


Exploration of Gene Therapy for Alport Syndrome

Abstract
:1. Overview of Alport Syndrome
2. What Are Gene Therapy and Gene Therapy Vectors?
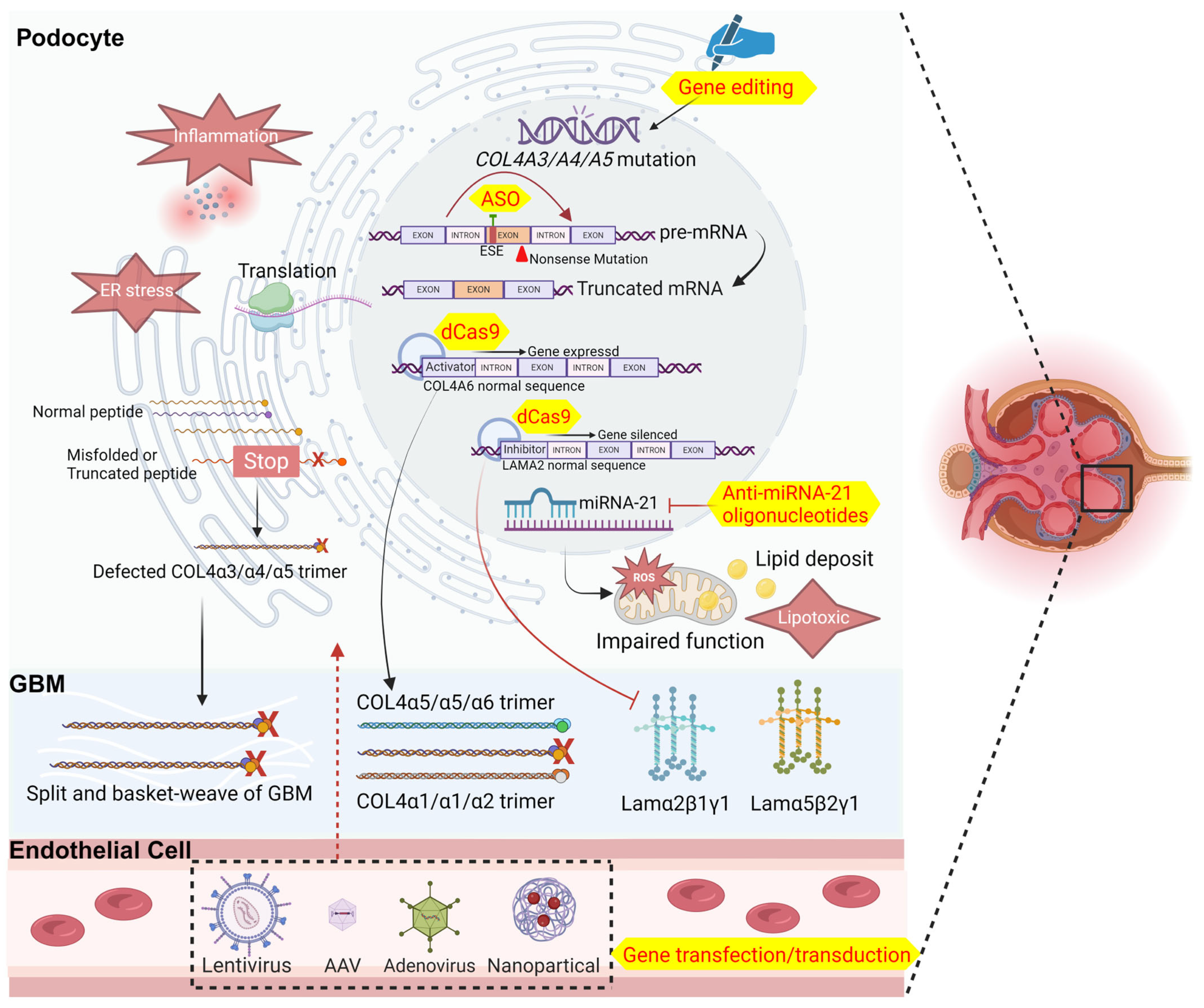
3. Available Attempts at Gene Therapy for Alport Syndrome
4. Difficulties Facing Gene Therapy for Kidney Diseases
5. Summary and Prospects
Funding
Conflicts of Interest
References
- Kalluri, R.; Shield, C.F.; Todd, P.; Hudson, B.G.; Neilson, E.G. Isoform switching of type IV collagen is developmentally arrested in X-linked Alport syndrome leading to increased susceptibility of renal basement membranes to endoproteolysis. J. Clin. Investig. 1997, 99, 2470–2478. [Google Scholar] [CrossRef]
- Mahrous, N.N.; Jamous, Y.F.; Almatrafi, A.M.; Fallatah, D.I.; Theyab, A.; Alanati, B.H.; Alsagaby, S.A.; Alenazi, M.K.; Khan, M.I.; Hawsawi, Y.M. A Current Landscape on Alport Syndrome Cases: Characterization, Therapy and Management Perspectives. Biomedicines 2023, 11, 2762. [Google Scholar] [CrossRef] [PubMed]
- Suh, J.H.; Miner, J.H. The glomerular basement membrane as a barrier to albumin. Nat. Rev. Nephrol. 2013, 9, 470–477. [Google Scholar] [CrossRef]
- Boutaud, A.; Borza, D.B.; Bondar, O.; Gunwar, S.; Netzer, K.O.; Singh, N.; Ninomiya, Y.; Sado, Y.; Noelken, M.E.; Hudson, B.G. Type IV collagen of the glomerular basement membrane. Evidence that the chain specificity of network assembly is encoded by the noncollagenous NC1 domains. J. Biol. Chem. 2000, 275, 30716–30724. [Google Scholar] [CrossRef]
- Gregorio, V.; Caparali, E.B.; Shojaei, A.; Ricardo, S.; Barua, M. Alport Syndrome: Clinical Spectrum and Therapeutic Advances. Kidney Med. 2023, 5, 100631. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Suh, J.H.; Go, G.; Miner, J.H. Feasibility of repairing glomerular basement membrane defects in Alport syndrome. J. Am. Soc. Nephrol. 2014, 25, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Warady, B.A.; Agarwal, R.; Bangalore, S.; Chapman, A.; Levin, A.; Stenvinkel, P.; Toto, R.D.; Chertow, G.M. Alport Syndrome Classification and Management. Kidney Med. 2020, 2, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Hasstedt, S.J.; Atkin, C.L. X-linked inheritance of Alport syndrome: Family P revisited. Am. J. Hum. Genet. 1983, 35, 1241–1251. [Google Scholar] [PubMed]
- Persson, U.; Hertz, J.M.; Wieslander, J.; Segelmark, M. Alport syndrome in southern Sweden. Clin. Nephrol. 2005, 64, 85–90. [Google Scholar] [CrossRef]
- Pajari, H.; Kaariainen, H.; Muhonen, T.; Koskimies, O. Alport’s syndrome in 78 patients: Epidemiological and clinical study. Acta Paediatr. 1996, 85, 1300–1306. [Google Scholar] [CrossRef]
