
Mild Disease Course of Experimental Autoimmune Encephalomyelitis without Pertussis Toxin: Brain Transcriptome Analysis Reveals Similar Signaling to Active Lesions in Multiple Sclerosis


Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Animals
2.3. Induction and Assessment of EAE
2.4. MS and Control Brain Tissue Transcriptomic Analysis
2.5. Ingenuity Pathway Analysis
2.6. RNA Extraction and RT-qPCR
2.7. Statistics
3. Results
3.1. Mild EAE Clinical Scores Exhibit Variability
3.2. EAE Mice Present with Numerous DEGs and Immune-Related Canonical Pathways
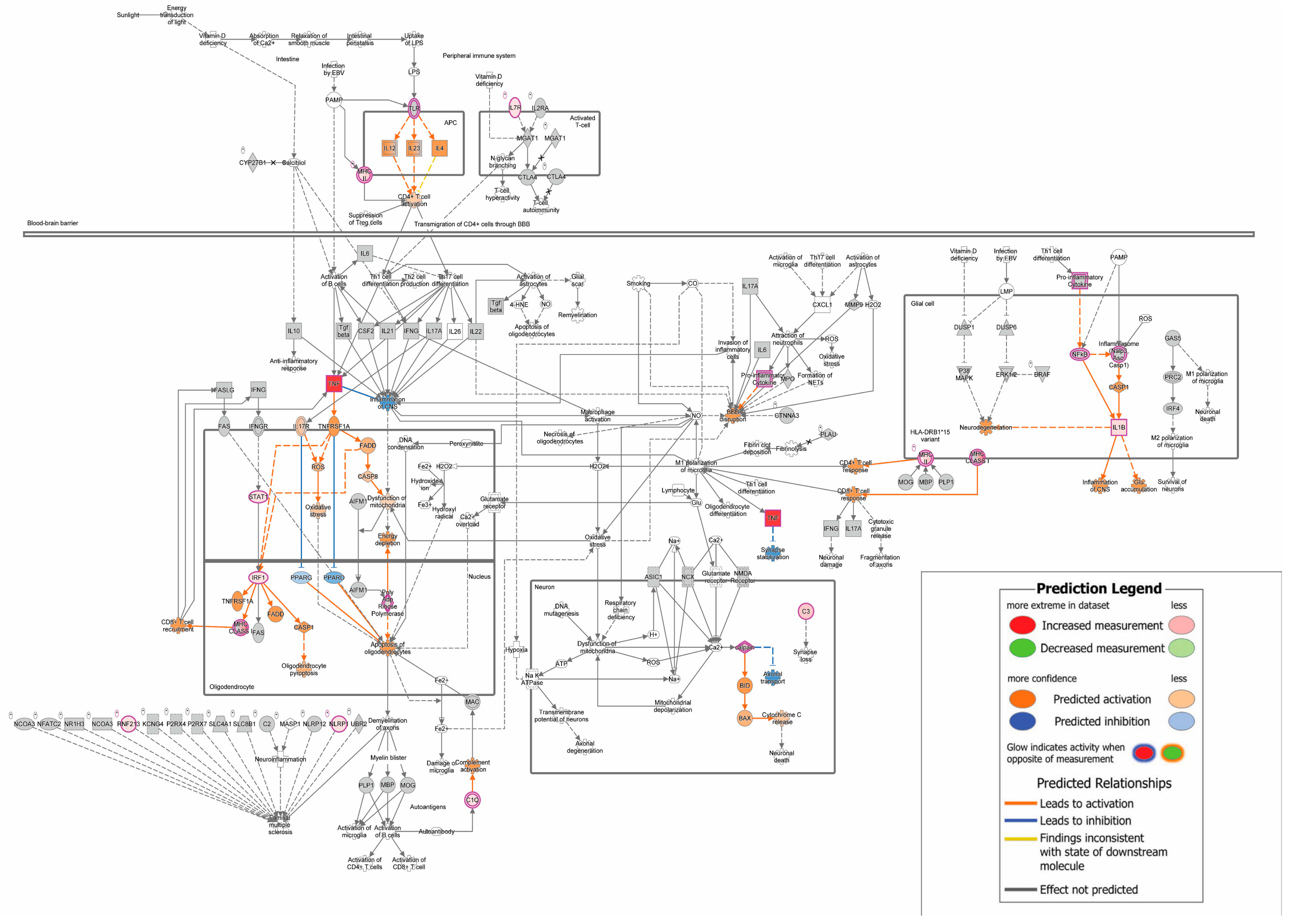
3.3. Mild EAE Brain Presents Share Numerous Canonical Pathways with MS Lesions
3.4. Neuroinflammatory Genes Expression Increased in EAE Symptomatic Mouse Brains
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Robinson, A.P.; Harp, C.T.; Noronha, A.; Miller, S.D. The experimental autoimmune encephalomyelitis (EAE) model of MS: Utility for understanding disease pathophysiology and treatment. Handb. Clin. Neurol. 2014, 122, 173–189. [Google Scholar] [PubMed]
- Weinstock-Guttman, B.; Nair, K.V.; Glajch, J.L.; Ganguly, T.C.; Kantor, D. Two decades of glatiramer acetate: From initial discovery to the current development of generics. J. Neurol. Sci. 2017, 376, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Neuhaus, O.; Kieseier, B.C.; Hartung, H.P. Therapeutic role of mitoxantrone in multiple sclerosis. Pharmacol. Ther. 2006, 109, 198–209. [Google Scholar] [CrossRef]
- TNF neutralization in MS: Results of a randomized, placebo-controlled multicenter study. The Lenercept Multiple Sclerosis Study Group and The University of British Columbia MS/MRI Analysis Group. Neurology 1999, 53, 457–465. [CrossRef]
- Bjelobaba, I.; Begovic-Kupresanin, V.; Pekovic, S.; Lavrnja, I. Animal models of multiple sclerosis: Focus on experimental autoimmune encephalomyelitis. J. Neurosci. Res. 2018, 96, 1021–1042. [Google Scholar] [CrossRef] [PubMed]
- Mangmool, S.; Kurose, H. G(i/o) protein-dependent and -independent actions of Pertussis Toxin (PTX). Toxins 2011, 3, 884–899. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, L.A.; Lolait, S.J.; Brownstein, M.J.; Young, A.C.; Bonner, T.I. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 1990, 346, 561–564. [Google Scholar] [CrossRef] [PubMed]
- Munro, S.; Thomas, K.L.; Abu-Shaar, M. Molecular characterization of a peripheral receptor for cannabinoids. Nature 1993, 365, 61–65. [Google Scholar] [CrossRef]
- Persia, D.; Mangiavacchi, F.; Marcotullio, M.C.; Rosati, O. Cannabinoids as multifaceted compounds. Phytochemistry 2023, 212, 113718. [Google Scholar] [CrossRef] [PubMed]
- Dopkins, N.; Miranda, K.; Wilson, K.; Holloman, B.L.; Nagarkatti, P.; Nagarkatti, M. Effects of Orally Administered Cannabidiol on Neuroinflammation and Intestinal Inflammation in the Attenuation of Experimental Autoimmune Encephalomyelitis. J. Neuroimmune Pharmacol. 2022, 17, 15–32. [Google Scholar] [CrossRef]
