

SARS-CoV-2 Spike Protein 1 Causes Aggregation of α-Synuclein via Microglia-Induced Inflammation and Production of Mitochondrial ROS: Potential Therapeutic Applications of Metformin

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Method
2.1. Reagents
2.2. Animals and SARS-CoV-2 Spike Protein 1 Administration
2.3. Immunohistochemistry
2.4. BE(2)-M17 Cell Culture
2.5. BV-2 Microglial Cell Culture and Conditioned Media (CM) Preparation
2.6. Immunoblot
2.7. Multiplex Analysis of Cytokines in BV-2 CM
2.8. Cell Transfection
2.9. Measurement of Mitochondrial Redox Status and Damage
2.10. Statical Analysis
3. Results
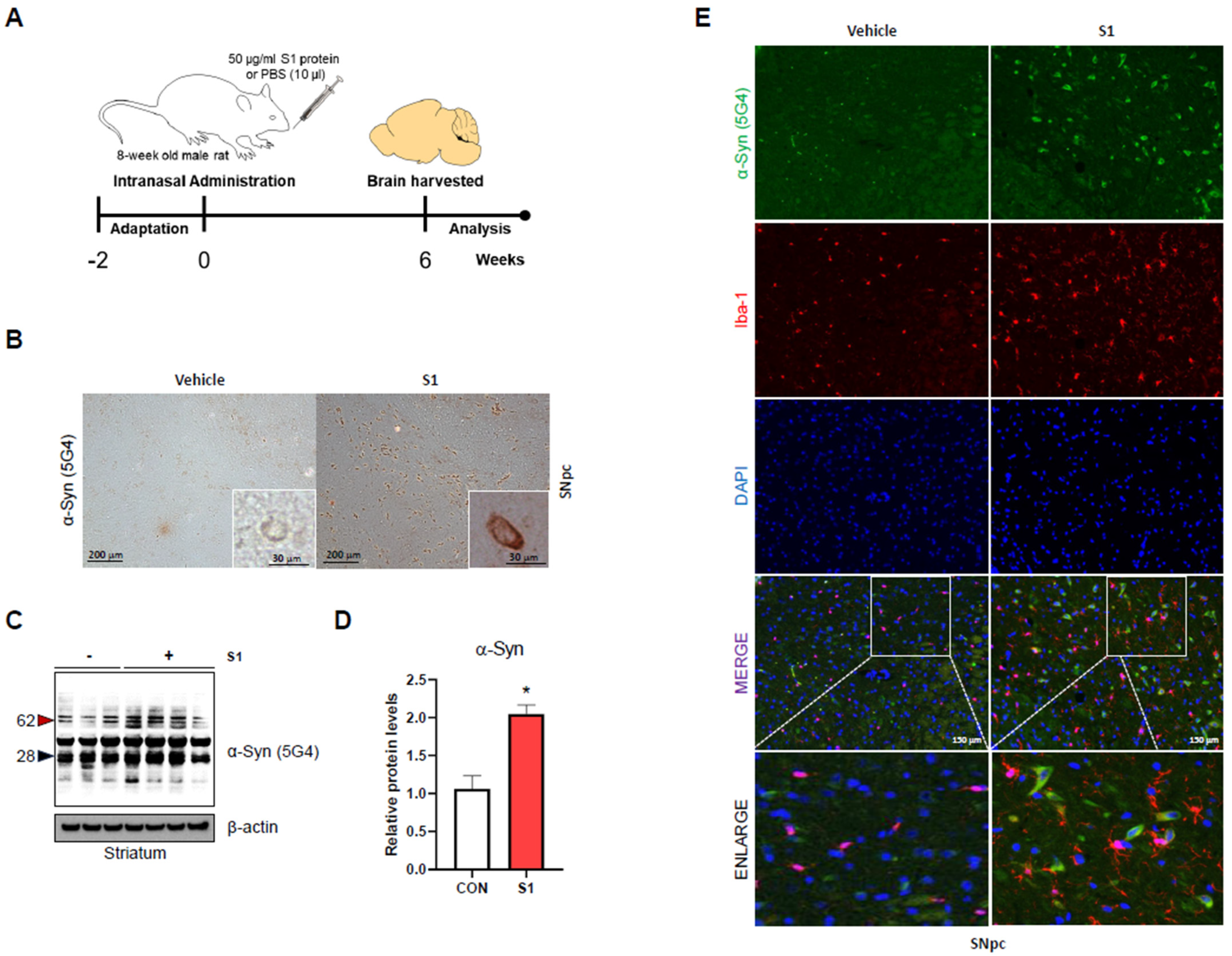
3.1. SARS-CoV-2 S1 Increases α-Syn Aggregation and Microglial Cell Activation in Rat Brain
3.2. SARS-CoV-2 S1 Increases Aggregation, Phosphorylation, and Monomer Levels of α-Syn via the Microglial Pro-Inflammatory Response
3.3. SARS-CoV-2 S1 Directly Increases Aggregation of α-Syn Only if α-Syn Levels Are Sufficient
3.4. SARS-CoV-2 S1-Induced Microglial Activation Enhances MPP+-Induced Cytotoxicity and α-Syn Aggregation and the Attenuating Effects of Metformin
3.5. SARS-CoV-2 S1 Directly Enhances MPP+-Induced Cytotoxicity and α-Syn Aggregation, with Attenuating Effects of Metformin
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Alafuzoff, I.; Hartikainen, P. Chapter 24—Alpha-synucleinopathies. In Handbook of Clinical Neurology; Kovacs, G.G., Alafuzoff, I., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 339–353. Available online: https://www.sciencedirect.com/science/article/pii/B9780128023952000249 (accessed on 14 December 2023).
- McCann, H.; Stevens, C.H.; Cartwright, H.; Halliday, G.M. α-Synucleinopathy phenotypes. Park. Relat. Disord. 2014, 20, S62–S67. [Google Scholar] [CrossRef] [PubMed]
- Stefanis, L. α-Synuclein in Parkinson’s Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009399. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. Neurodegenerative Diseases and Prions. N. Engl. J. Med. 2001, 344, 1516–1526. [Google Scholar] [CrossRef]
- Deleidi, M.; Gasser, T. The role of inflammation in sporadic and familial Parkinson’s disease. Cell. Mol. Life Sci. 2013, 70, 4259–4273. [Google Scholar] [CrossRef]
- Vlajinac, H.; Dzoljic, E.; Maksimovic, J.; Marinkovic, J.; Sipetic, S.; Kostic, V. Infections as a risk factor for Parkinson’s disease: A case–control study. Int. J. Neurosci. 2013, 123, 329–332. [Google Scholar] [CrossRef] [PubMed]
- Harris, M.A.; Tsui, J.K.; Marion, S.A.; Shen, H.; Teschke, K. Association of Parkinson’s disease with infections and occupational exposure to possible vectors. Mov. Disord. 2012, 27, 1111–1117. Available online: https://movementdisorders.onlinelibrary.wiley.com/doi/abs/10.1002/mds.25077 (accessed on 14 December 2023). [CrossRef]