- Chakravarti, S.; Enzo, E.; Rocha Monteiro de Barros, M.; Maffezzoni, M.B.R.; Pellegrini, G. Genetic Disorders of the Extracellular Matrix: From Cell and Gene Therapy to Future Applications in Regenerative Medicine. Annu. Rev. Genom. Hum. Genet. 2022, 23, 193–222. [Google Scholar] [CrossRef]
- Gibson, J.; Fieldhouse, R.; Chan, M.M.Y.; Sadeghi-Alavijeh, O.; Burnett, L.; Izzi, V.; Persikov, A.V.; Gale, D.P.; Storey, H.; Savige, J.; et al. Prevalence Estimates of Predicted Pathogenic COL4A3-COL4A5 Variants in a Population Sequencing Database and Their Implications for Alport Syndrome. J. Am. Soc. Nephrol. 2021, 32, 2273–2290. [Google Scholar] [CrossRef] [PubMed]
- Groopman, E.E.; Marasa, M.; Cameron-Christie, S.; Petrovski, S.; Aggarwal, V.S.; Milo-Rasouly, H.; Li, Y.; Zhang, J.; Nestor, J.; Krithivasan, P.; et al. Diagnostic Utility of Exome Sequencing for Kidney Disease. N. Engl. J. Med. 2019, 380, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Leenen, E.; Erger, F.; Altmuller, J.; Wenzel, A.; Thiele, H.; Harth, A.; Tschernoster, N.; Lokhande, S.; Joerres, A.; Becker, J.U.; et al. Alport syndrome and autosomal dominant tubulointerstitial kidney disease frequently underlie end-stage renal disease of unknown origin-a single-center analysis. Nephrol. Dial. Transplant. 2022, 37, 1895–1905. [Google Scholar] [CrossRef]
- Xie, J.Y.; Wu, X.X.; Ren, H.; Wang, W.X.; Wang, Z.H.; Pan, X.X.; Hao, X.; Tong, J.; Ma, J.; Ye, Z.B.; et al. COL4A3 mutations cause focal segmental glomerulosclerosis. J. Mol. Cell Biol. 2014, 6, 498–505. [Google Scholar] [CrossRef]
- Pieri, M.; Stefanou, C.; Zaravinos, A.; Erguler, K.; Stylianou, K.; Lapathitis, G.; Karaiskos, C.; Savva, I.; Paraskeva, R.; Dweep, H.; et al. Evidence for activation of the unfolded protein response in collagen IV nephropathies. J. Am. Soc. Nephrol. 2014, 25, 260–275. [Google Scholar] [CrossRef]
- Rubel, D.; Frese, J.; Martin, M.; Leibnitz, A.; Girgert, R.; Miosge, N.; Eckes, B.; Muller, G.A.; Gross, O. Collagen receptors integrin alpha2beta1 and discoidin domain receptor 1 regulate maturation of the glomerular basement membrane and loss of integrin alpha2beta1 delays kidney fibrosis in COL4A3 knockout mice. Matrix Biol. 2014, 34, 13–21. [Google Scholar] [CrossRef]
- Gross, O.; Girgert, R.; Beirowski, B.; Kretzler, M.; Kang, H.G.; Kruegel, J.; Miosge, N.; Busse, A.C.; Segerer, S.; Vogel, W.F.; et al. Loss of collagen-receptor DDR1 delays renal fibrosis in hereditary type IV collagen disease. Matrix Biol. 2010, 29, 346–356. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.J.; David, J.M.; Wilbon, S.S.; Santos, J.V.; Patel, D.M.; Ahmad, A.; Mitrofanova, A.; Liu, X.; Mallela, S.K.; Ducasa, G.M.; et al. Discoidin domain receptor 1 activation links extracellular matrix to podocyte lipotoxicity in Alport syndrome. EBioMedicine 2021, 63, 103162. [Google Scholar] [CrossRef]
