
In-Cell Testing of Zinc-Dependent Histone Deacetylase Inhibitors in the Presence of Class-Selective Fluorogenic Substrates: Potential and Limitations of the Method

Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Inhibitors, Substrates and Antibodies
2.3. Synthesis of Fluorogenic Substrates Boc-Lys(Cro/Pro)-AMC
2.4. HDAC Selectivity Assay
2.5. Cell Viability Assay
2.6. Western Blotting
2.7. Calculation of Correlation and Reliability
2.8. Statistical Data Analysis
3. Results and Discussion
3.1. Selection and Synthesis of Fluorogenic Substrates of HDACs
3.2. Development of Three-Substrate Cell-Test System (s3CTS)
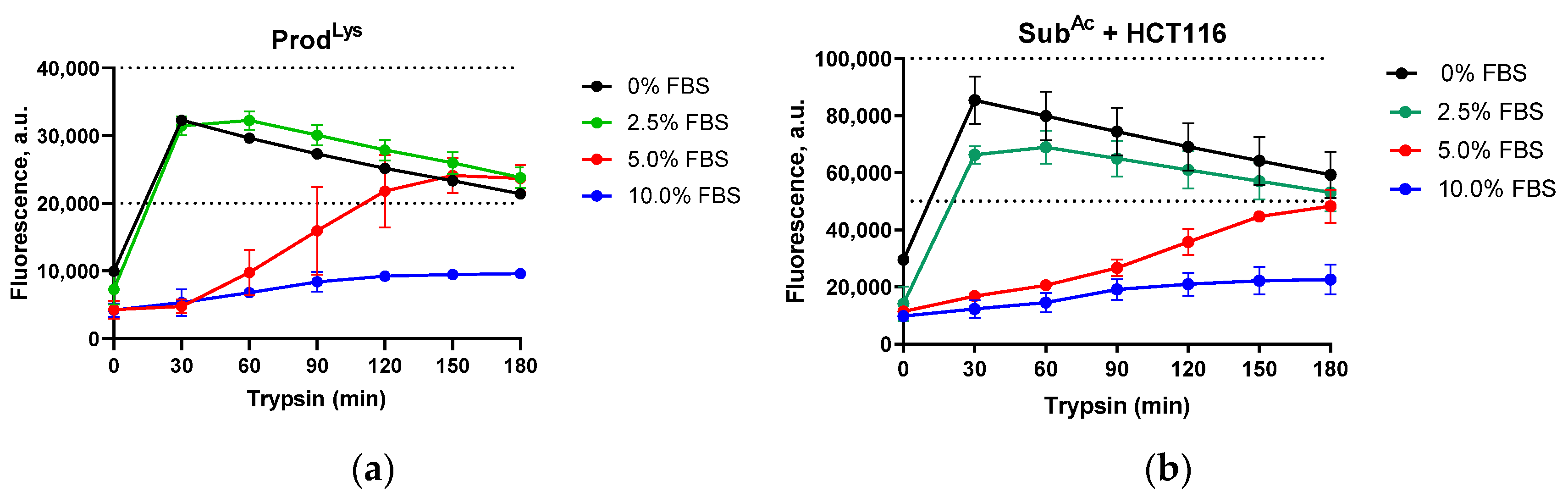
3.2.1. Optimization of Conditions for Determining HDAC in Cell Activity
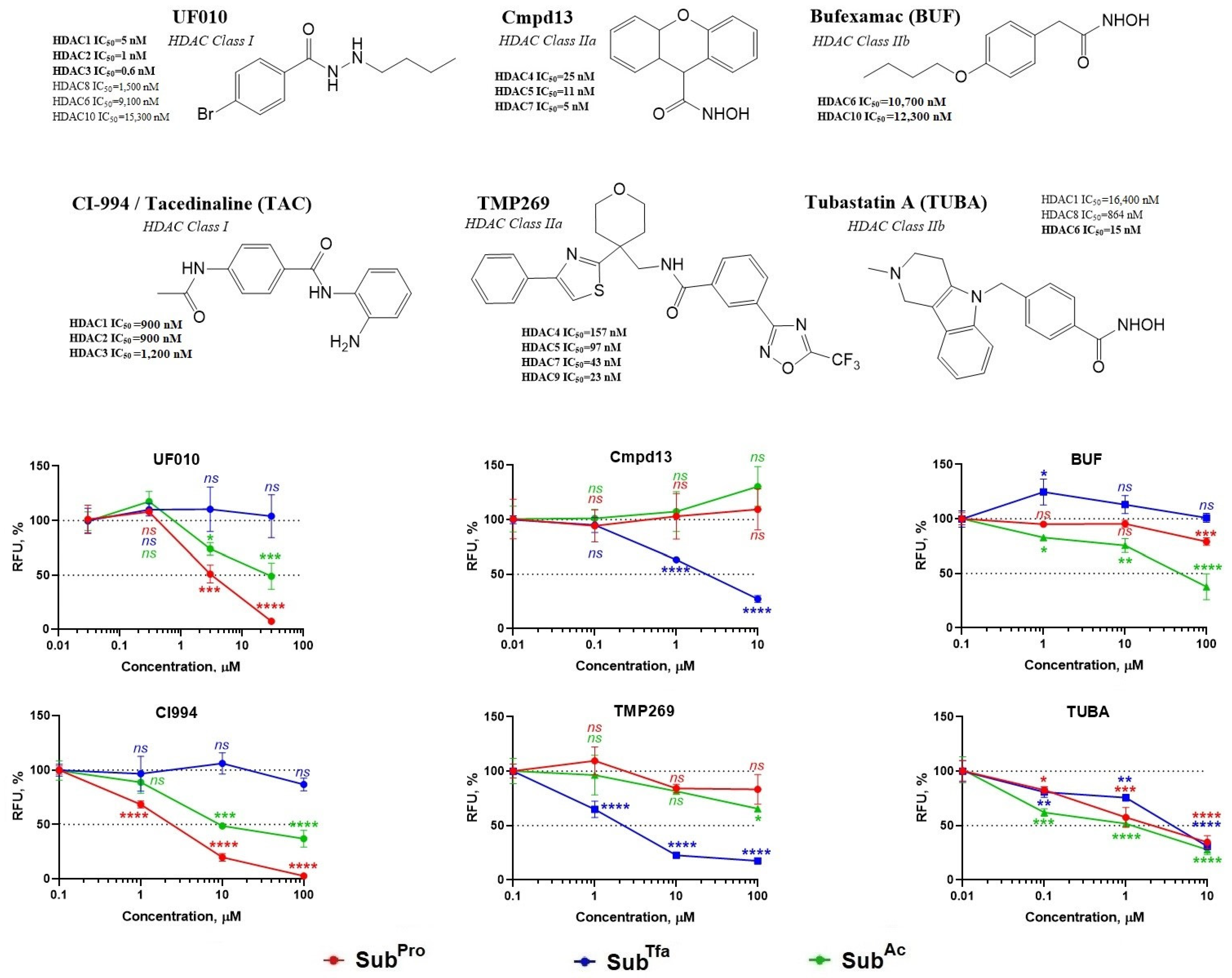
3.2.2. Validation of s3CTS by Testing HDACi of Known Selectivity
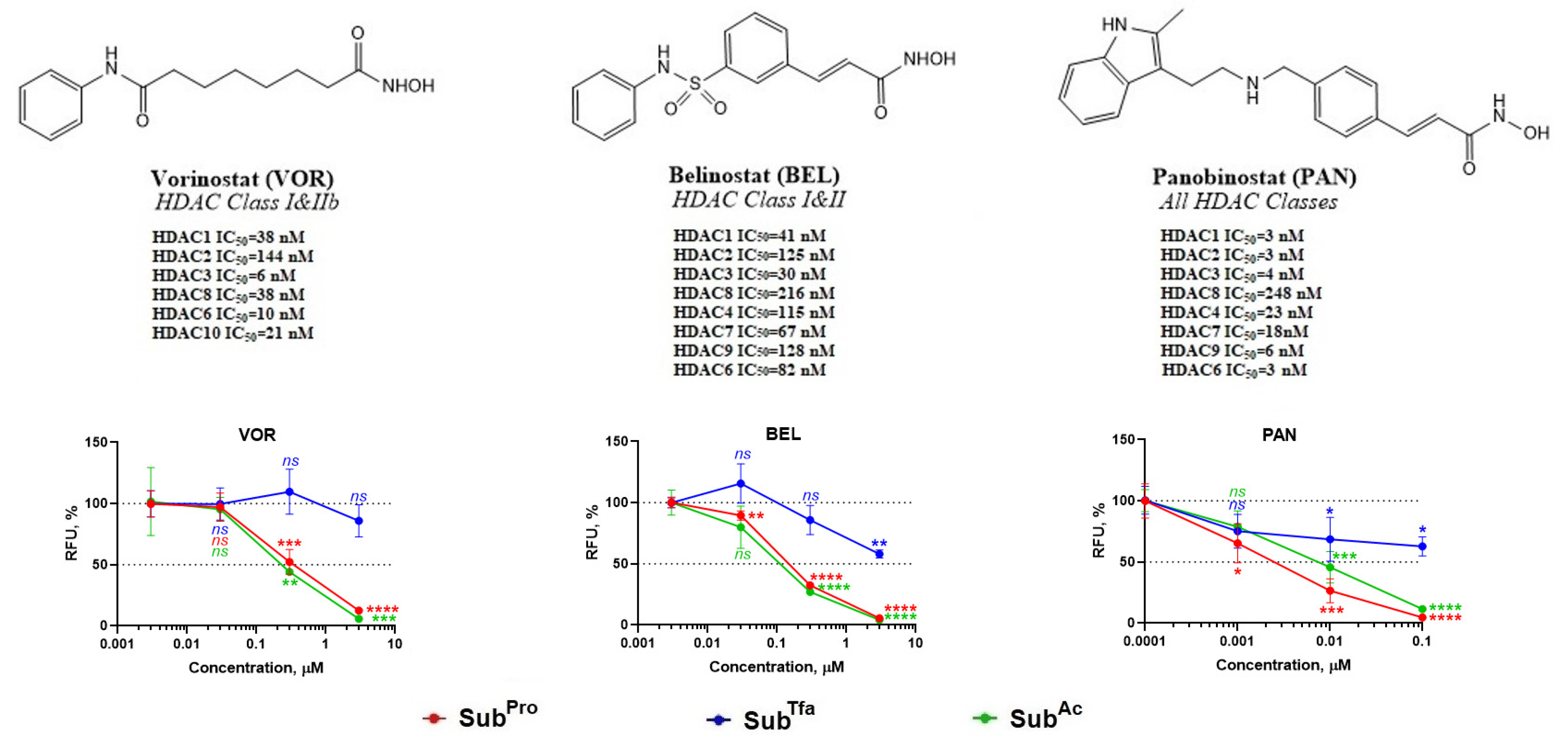
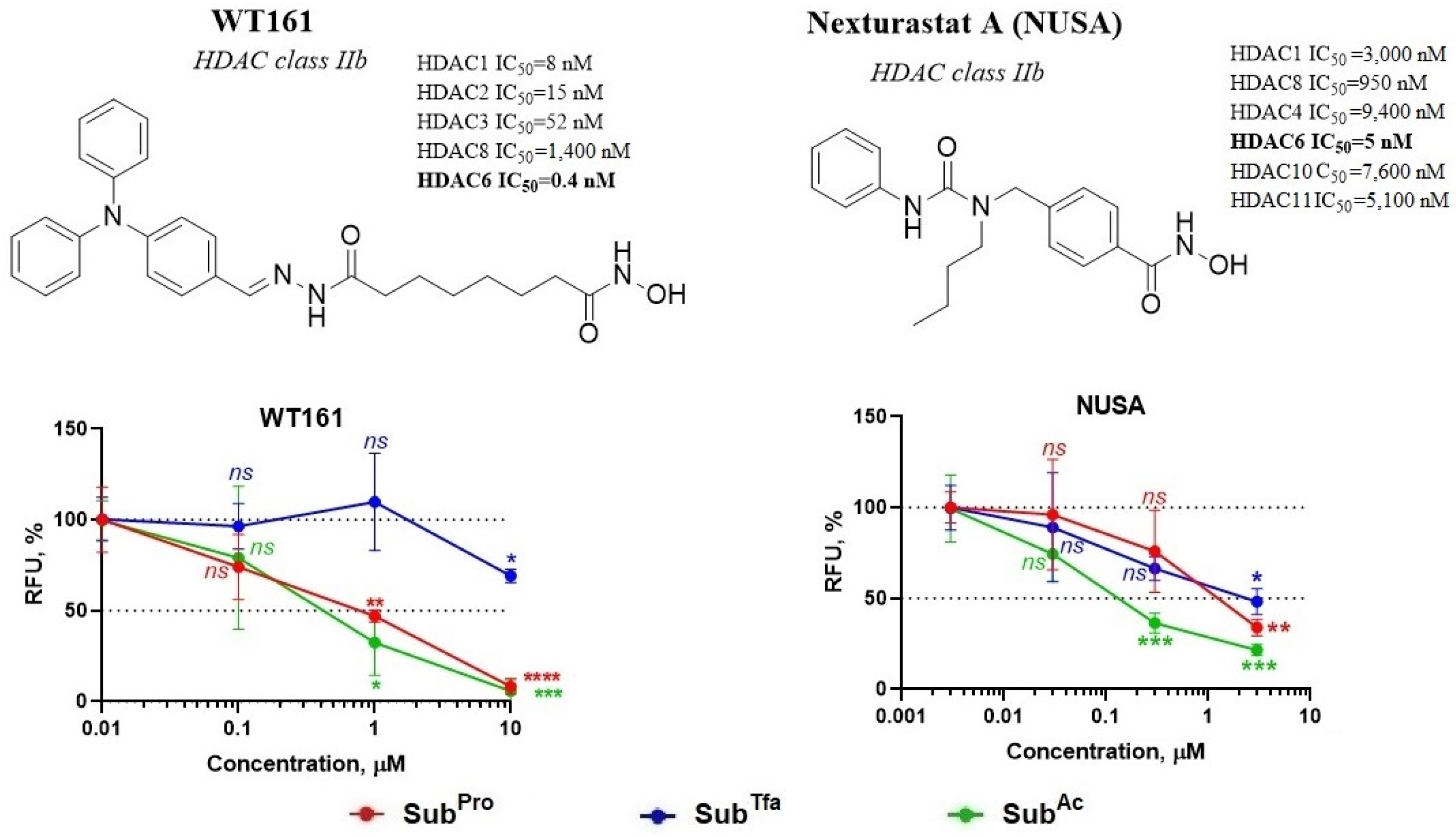
3.2.3. Validation of s3CTS Using Non-Selective HDACi
3.3. Studying the Possible Causes of Errors in s3CTS Performance
3.3.1. The Combination of TUBA and SubAcyl Does Not Cause Inhibition of HDACs Class I
3.3.2. MBLAC2 Activity Affects Performance of s3CTS
3.3.3. Correlation between s3CTS Performance and MBLAC2 Inhibition
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baldensperger, T.; Glomb, M.A. Pathways of non-enzymatic lysine acylation. Front. Cell Dev. Biol. 2021, 9, 664553. [Google Scholar] [CrossRef]
- Scott, I.; Wang, L.; Wu, K.; Thapa, D.; Sack, M.N. GCN5L1/BLOS1 links acetylation, organelle remodeling, and metabolism. Trends Cell Biol. 2018, 28, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Millard, C.J.; Watson, P.J.; Fairall, L.; Schwabe, J.W.R. Targeting class I histone deacetylases in a “complex” environment. Trends Pharmacol. Sci. 2017, 38, 363–377. [Google Scholar] [CrossRef]