- Mccall, S.; Henry, J.M.; Reid, A.H.; Taubenberger, J.K. Influenza RNA not Detected in Archival Brain Tissues from Acute Encephalitis Lethargica Cases or in Postencephalitic Parkinson Cases. J. Neuropathol. Exp. Neurol. 2001, 60, 696–704. [Google Scholar] [CrossRef]
- Sheng, Z.-M.; Chertow, D.S.; Ambroggio, X.; McCall, S.; Przygodzki, R.M.; Cunningham, R.E.; Maximova, O.A.; Kash, J.C.; Morens, D.M.; Taubenberger, J.K. Autopsy series of 68 cases dying before and during the 1918 influenza pandemic peak. Proc. Natl. Acad. Sci. USA 2011, 108, 16416–16421. [Google Scholar] [CrossRef]
- Marreiros, R.; Müller-Schiffmann, A.; Trossbach, S.V.; Prikulis, I.; Hänsch, S.; Weidtkamp-Peters, S.; Moreira, A.R.; Sahu, S.; Soloviev, I.; Selvarajah, S.; et al. Disruption of cellular proteostasis by H1N1 influenza A virus causes α-synuclein aggregation. Proc. Natl. Acad. Sci. USA 2020, 117, 6741–6751. [Google Scholar] [CrossRef]
- Jang, H.; Boltz, D.; Sturm-Ramirez, K.; Shepherd, K.R.; Jiao, Y.; Webster, R.; Smeyne, R.J. Highly pathogenic H5N1 influenza virus can enter the central nervous system and induce neuroinflammation and neurodegeneration. Proc. Natl. Acad. Sci. USA 2009, 106, 14063–14068. [Google Scholar] [CrossRef]
- Lai, C.-C.; Shih, T.-P.; Ko, W.-C.; Tang, H.-J.; Hsueh, P.-R. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and coronavirus disease-2019 (COVID-19): The epidemic and the challenges. Int. J. Antimicrob. Agents 2020, 55, 105924. [Google Scholar] [CrossRef]
- Davis, H.E.; McCorkell, L.; Vogel, J.M.; Topol, E.J. Long COVID: Major findings, mechanisms and recommendations. Nat. Rev. Microbiol. 2023, 21, 133–146. [Google Scholar] [CrossRef]
- Theoharides, T.C. Could SARS-CoV-2 Spike Protein Be Responsible for Long-COVID Syndrome? Mol. Neurobiol. 2022, 59, 1850–1861. [Google Scholar] [CrossRef] [PubMed]
- Shalash, A.; Helmy, A.; Salama, M.; Gaber, A.; El-Belkimy, M.; Hamid, E. A 6-month longitudinal study on worsening of Parkinson’s disease during the COVID-19 pandemic. Npj Park. Dis. 2022, 8, 111. [Google Scholar] [CrossRef]
- Xu, E.; Xie, Y.; Al-Aly, Z. Long-term neurologic outcomes of COVID-19. Nat. Med. 2022, 28, 2406–2415. [Google Scholar] [CrossRef] [PubMed]
- Fearon, C.; Fasano, A. Parkinson’s Disease and the COVID-19 Pandemic. J. Park. Dis. 2021, 11, 431–444. [Google Scholar] [CrossRef]
- Rao, A.R.; Hidayathullah, S.M.; Hegde, K.; Adhikari, P. Parkinsonism: An emerging post COVID sequelae. IDCases 2022, 27, e01388. [Google Scholar] [CrossRef]
- Wu, L.; Shen, J.; Jiang, Y.; Shen, X.; Wang, P.; Nie, X.; Kang, W.; Liu, J.; Chen, W. Clinical characteristics and outcome of COVID-19 patients with Parkinson’s disease: A hospital-based case–control study in Shanghai, China. Front. Aging Neurosci. 2023, 15, 1138418. [Google Scholar] [CrossRef] [PubMed]