- Ding, W.; Yousefi, K.; Goncalves, S.; Goldstein, B.J.; Sabater, A.L.; Kloosterboer, A.; Ritter, P.; Lambert, G.; Mendez, A.J.; Shehadeh, L.A. Osteopontin deficiency ameliorates Alport pathology by preventing tubular metabolic deficits. JCI Insight 2018, 3, e94818. [Google Scholar] [CrossRef]
- Tong, J.; Zheng, Q.M.; Gu, X.C.; Weng, Q.J.; Yu, S.W.; Fang, Z.Y.; Hafiz, M.; Xu, J.; Ren, H.; Chen, N.; et al. COL4A3 Mutation Induced Podocyte Apoptosis by Dysregulation of NADPH Oxidase 4 and MMP-2. Kidney Int. Rep. 2023, 8, 1864–1874. [Google Scholar] [CrossRef] [PubMed]
- Stenvinkel, P.; Chertow, G.M.; Devarajan, P.; Levin, A.; Andreoli, S.P.; Bangalore, S.; Warady, B.A. Chronic Inflammation in Chronic Kidney Disease Progression: Role of Nrf2. Kidney Int. Rep. 2021, 6, 1775–1787. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, C.; Jayasinghe, K. Bardoxolone Methyl for Alport Syndrome: Opportunities and Challenges. Clin. J. Am. Soc. Nephrol. 2022, 17, 1713–1715. [Google Scholar] [CrossRef]
- Gross, O.; Tonshoff, B.; Weber, L.T.; Pape, L.; Latta, K.; Fehrenbach, H.; Lange-Sperandio, B.; Zappel, H.; Hoyer, P.; Staude, H.; et al. A multicenter, randomized, placebo-controlled, double-blind phase 3 trial with open-arm comparison indicates safety and efficacy of nephroprotective therapy with ramipril in children with Alport’s syndrome. Kidney Int. 2020, 97, 1275–1286. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.D.; Huang, J.N.; Liu, Y.Z.; Ren, H.; Xie, J.Y.; Chen, N. Endoplasmic reticulum stress and proteasome pathway involvement in human podocyte injury with a truncated COL4A3 mutation. Chin. Med. J. 2019, 132, 1823–1832. [Google Scholar] [CrossRef]
- Daga, S.; Ding, J.; Deltas, C.; Savige, J.; Lipska-Ziętkiewicz, B.S.; Hoefele, J.; Flinter, F.; Gale, D.P.; Aksenova, M.; Kai, H.; et al. The 2019 and 2021 International Workshops on Alport Syndrome. Eur. J. Hum. Genet. 2022, 30, 507–516. [Google Scholar] [CrossRef]
- Kumar, S.R.; Markusic, D.M.; Biswas, M.; High, K.A.; Herzog, R.W. Clinical development of gene therapy: Results and lessons from recent successes. Mol. Ther. Methods Clin. Dev. 2016, 3, 16034. [Google Scholar] [CrossRef]
- Gene therapies should be for all. Nat. Med. 2021, 27, 1311. [CrossRef]
- Munis, A.M. Gene Therapy Applications of Non-Human Lentiviral Vectors. Viruses 2020, 12, 1106. [Google Scholar] [CrossRef]