- Milazzo, G.; Mercatelli, D.; Di Muzio, G.; Triboli, L.; De Rosa, P.; Perini, G.; Giorgi, F.M. Histone deacetylases (HDACs): Evolution, specificity, role in transcriptional complexes, and pharmacological actionability. Genes 2020, 11, 556. [Google Scholar] [CrossRef] [PubMed]
- Clocchiatti, A.; Di Giorgio, E.; Ingrao, S.; Meyer-Almes, F.J.; Tripodo, C.; Brancolini, C. Class IIa HDACs repressive activities on MEF2-depedent transcription are associated with poor prognosis of ER+ breast tumors. FASEB J. 2013, 27, 942–954. [Google Scholar] [CrossRef] [PubMed]
- Hudson, G.M.; Watson, P.J.; Fairall, L.; Jamieson, A.G.; Schwabe, J.W.R. Insights into the recruitment of class IIa histone deacetylases (HDACs) to the SMRT/NCoR transcriptional repression complex. J. Mol. Biol. 2015, 290, 18237–18244. [Google Scholar] [CrossRef]
- Hubbert, C.; Guardiola, A.; Shao, R.; Kawaguchi, Y.; Ito, A.; Nixon, A.; Yoshida, M.; Wang, X.F.; Yao, T.P. HDAC6 is a microtubule-associated deacetylase. Nature 2002, 417, 455–458. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yuan, Z.; Zhang, Y.; Yong, S.; Salas-Burgos, A.; Koomen, J.; Olashaw, N.; Parsons, J.T.; Yang, X.J.; Dent, S.R.; et al. HDAC6 modulates cell motility by altering the acetylation level of cortactin. Mol. Cell 2007, 27, 197–213. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Xiao, J.; Wang, Y.; Song, X.; Huang, L.; Ren, Z.; Kitazato, K.; Wang, Y. Posttranslational modification and beyond: Interplay between histone deacetylase 6 and heat-shock protein 90. Mol. Med. 2021, 27, 110. [Google Scholar] [CrossRef]
- Hai, Y.; Shinsky, S.; Porter, N.; Christianson, D.W. Histone deacetylase 10 structure and molecular function as a polyamine deacetylase. Nat. Commun. 2017, 8, 15368. [Google Scholar] [CrossRef]