- VanElzakker, M.B.; Bues, H.F.; Brusaferri, L.; Kim, M.; Saadi, D.; Ratai, E.-M.; Dougherty, D.D.; Loggia, M.L. Neuroinflammation in post-acute sequelae of COVID-19 (PASC) as assessed by [11C]PBR28 PET correlates with vascular disease measures. bioRxiv 2023. [Google Scholar] [CrossRef]
- Patterson, B.K.; Francisco, E.B.; Yogendra, R.; Long, E.; Pise, A.; Rodrigues, H.; Hall, E.; Herrera, M.; Parikh, P.; Guevara-Coto, J.; et al. Persistence of SARS CoV-2 S1 Protein in CD16+ Monocytes in Post-Acute Sequelae of COVID-19 (PASC) up to 15 Months Post-Infection. Front. Immunol. 2022, 12, 746021. [Google Scholar] [CrossRef]
- Craddock, V.; Mahajan, A.; Krishnamachary, B.; Spikes, L.; Chalise, P.; Dhillon, N.K. Persistent Presence of Spike protein and Viral RNA in the Circulation of Individuals with Post-Acute Sequelae of COVID-19. medRxiv 2022. [Google Scholar] [CrossRef]
- Rong, Z.; Mai, H.; Kapoor, S.; Puelles, V.G.; Czogalla, J.; Schädler, J.; Vering, J.; Delbridge, C.; Steinke, H.; Frenzel, H.; et al. SARS-CoV-2 Spike Protein Accumulation in the Skull-Meninges-Brain Axis: Potential Implications for Long-Term Neurological Complications in post-COVID-19. bioRxiv 2023. [Google Scholar] [CrossRef]
- Fontes-Dantas, F.L.; Fernandes, G.G.; Gutman, E.G.; De Lima, E.V.; Antonio, L.S.; Hammerle, M.B.; Mota-Araujo, H.P.; Colodeti, L.C.; Araújo, S.M.B.; Froz, G.M.; et al. SARS-CoV-2 Spike protein induces TLR4-mediated long-term cognitive dysfunction recapitulating post-COVID-19 syndrome in mice. Cell Rep. 2023, 42, 112189. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.; Cho, W.-H.; Barcelon, E.; Kim, K.H.; Hong, J.; Lee, S.J. SARS-CoV-2 spike protein induces cognitive deficit and anxiety-like behavior in mouse via non-cell autonomous hippocampal neuronal death. Sci. Rep. 2022, 12, 5496. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.S.; Koh, S.-H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 42. [Google Scholar] [CrossRef] [PubMed]
- Allen Reish, H.E.; Standaert, D.G. Role of α-synuclein in inducing innate and adaptive immunity in Parkinson disease. J. Park. Dis. 2015, 5, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Faustini, G.; Bono, F.; Valerio, A.; Pizzi, M.; Spano, P.; Bellucci, A. Mitochondria and α-Synuclein: Friends or Foes in the Pathogenesis of Parkinson’s Disease? Genes 2017, 8, 377. [Google Scholar] [CrossRef] [PubMed]
- Huynh, T.V.; Rethi, L.; Lee, T.-W.; Higa, S.; Kao, Y.-H.; Chen, Y.-J. Spike Protein Impairs Mitochondrial Function in Human Cardiomyocytes: Mechanisms Underlying Cardiac Injury in COVID-19. Cells 2023, 12, 877. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Nguyen, V.; Tsai, J.; Gao, C.; Tian, Y.; Zhang, Y.; Carver, W.; Kiaris, H.; Cui, T.; Tan, W. The SARS-CoV-2 spike protein induces long-term transcriptional perturbations of mitochondrial metabolic genes, causes cardiac fibrosis, and reduces myocardial contractile in obese mice. Mol. Metab. 2023, 74, 101756. [Google Scholar] [CrossRef]