- Jiang, Z.; Dalby, P.A. Challenges in scaling up AAV-based gene therapy manufacturing. Trends Biotechnol. 2023, 41, 1268–1281. [Google Scholar] [CrossRef]
- Crystal, R.G. Adenovirus: The first effective in vivo gene delivery vector. Hum. Gene Ther. 2014, 25, 3–11. [Google Scholar] [CrossRef]
- Sato-Dahlman, M.; LaRocca, C.J.; Yanagiba, C.; Yamamoto, M. Adenovirus and Immunotherapy: Advancing Cancer Treatment by Combination. Cancers 2020, 12, 1295. [Google Scholar] [CrossRef]
- Somanathan, S.; Calcedo, R.; Wilson, J.M. Adenovirus-Antibody Complexes Contributed to Lethal Systemic Inflammation in a Gene Therapy Trial. Mol. Ther. 2020, 28, 784–793. [Google Scholar] [CrossRef]
- Wang, A.Y.; Peng, P.D.; Ehrhardt, A.; Storm, T.A.; Kay, M.A. Comparison of adenoviral and adeno-associated viral vectors for pancreatic gene delivery in vivo. Hum. Gene Ther. 2004, 15, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Jacob-Dolan, C.; Barouch, D.H. COVID-19 Vaccines: Adenoviral Vectors. Annu. Rev. Med. 2022, 73, 41–54. [Google Scholar] [CrossRef]
- Cavalieri, V.; Baiamonte, E.; Lo Iacono, M. Non-Primate Lentiviral Vectors and Their Applications in Gene Therapy for Ocular Disorders. Viruses 2018, 10, 316. [Google Scholar] [CrossRef] [PubMed]
- Gene therapy needs a long-term approach. Nat. Med. 2021, 27, 563. [CrossRef]
- Goyal, S.; Tisdale, J.; Schmidt, M.; Kanter, J.; Jaroscak, J.; Whitney, D.; Bitter, H.; Gregory, P.D.; Parsons, G.; Foos, M.; et al. Acute Myeloid Leukemia Case after Gene Therapy for Sickle Cell Disease. N. Engl. J. Med. 2022, 386, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Gandara, C.; Affleck, V.; Stoll, E.A. Manufacture of Third-Generation Lentivirus for Preclinical Use, with Process Development Considerations for Translation to Good Manufacturing Practice. Hum. Gene Ther. Methods 2018, 29, 1–15. [Google Scholar] [CrossRef]
- Gurumoorthy, N.; Nordin, F.; Tye, G.J.; Wan Kamarul Zaman, W.S.; Ng, M.H. Non-Integrating Lentiviral Vectors in Clinical Applications: A Glance Through. Biomedicines 2022, 10, 107. [Google Scholar] [CrossRef]
- Parker, C.L.; Jacobs, T.M.; Huckaby, J.T.; Harit, D.; Lai, S.K. Efficient and Highly Specific Gene Transfer Using Mutated Lentiviral Vectors Redirected with Bispecific Antibodies. mBio 2020, 11, e02990-19. [Google Scholar] [CrossRef] [PubMed]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef] [PubMed]