- Nunez-Alvarez, Y.; Suelves, M. HDAC11: A multifaceted histone deacetylase with proficient fatty deacylase activity and its roles in physiological processes. FEBS J. 2022, 289, 2771–2792. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Xie, C.; Chen, Q.; Zhuang, S. HDAC11, an emerging therapeutic target for metabolic disorders. Front. Endocrinol. 2022, 13, 989305. [Google Scholar] [CrossRef] [PubMed]
- Hamze, A. How do we improve histone deacetylase inhibitor drug discovery? Expert Opin. Drug Discov. 2020, 15, 527–529. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.C.S.; Chan, A.; Ganesan, A. Thirty years of HDAC inhibitors: 2020 insight and hindsight. J. Med. Chem. 2020, 63, 12460–12484. [Google Scholar] [CrossRef] [PubMed]
- Di Bello, E.; Noce, B.; Fioravanti, R.; Mai, A. Current HDAC inhibitors in clinical trials. Chimia 2022, 76, 448–453. [Google Scholar] [CrossRef] [PubMed]
- Heltweg, B.; Jung, M. A microplate reader-based nonisotopic histone deacetylase activity assay. Anal. Biochem. 2002, 302, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Wegener, D.; Wirsching, F.; Riester, D.; Schwienhorst, A. A fluorogenic histone deacetylase assay well suited for high-throughput activity screening. Chem. Biol. 2003, 10, 61–68. [Google Scholar] [CrossRef]
- Bradner, J.; West, N.; Grachan, M.; Greenberg, E.F.; Haggarty, S.J.; Warnow, T.; Mazitschek, R. Chemical phylogenetics of histone deacetylases. Nat. Chem. Biol. 2010, 6, 238–243. [Google Scholar] [CrossRef]
- Schweipert, M.; Amurthavasan, A.; Meyer-Almes, F.J. Continuous enzyme activity assay for high-throughput classification of histone deacetylase 8 inhibitors. Explor. Target. Anti-Tumor Ther. 2023, 4, 447–459. [Google Scholar] [CrossRef]