- Clough, E.; Inigo, J.; Chandra, D.; Chaves, L.; Reynolds, J.L.; Aalinkeel, R.; Schwartz, S.A.; Khmaladze, A.; Mahajan, S.D. Mitochondrial Dynamics in SARS-CoV2 Spike Protein Treated Human Microglia: Implications for Neuro-COVID. J. Neuroimmune Pharmacol. 2021, 16, 770–784. [Google Scholar] [CrossRef]
- Wang, J.; Dai, L.; Deng, M.; Xiao, T.; Zhang, Z.; Zhang, Z. SARS-CoV-2 Spike Protein S1 Domain Accelerates α-Synuclein Phosphorylation and Aggregation in Cellular Models of Synucleinopathy. Mol. Neurobiol. 2023, 61, 2446–2458. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Zhang, X.; Huang, Z.; Ma, K. SARS-CoV-2 Proteins Interact with Alpha Synuclein and Induce Lewy Body-like Pathology In Vitro. Int. J. Mol. Sci. 2022, 23, 3394. [Google Scholar] [CrossRef] [PubMed]
- Pernicova, M. Korbonits Metformin-mode of action and clinical implications for diabetes and cancer. Nat. Rev. Endocrinol. 2014, 10, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Xenos, D.; Mecocci, P.; Boccardi, V. A blast from the past: To tame time with metformin. Mech. Ageing Dev. 2022, 208, 111743. [Google Scholar] [CrossRef] [PubMed]
- Katila, N.; Bhurtel, S.; Shadfar, S.; Srivastav, S.; Neupane, S.; Ojha, U.; Jeong, G.-S.; Choi, D.-Y. Metformin lowers α-synuclein phosphorylation and upregulates neurotrophic factor in the MPTP mouse model of Parkinson’s disease. Neuropharmacology 2017, 125, 396–407. [Google Scholar] [CrossRef] [PubMed]
- Łabuzek, K.; Suchy, D.; Gabryel, B.; Bielecka, A.; Liber, S.; Okopień, B. Quantification of metformin by the HPLC method in brain regions, cerebrospinal fluid and plasma of rats treated with lipopolysaccharide. Pharmacol. Rep. 2010, 62, 956–965. [Google Scholar] [CrossRef]
- Jiang, T.; Yu, J.; Zhu, X.; Wang, H.; Tan, M.; Cao, L.; Zhang, Q.; Gao, L.; Shi, J.; Zhang, Y.; et al. Acute metformin preconditioning confers neuroprotection against focal cerebral ischaemia by pre-activation of AMPK-dependent autophagy. Br. J. Pharmacol. 2014, 171, 3146–3157. [Google Scholar] [CrossRef]
- Ng, T.P.; Feng, L.; Yap, K.B.; Lee, T.S.; Tan, C.H.; Winblad, B. Long-Term Metformin Usage and Cognitive Function among Older Adults with Diabetes. J. Alzheimer’s Dis. 2014, 41, 61–68. [Google Scholar] [CrossRef]
- Patil, S.P.; Jain, P.D.; Ghumatkar, P.J.; Tambe, R.; Sathaye, S. Neuroprotective effect of metformin in MPTP-induced Parkinson’s disease in mice. Neuroscience 2014, 277, 747–754. [Google Scholar] [CrossRef]
- Wahlqvist, M.L.; Lee, M.-S.; Hsu, C.-C.; Chuang, S.-Y.; Lee, J.-T.; Tsai, H.-N. Metformin-inclusive sulfonylurea therapy reduces the risk of Parkinson’s disease occurring with Type 2 diabetes in a Taiwanese population cohort. Park. Relat. Disord. 2012, 18, 753–758. [Google Scholar] [CrossRef]