- Elliott, D.M.; Singh, N.; Nagarkatti, M.; Nagarkatti, P.S. Cannabidiol Attenuates Experimental Autoimmune Encephalomyelitis Model of Multiple Sclerosis Through Induction of Myeloid-Derived Suppressor Cells. Front. Immunol. 2018, 9, 1782. [Google Scholar] [CrossRef] [PubMed]
- Giacoppo, S.; Soundara Rajan, T.; Galuppo, M.; Pollastro, F.; Grassi, G.; Bramanti, P.; Mazzon, E. Purified Cannabidiol, the main non-psychotropic component of Cannabis sativa, alone, counteracts neuronal apoptosis in experimental multiple sclerosis. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 4906–4919. [Google Scholar] [PubMed]
- Gonzalez-Garcia, C.; Torres, I.M.; Garcia-Hernandez, R.; Campos-Ruiz, L.; Esparragoza, L.R.; Coronado, M.J.; Grande, A.G.; Garcia-Merino, A.; Sanchez Lopez, A.J. Mechanisms of action of cannabidiol in adoptively transferred experimental autoimmune encephalomyelitis. Exp. Neurol. 2017, 298, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Kozela, E.; Lev, N.; Kaushansky, N.; Eilam, R.; Rimmerman, N.; Levy, R.; Ben-Nun, A.; Juknat, A.; Vogel, Z. Cannabidiol inhibits pathogenic T cells, decreases spinal microglial activation and ameliorates multiple sclerosis-like disease in C57BL/6 mice. Br. J. Pharmacol. 2011, 163, 1507–1519. [Google Scholar] [CrossRef] [PubMed]
- Mecha, M.; Feliu, A.; Inigo, P.M.; Mestre, L.; Carrillo-Salinas, F.J.; Guaza, C. Cannabidiol provides long-lasting protection against the deleterious effects of inflammation in a viral model of multiple sclerosis: A role for A2A receptors. Neurobiol. Dis. 2013, 59, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Nichols, J.M.; Kummari, E.; Sherman, J.; Yang, E.J.; Dhital, S.; Gilfeather, C.; Yray, G.; Morgan, T.; Kaplan, B.L.F. CBD Suppression of EAE Is Correlated with Early Inhibition of Splenic IFN-gamma + CD8+ T Cells and Modest Inhibition of Neuroinflammation. J. Neuroimmune Pharmacol. 2021, 16, 346–362. [Google Scholar] [CrossRef] [PubMed]
- Vitarelli da Silva, T.; Bernardes, D.; Oliveira-Lima, O.C.; Fernandes Pinto, B.; Filho, M.L.; Faraco, C.C.F.; Juliano, M.A.; Arantes, R.M.E.; Moreira, F.A.; Carvalho-Tavares, J. Cannabidiol Attenuates In Vivo Leukocyte Recruitment to the Spinal Cord Microvasculature at Peak Disease of Experimental Autoimmune Encephalomyelitis. Cannabis Cannabinoid Res. 2023, 9, 537–546. [Google Scholar] [CrossRef]
- Meuth, S.G.; Henze, T.; Essner, U.; Trompke, C.; Vila Silvan, C. Tetrahydrocannabinol and cannabidiol oromucosal spray in resistant multiple sclerosis spasticity: Consistency of response across subgroups from the SAVANT randomized clinical trial. Int. J. Neurosci. 2020, 130, 1199–1205. [Google Scholar] [CrossRef]
- Nicholas, J.; Lublin, F.; Klineova, S.; Berwaerts, J.; Chinnapongse, R.; Checketts, D.; Javaid, S.; Steinerman, J.R. Efficacy of nabiximols oromucosal spray on spasticity in people with multiple sclerosis: Treatment effects on Spasticity Numeric Rating Scale, muscle spasm count, and spastic muscle tone in two randomized clinical trials. Mult. Scler. Relat. Disord. 2023, 75, 104745. [Google Scholar] [CrossRef]