- Fan, J.; Alsarraf, O.; Chou, C.J.; Yates, P.W.; Goodwin, N.C.; Rice, D.S.; Crosson, C.E. Ischemic preconditioning, retinal neuroprotection and histone deacetylase activities. Exp. Eye Res. 2016, 146, 269–275. [Google Scholar] [CrossRef]
- Potluri, V.; Shandil, R.K.; Gavara, R.; Sambasivam, G.; Campo, B.; Wittlin, S.; Narayanan, S. Discovery of FNDR-20123, a histone deacetylase inhibitor for the treatment of Plasmodium falciparum malaria. Malar. J. 2020, 19, 365. [Google Scholar] [CrossRef] [PubMed]
- Ciossek, T.; Julius, H.; Wieland, H.; Maier, T.; Beckers, T. A homogeneous cellular histone deacetylase assay suitable for compound profiling and robotic screening. Anal. Biochem. 2008, 372, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Bonfils, C.; Kalita, A.; Dubay, M.; Siu, L.L.; Carducci, M.A.; Reid, G.; Martell, R.E.; Besterman, J.M.; Li, Z. Evaluation of the pharmacodynamic effects of MGCD0103 from preclinical models to human using a novel HDAC enzyme assay. Clin. Cancer Res. 2008, 14, 3441–3449. [Google Scholar] [CrossRef]
- Marek, L.; Hamacher, A.; Hansen, F.K.; Kuna, K.; Gohlke, H.; Kassack, M.U.; Kurz, T. Histone deacetylase (HDAC) inhibitors with a novel connecting unit linker region reveal a selectivity profile for HDAC4 and HDAC5 with improved activity against chemoresistant cancer cells. J. Med. Chem. 2013, 56, 427–436. [Google Scholar] [CrossRef]
- Lobera, M.; Madauss, K.P.; Pohlhaus, D.T.; Wright, Q.G.; Trocha, M.; Schmidt, D.R.; Baloglu, E.; Trump, R.P.; Head, M.S.; Hofmann, G.A.; et al. Selective class IIa histone deacetylase inhibition via a nonchelating zinc-binding group. Nat. Chem. Biol. 2013, 9, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.W.; Shou, D.; Huang, R.; Khuc, T.; Dai, S.; Zheng, W.; Klumpp-Thomas, C.; Xia, M. Identification of HDAC inhibitors using a cell-based HDAC I/II assay. J. Biomol. Screen. 2016, 21, 643–652. [Google Scholar] [CrossRef]
