Ono, M.; Takahashi, S.; et al. Modulation of methotrexate-induced intestinal mucosal injury by dietary factors. Hum. Exp. Toxicol. 2020, 39, 500–513. [Google Scholar] [CrossRef] [PubMed]
- Wardill, H.R.; da Silva Ferreira, A.R.; Cloo, S.L.; Havinga, R.; Harmsen, H.J.M.; Vermeij, W.P.; Tissing, W.J.E. Pre-therapy fasting slows epithelial turnover and modulates the microbiota but fails to mitigate methotrexate-induced gastrointestinal mucositis. Gut Microbes 2020, 12, 1809332. [Google Scholar] [CrossRef] [PubMed]
- Llorenç, V.; Nakamura, Y.; Metea, C.; Karstens, L.; Molins, B.; Lin, P. Antimetabolite Drugs Exhibit Distinctive Immunomodulatory Mechanisms and Effects on the Intestinal Microbiota in Experimental Autoimmune Uveitis. Investig. Opthalmol. Vis. Sci. 2022, 63, 30. [Google Scholar] [CrossRef]
- Nomura, M.; Nagatomo, R.; Doi, K.; Shimizu, J.; Baba, K.; Saito, T.; Matsumoto, S.; Inoue, K.; Muto, M. Association of Short-Chain Fatty Acids in the Gut Microbiome With Clinical Response to Treatment With Nivolumab or Pembrolizumab in Patients With Solid Cancer Tumors. JAMA Netw. Open 2020, 3, e202895. [Google Scholar] [CrossRef]
- Chen, S.-J.; Chen, C.-C.; Liao, H.-Y.; Lin, Y.-T.; Wu, Y.-W.; Liou, J.-M.; Wu, M.-S.; Kuo, C.-H.; Lin, C.-H. Association of Fecal and Plasma Levels of Short-Chain Fatty Acids With Gut Microbiota and Clinical Severity in Patients With Parkinson Disease. Neurology 2022, 98, e848–e858. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Predicted Pattern | Verified Genes | Cortex | Hippo | |
|---|---|---|---|---|
| (XTP vs. CTP) | (XTP vs. CTP) | (XTP vs. CTP) | ||
| SCFAs Gene | ||||
| Acads | - | Acads | - | - |
| Acat | ↓ | Acat1 | ↑↑ | ↓ |
| Acat2 | - | ↓↓ | ||
| Acot12 | - | Acot12 | ↑ | ↑ |
| Acss1/2 | ↑ | Acss1 | ↑ | ↑ |
| Acss2 | - | - | ||
| Acss3 | ↓ | Acss3 | ↓ | - |
| Ldha | ↓↓ | Ldha | - | - |
| Pkm | ↓ | Pkm | ↑↑ | ↓ |
| Glutamatergic and GABAergic synapse genes | ||||
| Pld1/2 | ↓ | Pld1 | ↓ | - |
| Pld2 | - | - | ||
| Gls | - | Gls | ↑ | - |
| Gls2 | ↑ | - | ||
| Glul | ↑ | Glul | ↑ | ↑↑ |
| Neurotransmitter genes | ||||
| Tyr | ↓ | Tyr | ↑↑ | - |
| Ddc | ↓↓ | Ddc | - | - |
| Ggt6/7 | ↓ | Ggt6 | ↑ | ↓ |
| Ggt7 | - | - | ||
| Gad1/2 | ↑ | Gad1 | ↑↑ | ↓↓ |
| Gad2 | ↑ | - | ||
| Glul | ↑ | Glul | ↑ | ↑↑ |
| Comt | ↓ | Comt | ↓ | - |
| Maoa | ↓ | Maoa | - | ↓ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.-C.; Hou, C.-Y.; Hsu, M.-H.; Huang, L.-T.; Hsiao, C.-C.; Sheen, J.-M. The Impact of Gut Microbiota Changes on Methotrexate-Induced Neurotoxicity in Developing Young Rats. Biomedicines 2024, 12, 908. https://doi.org/10.3390/biomedicines12040908
Chen Y-C, Hou C-Y, Hsu M-H, Huang L-T, Hsiao C-C, Sheen J-M. The Impact of Gut Microbiota Changes on Methotrexate-Induced Neurotoxicity in Developing Young Rats. Biomedicines. 2024; 12(4):908. https://doi.org/10.3390/biomedicines12040908
Chicago/Turabian StyleChen, Yu-Chieh, Chih-Yao Hou, Mei-Hsin Hsu, Li-Tung Huang, Chih-Cheng Hsiao, and Jiunn-Ming Sheen. 2024. "The Impact of Gut Microbiota Changes on Methotrexate-Induced Neurotoxicity in Developing Young Rats" Biomedicines 12, no. 4: 908. https://doi.org/10.3390/biomedicines12040908
APA StyleChen, Y. -C., Hou, C. -Y., Hsu, M. -H., Huang, L. -T., Hsiao, C. -C., & Sheen, J. -M. (2024). The Impact of Gut Microbiota Changes on Methotrexate-Induced Neurotoxicity in Developing Young Rats. Biomedicines, 12(4), 908. https://doi.org/10.3390/biomedicines12040908