- Patti, F.; Chisari, C.G.; Solaro, C.; Benedetti, M.D.; Berra, E.; Bianco, A.; Bruno Bossio, R.; Buttari, F.; Castelli, L.; Cavalla, P.; et al. Effects of THC/CBD oromucosal spray on spasticity-related symptoms in people with multiple sclerosis: Results from a retrospective multicenter study. Neurol. Sci. 2020, 41, 2905–2913. [Google Scholar] [CrossRef]
- de Almeida, D.L.; Devi, L.A. Diversity of molecular targets and signaling pathways for CBD. Pharmacol. Res. Perspect. 2020, 8, e00682. [Google Scholar] [CrossRef] [PubMed]
- Kummari, E.; Nichols, J.M.; Yang, E.J.; Kaplan, B.L.F. Neuroinflammation and B-Cell Phenotypes in Cervical and Lumbosacral Regions of the Spinal Cord in Experimental Autoimmune Encephalomyelitis in the Absence of Pertussis Toxin. Neuroimmunomodulation 2019, 26, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Kummari, E.; Rushing, E.; Nicaise, A.; McDonald, A.; Kaplan, B.L.F. TCDD attenuates EAE through induction of FasL on B cells and inhibition of IgG production. Toxicology 2021, 448, 152646. [Google Scholar] [CrossRef] [PubMed]
- McDonald, A.; Nicaise, A.; Sears, E.R.; Bell, A.; Kummari, E.; Kaplan, B.L.F. Potential for TCDD to induce regulatory functions in B cells as part of the mechanism for T cell suppression in EAE. Toxicol. Appl. Pharmacol. 2022, 454, 116259. [Google Scholar] [CrossRef] [PubMed]
- Nicaise, A.J.; McDonald, A.; Sears, E.R.; Sturgis, T.; Kaplan, B.L.F. TCDD Inhibition of IgG1 Production in Experimental Autoimmune Encephalomyelitis (EAE) and In Vitro. Antibodies 2022, 11, 4. [Google Scholar] [CrossRef] [PubMed]
- Yang, E.J.; Stokes, J.V.; Kummari, E.; Eells, J.; Kaplan, B.L. Immunomodulation By Subchronic Low Dose 2,3,7,8-Tetrachlorodibenzo-p-Dioxin in Experimental Autoimmune Encephalomyelitis in the Absence of Pertussis Toxin. Toxicol. Sci. 2016, 151, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Frisch, T.; Elkjaer, M.L.; Reynolds, R.; Michel, T.M.; Kacprowski, T.; Burton, M.; Kruse, T.A.; Thomassen, M.; Baumbach, J.; Illes, Z. Multiple Sclerosis Atlas: A Molecular Map of Brain Lesion Stages in Progressive Multiple Sclerosis. Netw. Syst. Med. 2020, 3, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Van Kaer, L.; Postoak, J.L.; Wang, C.; Yang, G.; Wu, L. Innate, innate-like and adaptive lymphocytes in the pathogenesis of MS and EAE. Cell Mol. Immunol. 2019, 16, 531–539. [Google Scholar] [CrossRef]
- Mews, I.; Bergmann, M.; Bunkowski, S.; Gullotta, F.; Bruck, W. Oligodendrocyte and axon pathology in clinically silent multiple sclerosis lesions. Mult. Scler. 1998, 4, 55–62. [Google Scholar] [CrossRef]
- Nichols, J.M.; Kaplan, B.L.F. The CB(1) Receptor Differentially Regulates IFN-gamma Production In Vitro and in Experimental Autoimmune Encephalomyelitis. Cannabis Cannabinoid Res. 2021, 6, 300–314. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.D.; Karpus, W.J. Experimental autoimmune encephalomyelitis in the mouse. Curr. Protoc. Immunol. 2007, 88, 15.1.1–15.1.18. [Google Scholar] [CrossRef] [PubMed]