- Lu, M.; Su, C.; Qiao, C.; Bian, Y.; Ding, J.; Hu, G. Metformin Prevents Dopaminergic Neuron Death in MPTP/P-Induced Mouse Model of Parkinson’s Disease via Autophagy and Mitochondrial ROS Clearance. Int. J. Neuropsychopharmacol. 2016, 19, pyw047. [Google Scholar] [CrossRef] [PubMed]
- Orihuela, R.; McPherson, C.A.; Harry, G.J. Microglial M1/M2 polarization and metabolic states. Br. J. Pharmacol. 2016, 173, 649–665. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.-K.; Bae, E.-J.; Jung, B.C.; Choi, M.; Shin, S.J.; Park, S.J.; Kim, J.T.; Jung, M.K.; Ulusoy, A.; Song, M.-Y.; et al. Inflammation promotes synucleinopathy propagation. Exp. Mol. Med. 2022, 54, 2148–2161. [Google Scholar] [CrossRef]
- Lim, S.; Chun, Y.; Lee, J.S.; Lee, S.-J. Neuroinflammation in Synucleinopathies. Brain Pathol. 2016, 26, 404. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Kato, T.; Arawaka, S. The role of Ser129 phosphorylation of α-synuclein in neurodegeneration of Parkinson’s disease: A review of in vivo models. Rev. Neurosci. 2013, 24, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Zekri-Nechar, K.; Zamorano-León, J.J.; Reche, C.; Giner, M.; López-de-Andrés, A.; Jiménez-García, R.; López-Farré, A.J.; Martínez-Martínez, C.H. Spike Protein Subunits of SARS-CoV-2 Alter Mitochondrial Metabolism in Human Pulmonary Microvascular Endothelial Cells: Involvement of Factor Xa. Dis. Markers 2022, 2022, 1118195. [Google Scholar] [CrossRef]
- Choi, M.L.; Chappard, A.; Singh, B.P.; Maclachlan, C.; Rodrigues, M.; Fedotova, E.I.; Berezhnov, A.V.; De, S.; Peddie, C.J.; Athauda, D.; et al. Pathological structural conversion of α-synuclein at the mitochondria induces neuronal toxicity. Nat. Neurosci. 2022, 25, 1134–1148. [Google Scholar] [CrossRef]
- Kowall, N.W.; Hantraye, P.; Brouillet, E.; Beal, M.F.; McKee, A.C.; Ferrante, R.J. MPTP induces alpha-synuclein aggregation in the substantia nigra of baboons. NeuroReport 2000, 11, 211. [Google Scholar] [CrossRef] [PubMed]
- Bramante, C.T.; Buse, J.B.; Liebovitz, D.M.; Nicklas, J.M.; Puskarich, M.A.; Cohen, K.; Belani, H.K.; Anderson, B.J.; Huling, J.D.; Tignanelli, C.J.; et al. Outpatient treatment of COVID-19 and incidence of post-COVID-19 condition over 10 months (COVID-OUT): A multicentre, randomised, quadruple-blind, parallel-group, phase 3 trial. Lancet Infect. Dis. 2023, 23, 1119–1129. [Google Scholar] [CrossRef]
- Bourget, C.; Adams, K.V.; Morshead, C.M. Reduced microglia activation following metformin administration or microglia ablation is sufficient to prevent functional deficits in a mouse model of neonatal stroke. J. Neuroinflamm. 2022, 19, 146. [Google Scholar] [CrossRef]
- DiBona, V.L.; Shah, M.K.; Krause, K.J.; Zhu, W.; Voglewede, M.M.; Smith, D.M.; Crockett, D.P.; Zhang, H. Metformin reduces neuroinflammation and improves cognitive functions after traumatic brain injury. Neurosci. Res. 2021, 172, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Limphaibool, N.; Iwanowski, P.; Holstad, M.J.V.; Kobylarek, D.; Kozubski, W. Infectious Etiologies of Parkinsonism: Pathomechanisms and Clinical Implications. Front. Neurol. 2019, 10, 652. [Google Scholar] [CrossRef] [PubMed]