- Asfaha, Y.; Skerhut, A.J.; Alves-Avelar, L.A.; Horstick-Muche, N.; Lungerich, B.; Klinken, S.; Kassack, M.U.; Kurz, T. Synthesis of thiazolyl-based hydroxamic acids as histone deacetylase inhibitors. Arkivoc 2020, vii, 228–241. [Google Scholar] [CrossRef]
- Kozlov, M.V.; Konduktorov, K.A.; Shcherbakova, A.S.; Kochetkov, S.N. Synthesis of N’-propylhydrazide analogs of hydroxamic inhibitors of histone deacetylases (HDACs) and evaluation of their impact on activities of HDACs and replication of hepatitis C virus (HCV). Bioorg. Med. Chem. Lett. 2019, 29, 2369–2374. [Google Scholar] [CrossRef]
- Ivanter, E.V.; Korosov, A.V. Elementary Biometrics: A Study Guide (In Russian); PetrSU: Petrozavodsk, Russia, 2005; pp. 71–73. [Google Scholar]
- McClure, J.J.; Inks, E.S.; Zhang, C.; Peterson, Y.K.; Li, J.; Chundru, K.; Lee, B.; Buchanan, A.; Miao, S.; Chou, C.J. Comparison of the deacylase and deacetylase activity of zinc-dependent HDACs. ACS Chem. Biol. 2017, 12, 1644–1655. [Google Scholar] [CrossRef]
- Howitz, K.T.; Zhang, Z.; Kisielewski, A.; Dale, E.; Patton, W.F.; Enzo Life Sciences Inc. Compounds and Methods for Detection of Enzymes That Remove Methyl Succinyl Groups from Epsilon-Amino Lysine Moieties. U.S. Patent 10,920,265, 16 February 2021. [Google Scholar]
- Smith, B.C.; Denu, J.M. Sir2 deacetylases exhibit nucleophilic participation of acetyl-lysine in NAD+ cleavage. J. Am. Chem. Soc. 2007, 129, 5802–5803. [Google Scholar] [CrossRef]