- Luther, S.A.; Cyster, J.G. Chemokines as regulators of T cell differentiation. Nat. Immunol. 2001, 2, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Dhaiban, S.; Al-Ani, M.; Elemam, N.M.; Maghazachi, A.A. Targeting Chemokines and Chemokine Receptors in Multiple Sclerosis and Experimental Autoimmune Encephalomyelitis. J. Inflamm. Res. 2020, 13, 619–633. [Google Scholar] [CrossRef] [PubMed]
- Sawabe, A.; Okazaki, S.; Nakamura, A.; Goitsuka, R.; Kaifu, T. The orphan G protein-coupled receptor 141 expressed in myeloid cells functions as an inflammation suppressor. J. Leukoc. Biol. 2024, 115, 935–945. [Google Scholar] [CrossRef]
- Maresz, K.; Pryce, G.; Ponomarev, E.D.; Marsicano, G.; Croxford, J.L.; Shriver, L.P.; Ledent, C.; Cheng, X.; Carrier, E.J.; Mann, M.K.; et al. Direct suppression of CNS autoimmune inflammation via the cannabinoid receptor CB1 on neurons and CB2 on autoreactive T cells. Nat. Med. 2007, 13, 492–497. [Google Scholar] [CrossRef] [PubMed]
- Kroenke, M.A.; Segal, B.M. IL-23 modulated myelin-specific T cells induce EAE via an IFNgamma driven, IL-17 independent pathway. Brain Behav. Immun. 2011, 25, 932–937. [Google Scholar] [CrossRef]
- Tichauer, J.E.; Arellano, G.; Acuna, E.; Gonzalez, L.F.; Kannaiyan, N.R.; Murgas, P.; Panadero-Medianero, C.; Ibanez-Vega, J.; Burgos, P.I.; Loda, E.; et al. Interferon-gamma ameliorates experimental autoimmune encephalomyelitis by inducing homeostatic adaptation of microglia. Front. Immunol. 2023, 14, 1191838. [Google Scholar] [CrossRef]
- Reynolds, R.; Roncaroli, F.; Nicholas, R.; Radotra, B.; Gveric, D.; Howell, O. The neuropathological basis of clinical progression in multiple sclerosis. Acta Neuropathol. 2011, 122, 155–170. [Google Scholar] [CrossRef]
- van der Valk, P.; De Groot, C.J. Staging of multiple sclerosis (MS) lesions: Pathology of the time frame of MS. Neuropathol. Appl. Neurobiol. 2000, 26, 2–10. [Google Scholar] [CrossRef]
- Dobson, R.; Giovannoni, G. Multiple sclerosis—A review. Eur. J. Neurol. 2019, 26, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Kornek, B.; Storch, M.K.; Weissert, R.; Wallstroem, E.; Stefferl, A.; Olsson, T.; Linington, C.; Schmidbauer, M.; Lassmann, H. Multiple sclerosis and chronic autoimmune encephalomyelitis: A comparative quantitative study of axonal injury in active, inactive, and remyelinated lesions. Am. J. Pathol. 2000, 157, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Calvi, A.; Haider, L.; Prados, F.; Tur, C.; Chard, D.; Barkhof, F. In vivo imaging of chronic active lesions in multiple sclerosis. Mult. Scler. 2022, 28, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Frodella, C.M.; Pruett, S.B.; Ross, M.K.; Kaplan, B.L. Oxytocin and Vasopressin Gene Expression in the Brain as Potential Biomarkers for Cannabidiol Exposure and Anti-inflammatory Efficacy. 2024, submitted.