- Feins, S.; Kong, W.; Williams, E.F.; Milone, M.C.; Fraietta, J.A. An introduction to chimeric antigen receptor (CAR) T-cell immunotherapy for human cancer. Am. J. Hematol. 2019, 94, S3–S9. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, N.L.; Berguig, G.Y.; Karim, O.A.; Cortesio, C.L.; De Angelis, R.; Khan, A.A.; Gold, D.; Maga, J.A.; Bhat, V.S. Comprehensive characterization and quantification of adeno associated vectors by size exclusion chromatography and multi angle light scattering. Sci. Rep. 2021, 11, 3012. [Google Scholar] [CrossRef]
- Kaplitt, M.G.; Xiao, X.; Samulski, R.J.; Li, J.; Ojamaa, K.; Klein, I.L.; Makimura, H.; Kaplitt, M.J.; Strumpf, R.K.; Diethrich, E.B. Long-term gene transfer in porcine myocardium after coronary infusion of an adeno-associated virus vector. Ann. Thorac. Surg. 1996, 62, 1669–1676. [Google Scholar] [CrossRef]
- Li, C.; Samulski, R.J. Engineering adeno-associated virus vectors for gene therapy. Nat. Rev. Genet. 2020, 21, 255–272. [Google Scholar] [CrossRef] [PubMed]
- Vandamme, C.; Adjali, O.; Mingozzi, F. Unraveling the Complex Story of Immune Responses to AAV Vectors Trial After Trial. Hum. Gene Ther. 2017, 28, 1061–1074. [Google Scholar] [CrossRef] [PubMed]
- Louis Jeune, V.; Joergensen, J.A.; Hajjar, R.J.; Weber, T. Pre-existing anti-adeno-associated virus antibodies as a challenge in AAV gene therapy. Hum. Gene Ther. Methods 2013, 24, 59–67. [Google Scholar] [CrossRef]
- Kimura, T.; Ferran, B.; Tsukahara, Y.; Shang, Q.; Desai, S.; Fedoce, A.; Pimentel, D.R.; Luptak, I.; Adachi, T.; Ido, Y.; et al. Production of adeno-associated virus vectors for in vitro and in vivo applications. Sci. Rep. 2019, 9, 13601. [Google Scholar] [CrossRef]
- Hinderer, C.; Katz, N.; Buza, E.L.; Dyer, C.; Goode, T.; Bell, P.; Richman, L.K.; Wilson, J.M. Severe Toxicity in Nonhuman Primates and Piglets Following High-Dose Intravenous Administration of an Adeno-Associated Virus Vector Expressing Human SMN. Hum. Gene Ther. 2018, 29, 285–298. [Google Scholar] [CrossRef]
- Koeberl, D.; Schulze, A.; Sondheimer, N.; Lipshutz, G.S.; Geberhiwot, T.; Li, L.; Saini, R.; Luo, J.; Sikirica, V.; Jin, L.; et al. Interim analyses of a first-in-human phase 1/2 mRNA trial for propionic acidaemia. Nature 2024, 628, 872–877. [Google Scholar] [CrossRef] [PubMed]
- Heidet, L.; Cai, Y.; Guicharnaud, L.; Antignac, C.; Gubler, M.C. Glomerular expression of type IV collagen chains in normal and X-linked Alport syndrome kidneys. Am. J. Pathol. 2000, 156, 1901–1910. [Google Scholar] [CrossRef] [PubMed]
- Harvey, S.J.; Zheng, K.; Jefferson, B.; Moak, P.; Sado, Y.; Naito, I.; Ninomiya, Y.; Jacobs, R.; Thorner, P.S. Transfer of the alpha 5(IV) collagen chain gene to smooth muscle restores in vivo expression of the alpha 6(IV) collagen chain in a canine model of Alport syndrome. Am. J. Pathol. 2003, 162, 873–885. [Google Scholar] [CrossRef] [PubMed]