- Chi, H.; Takemoto, Y.; Nsiama, T.K.; Kato, T.; Nishino, N.; Ito, A.; Yoshida, M. Design and synthesis of peptide-MCA substrates for a novel assay of histone methyltransferases and their inhibitors. Bioorg. Med. Chem. 2014, 22, 1268–1275. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Stowe, R.L.; Pinello, C.E.; Tian, G.; Madoux, F.; Li, D.; Zhao, L.Y.; Li, J.L.; Wang, Y.; Wang, Y.; et al. Identification of histone deacetylase inhibitors with benzoylhydrazide scaffold that selectively inhibit class I histone deacetylases. Chem. Biol. 2015, 22, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Moradei, O.M.; Mallais, T.C.; Frechette, S.; Paquin, I.; Tessier, P.E.; Leit, S.M.; Fournel, M.; Bonfils, C.; Trachy-Bourget, M.C.; Liu, J.; et al. Novel aminophenyl benzamide-type histone deacetylase inhibitors with enhanced potency and selectivity. J. Med. Chem. 2007, 50, 5543–5546. [Google Scholar] [CrossRef] [PubMed]
- Tessier, P.; Smil, D.V.; Wahhab, A.; Leit, S.; Rahil, J.; Li, Z.; Deziel, R.; Besterman, J.M. Diphenylmethylene hydroxamic acids as selective class IIa histone deacetylase inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 5684–5688. [Google Scholar] [CrossRef]
- Bantscheff, M.; Hopf, C.; Savitski, M.; Dittmann, A.; Grandi, P.; Michon, A.M.; Schlegl, J.; Abraham, Y.; Becher, I.; Bergamini, G.; et al. Chemoproteomics profiling of HDAC inhibitors reveals selective targeting of HDAC complexes. Nat. Biotechnol. 2011, 29, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Butler, K.V.; Kalin, J.; Brochier, C.; Vistoli, G.; Langley, B.; Kozikowski, A.P. Rational design and simple chemistry yield a superior, neuroprotective HDAC6 inhibitor, tubastatin A. J. Am. Chem. Soc. 2010, 132, 10842–10846. [Google Scholar] [CrossRef] [PubMed]
- Rana, Z.; Diermeier, S.; Hanif, M.; Rosengren, R.J. Understanding failure and improving treatment using HDAC inhibitors for prostate cancer. Biomedicines 2020, 8, 22. [Google Scholar] [CrossRef] [PubMed]