- Almutairi, M.M.; Sivandzade, F.; Albekairi, T.H.; Alqahtani, F.; Cucullo, L. Neuroinflammation and Its Impact on the Pathogenesis of COVID-19. Front. Med. 2021, 8, 745789. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.; Soung, A.; Sissoko, C.; Nordvig, A.; Canoll, P.; Mariani, M.; Jiang, X.; Bricker, T.; Goldman, J.; Rosoklija, G.; et al. COVID-19 induces neuroinflammation and loss of hippocampal neurogenesis. Res. Sq. 2021. [Google Scholar] [CrossRef] [PubMed]
- Lenz, K.M.; Nelson, L.H. Microglia and Beyond: Innate Immune Cells As Regulators of Brain Development and Behavioral Function. Front. Immunol. 2018, 9, 698. [Google Scholar] [CrossRef] [PubMed]
- Schultheiß, C.; Willscher, E.; Paschold, L.; Gottschick, C.; Klee, B.; Henkes, S.-S.; Bosurgi, L.; Dutzmann, J.; Sedding, D.; Frese, T.; et al. From online data collection to identification of disease mechanisms: The IL-1ß, IL-6 and TNF-α cytokine triad is associated with post-acute sequelae of COVID-19 in a digital research cohort. medRxiv 2021. [Google Scholar]
- Patterson, B.K.; Guevara-Coto, J.; Yogendra, R.; Francisco, E.B.; Long, E.; Pise, A.; Rodrigues, H.; Parikh, P.; Mora, J.; Mora-Rodríguez, R.A. Immune-Based Prediction of COVID-19 Severity and Chronicity Decoded Using Machine Learning. Front. Immunol. 2021, 12, 700782. [Google Scholar] [CrossRef] [PubMed]
- Menza, M.; DeFronzo Dobkin, R.; Marin, H.; Mark, M.H.; Gara, M.; Bienfait, K.; Dicke, A.; Kusnekov, A. The Role of Inflammatory Cytokines in Cognition and Other Non-Motor Symptoms of Parkinson’s Disease. Psychosomatics 2010, 51, 474–479. [Google Scholar] [PubMed]
- Tang, P.; Chong, L.; Li, X.; Liu, Y.; Liu, P.; Hou, C.; Li, R. Correlation between Serum RANTES Levels and the Severity of Parkinson’s Disease. Oxidative Med. Cell. Longev. 2014, 2014, 208408. [Google Scholar] [CrossRef]
- Festa, B.P.; Siddiqi, F.H.; Jimenez-Sanchez, M.; Won, H.; Rob, M.; Djajadikerta, A.; Stamatakou, E.; Rubinsztein, D.C. Microglial-to-neuronal CCR5 signaling regulates autophagy in neurodegeneration. Neuron 2023, 111, 2021–2037.e12. [Google Scholar] [CrossRef]
- Zhao, J.; An, K.; Mao, Z.; Qu, Y.; Wang, D.; Li, J.; Min, Z.; Xue, Z. CCL5 promotes LFA-1 expression in Th17 cells and induces LCK and ZAP70 activation in a mouse model of Parkinson’s disease. Front. Aging Neurosci. 2023, 15, 1250685. [Google Scholar] [CrossRef] [PubMed]
- Bae, E.-J.; Choi, M.; Kim, J.T.; Kim, D.-K.; Jung, M.K.; Kim, C.; Kim, T.-K.; Lee, J.S.; Jung, B.C.; Shin, S.J.; et al. TNF-α promotes α-synuclein propagation through stimulation of senescence-associated lysosomal exocytosis. Exp. Mol. Med. 2022, 54, 788–800. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, J.C.; Moreira, T.C.L.; De Araújo, A.L.; Imamura, M.; Damiano, R.F.; Garcia, M.L.; Sawamura, M.V.; Pinna, F.R.; Guedes, B.F.; Gonçalves, F.A.R.; et al. Clinical, sociodemographic and environmental factors impact post-COVID-19 syndrome. J. Glob. Health 2022, 12, 05029. [Google Scholar] [CrossRef]