- Heikkila, P.; Parpala, T.; Lukkarinen, O.; Weber, M.; Tryggvason, K. Adenovirus-mediated gene transfer into kidney glomeruli using an ex vivo and in vivo kidney perfusion system—First steps towards gene therapy of Alport syndrome. Gene Ther. 1996, 3, 21–27. [Google Scholar] [PubMed]
- Heikkila, P.; Tibell, A.; Morita, T.; Chen, Y.; Wu, G.; Sado, Y.; Ninomiya, Y.; Pettersson, E.; Tryggvason, K. Adenovirus-mediated transfer of type IV collagen alpha5 chain cDNA into swine kidney in vivo: Deposition of the protein into the glomerular basement membrane. Gene Ther. 2001, 8, 882–890. [Google Scholar] [CrossRef] [PubMed]
- Gomez, I.G.; MacKenna, D.A.; Johnson, B.G.; Kaimal, V.; Roach, A.M.; Ren, S.; Nakagawa, N.; Xin, C.; Newitt, R.; Pandya, S.; et al. Anti-microRNA-21 oligonucleotides prevent Alport nephropathy progression by stimulating metabolic pathways. J. Clin. Investig. 2015, 125, 141–156. [Google Scholar] [CrossRef]
- Guo, J.; Song, W.; Boulanger, J.; Xu, E.Y.; Wang, F.; Zhang, Y.; He, Q.; Wang, S.; Yang, L.; Pryce, C.; et al. Dysregulated Expression of microRNA-21 and Disease-Related Genes in Human Patients and in a Mouse Model of Alport Syndrome. Hum. Gene Ther. 2019, 30, 865–881. [Google Scholar] [CrossRef] [PubMed]
- Yamamura, T.; Horinouchi, T.; Adachi, T.; Terakawa, M.; Takaoka, Y.; Omachi, K.; Takasato, M.; Takaishi, K.; Shoji, T.; Onishi, Y.; et al. Development of an exon skipping therapy for X-linked Alport syndrome with truncating variants in COL4A5. Nat. Commun. 2020, 11, 2777. [Google Scholar] [CrossRef]
- Daga, S.; Baldassarri, M.; Lo Rizzo, C.; Fallerini, C.; Imperatore, V.; Longo, I.; Frullanti, E.; Landucci, E.; Massella, L.; Pecoraro, C.; et al. Urine-derived podocytes-lineage cells: A promising tool for precision medicine in Alport Syndrome. Hum. Mutat. 2018, 39, 302–314. [Google Scholar] [CrossRef]
- Daga, S.; Donati, F.; Capitani, K.; Croci, S.; Tita, R.; Giliberti, A.; Valentino, F.; Benetti, E.; Fallerini, C.; Niccheri, F.; et al. New frontiers to cure Alport syndrome: COL4A3 and COL4A5 gene editing in podocyte-lineage cells. Eur. J. Hum. Genet. 2020, 28, 480–490. [Google Scholar] [CrossRef]
- Omachi, K.; Miner, J.H. Comparative analysis of dCas9-VP64 variants and multiplexed guide RNAs mediating CRISPR activation. PLoS ONE 2022, 17, e0270008. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Le, Y.; Zhang, Z.; Nian, X.; Liu, B.; Yang, X. Viral Vector-Based Gene Therapy. Int. J. Mol. Sci. 2023, 24, 7736. [Google Scholar] [CrossRef]