- Malgapo, M.I.P.; Safadi, J.M.; Linder, M.E. Metallo-β-lactamase domain-containing protein 2 is S-palmitoylated and exhibits acyl-CoA hydrolase activity. J. Biol. Chem. 2021, 296, 100106. [Google Scholar] [CrossRef]
- Lechner, S.; Malgapo, M.I.P.; Grätz, C.; Steimbach, R.R.; Baron, A.; Ruther, P.; Nadal, S.; Stumpf, C.; Loos, C.; Ku, X.; et al. Target deconvolution of HDAC pharmacopoeia reveals MBLAC2 as common off-target. Nat. Chem. Biol. 2022, 18, 812–820. [Google Scholar] [CrossRef]
- Hideshima, T.; Qi, J.; Paranal, R.M.; Tang, W.; Greenberg, E.; West, N.; Colling, M.E.; Estiu, G.; Mazitschek, R.; Perry, J.A.; et al. Discovery of selective small-molecule HDAC6 inhibitor for overcoming proteasome inhibitor resistance in multiple myeloma. Proc. Natl. Acad. Sci. USA 2016, 113, 13162–13167. [Google Scholar] [CrossRef]
- Bergman, J.A.; Woan, K.; Perez-Villarroel, P.; Villagra, A.; Sotomayor, E.M.; Kozikowski, A.P. Selective histone deacetylase 6 inhibitors bearing substituted urea linkers inhibit melanoma cell growth. J. Med. Chem. 2012, 55, 9891–9899. [Google Scholar] [CrossRef] [PubMed]
- Valdez, B.C.; Li, Y.; Murray, D.; Brammer, J.E.; Liu, Y.; Hosing, C.; Nieto, Y.; Champlin, R.E.; Andersson, B.S. Differential effects of histone deacetylase inhibitors on cellular drug transporters and their implications for using epigenetic modifiers in combination chemotherapy. Oncotarget 2016, 7, 63829–63838. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Bai, X.; Ni, J.; Zhang, H.; Duan, W.; Graham, P.; Li, Y. Exosomes and breast cancer drug resistance. Cell Death Dis. 2020, 11, 987. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yeung, B.Z.; Wientjes, M.G.; Cui, M.; Peer, C.J.; Lu, Z.; Figg, W.D.; Woo, S.; Au, J.L. A quantitative pharmacology model of exosome-mediated drug efflux and perturbation-induced synergy. Pharmaceutics 2021, 13, 997. [Google Scholar] [CrossRef]