- Langston, J.W. The MPTP Story. J. Park. Dis. 2017, 7, S11–S19. [Google Scholar] [CrossRef]
- Sadasivan, S.; Sharp, B.; Schultz-Cherry, S.; Smeyne, R.J. Synergistic effects of influenza and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) can be eliminated by the use of influenza therapeutics: Experimental evidence for the multi-hit hypothesis. Npj Park. Dis. 2017, 3, 18. [Google Scholar] [CrossRef] [PubMed]
- Parthasarathy, H.; Tandel, D.; Siddiqui, A.H.; Harshan, K.H. Metformin suppresses SARS-CoV-2 in cell culture. Virus Res. 2023, 323, 199010. [Google Scholar] [CrossRef]
- Ventura-López, C.; Cervantes-Luevano, K.; Aguirre-Sánchez, J.S.; Flores-Caballero, J.C.; Alvarez-Delgado, C.; Bernaldez-Sarabia, J.; Sánchez-Campos, N.; Lugo-Sánchez, L.A.; Rodríguez-Vázquez, I.C.; Sander-Padilla, J.G.; et al. Treatment with metformin glycinate reduces SARS-CoV-2 viral load: An in vitro model and randomized, double-blind, Phase IIb clinical trial. Biomed. Pharmacother. 2022, 152, 113223. [Google Scholar] [CrossRef]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef]
- Schaller, M.A.; Sharma, Y.; Dupee, Z.; Nguyen, D.; Urueña, J.; Smolchek, R.; Loeb, J.C.; Machuca, T.N.; Lednicky, J.A.; Odde, D.J.; et al. Ex vivo SARS-CoV-2 infection of human lung reveals heterogeneous host defense and therapeutic responses. JCI Insight 2021, 6, e148003. [Google Scholar] [CrossRef]
- Andrzejewski, S.; Gravel, S.-P.; Pollak, M.; St-Pierre, J. Metformin directly acts on mitochondria to alter cellular bioenergetics. Cancer Metab. 2014, 2, 12. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, M.H.; Park, J.H.; Lee, H.K.; Choi, J.Y.; Koh, Y.H. SARS-CoV-2 Spike Protein 1 Causes Aggregation of α-Synuclein via Microglia-Induced Inflammation and Production of Mitochondrial ROS: Potential Therapeutic Applications of Metformin. Biomedicines 2024, 12, 1223. https://doi.org/10.3390/biomedicines12061223
Chang MH, Park JH, Lee HK, Choi JY, Koh YH. SARS-CoV-2 Spike Protein 1 Causes Aggregation of α-Synuclein via Microglia-Induced Inflammation and Production of Mitochondrial ROS: Potential Therapeutic Applications of Metformin. Biomedicines. 2024; 12(6):1223. https://doi.org/10.3390/biomedicines12061223
Chicago/Turabian StyleChang, Moon Han, Jung Hyun Park, Hye Kyung Lee, Ji Young Choi, and Young Ho Koh. 2024. "SARS-CoV-2 Spike Protein 1 Causes Aggregation of α-Synuclein via Microglia-Induced Inflammation and Production of Mitochondrial ROS: Potential Therapeutic Applications of Metformin" Biomedicines 12, no. 6: 1223. https://doi.org/10.3390/biomedicines12061223