- Peek, J.L.; Wilson, M.H. Cell and gene therapy for kidney disease. Nat. Rev. Nephrol. 2023, 19, 451–462. [Google Scholar] [CrossRef]
- Chen, S.; Agarwal, A.; Glushakova, O.Y.; Jorgensen, M.S.; Salgar, S.K.; Poirier, A.; Flotte, T.R.; Croker, B.P.; Madsen, K.M.; Atkinson, M.A.; et al. Gene delivery in renal tubular epithelial cells using recombinant adeno-associated viral vectors. J. Am. Soc. Nephrol. 2003, 14, 947–958. [Google Scholar] [CrossRef] [PubMed]
- Funk, S.D.; Bayer, R.H.; Miner, J.H. Endothelial cell-specific collagen type IV-alpha(3) expression does not rescue Alport syndrome in Col4a3(-)(/-) mice. Am. J. Physiol. Renal Physiol. 2019, 316, F830–F837. [Google Scholar] [CrossRef]
- Ding, W.Y.; Kuzmuk, V.; Hunter, S.; Lay, A.; Hayes, B.; Beesley, M.; Rollason, R.; Hurcombe, J.A.; Barrington, F.; Masson, C.; et al. Adeno-associated virus gene therapy prevents progression of kidney disease in genetic models of nephrotic syndrome. Sci. Transl. Med. 2023, 15, eabc8226. [Google Scholar] [CrossRef]
- Riedmayr, L.M.; Hinrichsmeyer, K.S.; Thalhammer, S.B.; Mittas, D.M.; Karguth, N.; Otify, D.Y.; Bohm, S.; Weber, V.J.; Bartoschek, M.D.; Splith, V.; et al. mRNA trans-splicing dual AAV vectors for (epi)genome editing and gene therapy. Nat. Commun. 2023, 14, 6578. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Wang, H.; Wang, S.; Hu, S.W.; Lv, J.; Xun, M.; Gao, K.; Wang, F.; Chen, Y.; Wang, D.; et al. Hearing of Otof-deficient mice restored by trans-splicing of N- and C-terminal otoferlin. Hum. Genet. 2023, 142, 289–304. [Google Scholar] [CrossRef]
- Qi, J.; Tan, F.; Zhang, L.; Lu, L.; Zhang, S.; Zhai, Y.; Lu, Y.; Qian, X.; Dong, W.; Zhou, Y.; et al. AAV-Mediated Gene Therapy Restores Hearing in Patients with DFNB9 Deafness. Adv. Sci. 2024, 11, e2306788. [Google Scholar] [CrossRef]
- Lv, J.; Wang, H.; Cheng, X.; Chen, Y.; Wang, D.; Zhang, L.; Cao, Q.; Tang, H.; Hu, S.; Gao, K.; et al. AAV1-hOTOF gene therapy for autosomal recessive deafness 9: A single-arm trial. Lancet, 2024; online ahead of print. [Google Scholar] [CrossRef]
- Zhu, X.; Yin, L.; Theisen, M.; Zhuo, J.; Siddiqui, S.; Levy, B.; Presnyak, V.; Frassetto, A.; Milton, J.; Salerno, T.; et al. Systemic mRNA Therapy for the Treatment of Fabry Disease: Preclinical Studies in Wild-Type Mice, Fabry Mouse Model, and Wild-Type Non-human Primates. Am. J. Hum. Genet. 2019, 104, 625–637. [Google Scholar] [CrossRef]
- Granata, S.; Stallone, G.; Zaza, G. mRNA as a medicine in nephrology: The future is now. Clin. Kidney J. 2023, 16, 2349–2356. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Characteristic | AAV | Adenovirus | Lentivirus | Nanoparticle |