- Bai, L.; Yang, P.; Kong, L. Progress of acyl-Coenzyme A thioester hydrolase family in cancer. Front. Oncol. 2024, 14, 1374094. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HDACi | pKdapp * | Rank 1 | Incorrect | Rank 2 |
|---|---|---|---|---|
| BUF | <4.52 | 2.5 | 0 | 3 |
| TAC | <4.52 | 2.5 | 0 | 3 |
| VOR | <4.52 | 2.5 | 0 | 3 |
| BEL | <4.52 | 2.5 | 0 | 3 |
| PAN | 5.86 | 5 | 0 | 3 |
| WT161 | 6.04 | 6 | 1 | 7 |
| TUBA | 6.31 | 7 | 1 | 7 |
| NUSA | 7.59 | 8 | 1 | 7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kleymenova, A.; Zemskaya, A.; Kochetkov, S.; Kozlov, M. In-Cell Testing of Zinc-Dependent Histone Deacetylase Inhibitors in the Presence of Class-Selective Fluorogenic Substrates: Potential and Limitations of the Method. Biomedicines 2024, 12, 1203. https://doi.org/10.3390/biomedicines12061203
Kleymenova A, Zemskaya A, Kochetkov S, Kozlov M. In-Cell Testing of Zinc-Dependent Histone Deacetylase Inhibitors in the Presence of Class-Selective Fluorogenic Substrates: Potential and Limitations of the Method. Biomedicines. 2024; 12(6):1203. https://doi.org/10.3390/biomedicines12061203
Chicago/Turabian StyleKleymenova, Alla, Anastasia Zemskaya, Sergey Kochetkov, and Maxim Kozlov. 2024. "In-Cell Testing of Zinc-Dependent Histone Deacetylase Inhibitors in the Presence of Class-Selective Fluorogenic Substrates: Potential and Limitations of the Method" Biomedicines 12, no. 6: 1203. https://doi.org/10.3390/biomedicines12061203
APA StyleKleymenova, A., Zemskaya, A., Kochetkov, S., & Kozlov, M. (2024). In-Cell Testing of Zinc-Dependent Histone Deacetylase Inhibitors in the Presence of Class-Selective Fluorogenic Substrates: Potential and Limitations of the Method. Biomedicines, 12(6), 1203. https://doi.org/10.3390/biomedicines12061203