{kind=link}
{kind=link}
{kind=link}
| EAE/CO Mouse ID 1 | Clinical Score |
|---|---|
| 21A | 2 |
| 22A | 0 |
| 23A | 0 |
| 24A | 2.5 |
| 25A | 0 |
| 21B | 2.25 |
| 22B | 0 |
| 23B | 0 |
| 24B | 2 |
| 25B | 2.25 |
| EAE Canonical Pathways 1 | −log (p-Value) | z-Score |
|---|---|---|
| Pathogen-Induced Cytokine Storm Signaling Pathway | 27.4 | 6.708 |
| Multiple Sclerosis Signaling Pathway | 21.8 | 5.657 |
| Macrophage Classical Activation Signaling Pathway | 21.7 | 4.747 |
| Th1 Pathway | 17.3 | 4.264 |
| Role of Hypercytokinemia/hyperchemokinemia in the Pathogenesis of Influenza | 16.8 | 4.359 |
| Neuroinflammation Signaling Pathway | 15.3 | 5 |
| Th2 Pathway | 15 | 2.982 |
| Interferon Signaling | 13.2 | 2.714 |
| Role of Pattern Recognition Receptors in Recognition of Bacteria and Viruses | 12.8 | 3.464 |
| Acute Phase Response Signaling | 12.4 | 3.051 |
| Pyroptosis Signaling Pathway | 12.4 | 4 |
| IL-10 Signaling | 11.9 | −2.524 |
| Crosstalk between Dendritic Cells and Natural Killer Cells | 11.4 | 3.742 |
| PD-1, PD-L1 cancer immunotherapy pathway | 11.4 | −2.5 |
| Dendritic Cell Maturation | 10.9 | 4.642 |
| Natural Killer Cell Signaling | 10.9 | 2.524 |
| Production of Nitric Oxide and Reactive Oxygen Species in Macrophages | 10.2 | 4 |
| TREM1 Signaling | 10.1 | 3.606 |
| Phagosome Formation | 9.68 | 5.916 |
| EAE Canonical Pathways | −log (p-Value) | z-Score |
|---|---|---|
| Interferon Signaling | 13.2 | 2.714 |
| IL-10 Signaling | 11.9 | −2.524 |
| IL-17 Signaling | 5.41 | 3.606 |
| IL-33 Signaling Pathway | 4.71 | 2.714 |
| HMGB1 Signaling | 4.45 | 2.828 |
| IL-6 Signaling | 3.93 | 2.333 |
| IL-15 Production | 3.35 | 2.828 |
| IL-9 Signaling | 2.79 | 2 |
| IL-4 Signaling | 2.66 | 3.3 |
| IL-8 Signaling | 2.42 | 2.828 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frodella, C.M.; Pruett, S.B.; Kaplan, B.L.F. Mild Disease Course of Experimental Autoimmune Encephalomyelitis without Pertussis Toxin: Brain Transcriptome Analysis Reveals Similar Signaling to Active Lesions in Multiple Sclerosis. Biomedicines 2024, 12, 1215. https://doi.org/10.3390/biomedicines12061215
Frodella CM, Pruett SB, Kaplan BLF. Mild Disease Course of Experimental Autoimmune Encephalomyelitis without Pertussis Toxin: Brain Transcriptome Analysis Reveals Similar Signaling to Active Lesions in Multiple Sclerosis. Biomedicines. 2024; 12(6):1215. https://doi.org/10.3390/biomedicines12061215
Chicago/Turabian StyleFrodella, Christa M., Stephen B. Pruett, and Barbara L. F. Kaplan. 2024. "Mild Disease Course of Experimental Autoimmune Encephalomyelitis without Pertussis Toxin: Brain Transcriptome Analysis Reveals Similar Signaling to Active Lesions in Multiple Sclerosis" Biomedicines 12, no. 6: 1215. https://doi.org/10.3390/biomedicines12061215
APA StyleFrodella, C. M., Pruett, S. B., & Kaplan, B. L. F. (2024). Mild Disease Course of Experimental Autoimmune Encephalomyelitis without Pertussis Toxin: Brain Transcriptome Analysis Reveals Similar Signaling to Active Lesions in Multiple Sclerosis. Biomedicines, 12(6), 1215. https://doi.org/10.3390/biomedicines12061215