|---|---|---|---|---|
| Size | 25 nm | 60–90 nm | 90–100 nm | Varies |
| Genome type | DNA (single-stranded) | DNA (double-stranded) | RNA (double-stranded) | DNA/RNA |
| Envelope | No | No | Yes | Yes |
| Genome size limitation | 4.7 kb | ~40 kb | ~10 kb | Varies |
| Immunogenicity | Very low | Strong | Low | Low |
| Targeting | Yes | Yes | Yes | Yes |
| Integration | Rare (2%) | No | Yes | Moderate |
| Endurance | Months to years | Weeks | Months | Days to weeks |
| Approach | Subjects | Treatment | Administration | Results | Reference |
|---|---|---|---|---|---|
| Gene transfection/transduction | AS dog | Adenovirus containing COL4A5 | Bladder injection | α5 chain express in bladder smooth muscle and deposit in the basement membrane | [53] |
| Normal pig | Adenovirus containing COL4A5 | Renal artery perfusion | α5 chain express in porcine glomeruli and deposit in the GBM | [54,55] | |
| Anti-miRNA-21 oligonucleotides | AS mice | anti-miRNA-21 oligonucleotide | Subcutaneous injection | Reduce renal fibrosis and urine protein, preserve renal function, and increase the survival rate of mice; but failed in clinic trial (NCT02855268) | [56] |
| ASO | AS mice | ASO | Subcutaneous injection | Reduce urinary protein and creatinine, improve renal pathology, and partial restore α5 chain expression | [58] |
| Gene editing by CRISPR/Cas9 | Podocytes from AS patients | CRISPR/Cas9 carried by AAV | Cell transfection in vitro | Moderate gene correction rates | [59,60] |
| dCas9 targeting activator | HEK293T cells | CRISPRa/dCas9 carried by Lipofectamine 3000 | Cell transfection in vitro | Replace the missing α3α4α5(IV) network with the α5α5α6(IV) network | [61] |
| dCas9 targeting inhibitor | HEK293T cells | CRISPRi/dCas9 carried by Lipofectamine 3000 | Cell transfection in vitro | Reduce laminin α2 in the GBM | [26,61] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, Y.; Zheng, Q.; Xie, J. Exploration of Gene Therapy for Alport Syndrome. Biomedicines 2024, 12, 1159. https://doi.org/10.3390/biomedicines12061159
Zhao Y, Zheng Q, Xie J. Exploration of Gene Therapy for Alport Syndrome. Biomedicines. 2024; 12(6):1159. https://doi.org/10.3390/biomedicines12061159
Chicago/Turabian StyleZhao, Yafei, Qimin Zheng, and Jingyuan Xie. 2024. "Exploration of Gene Therapy for Alport Syndrome" Biomedicines 12, no. 6: 1159. https://doi.org/10.3390/biomedicines12061159
APA StyleZhao, Y., Zheng, Q., & Xie, J. (2024). Exploration of Gene Therapy for Alport Syndrome. Biomedicines, 12(6), 1159. https://doi.org/10.3390/biomedicines12061159