
APOA1/C3/A4/A5 Gene Cluster at 11q23.3 and Lipid Metabolism Disorders: From Epigenetic Mechanisms to Clinical Practices
Abstract
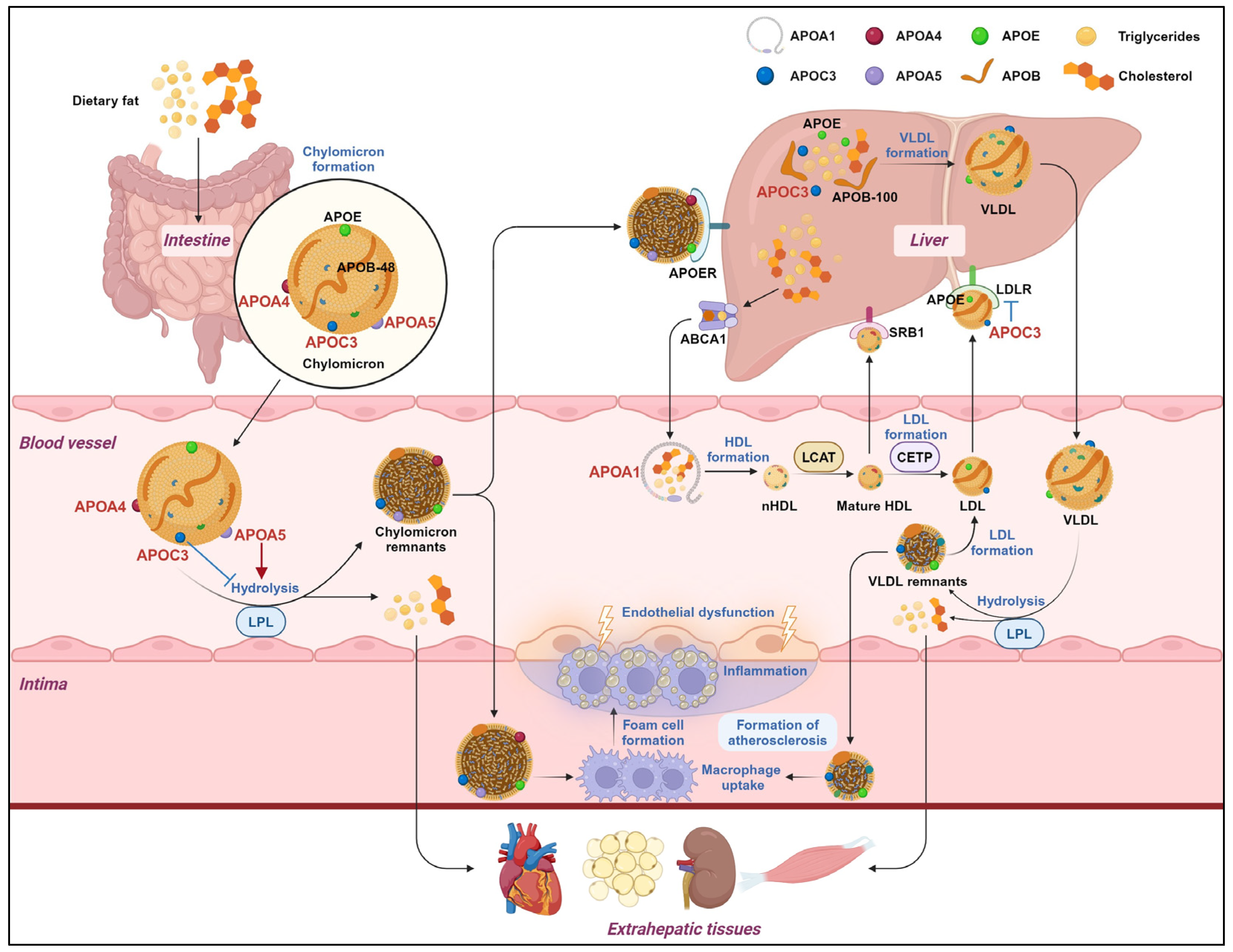
:1. Introduction
2. APOA1: Proteins with Therapeutic Potential for Lipid Metabolism Disorders
2.1. Function of APOA1
2.2. Epigenetic Regulation and Therapeutic Potential of APOA1
3. APOC3: An Emerging Target in the Field of Lipid-Lowering Therapy
3.1. Function of APOC3
3.2. Epigenetic Regulation and Therapeutic Potential of APOC3
4. APOA4: Biomarker for Disorders of Lipid Metabolism
4.1. Function of APOA4
4.2. The Epigenetic Regulation and Therapeutic Potential of APOA4
5. APOA5: A Regulator of Obesity and Metabolic Syndrome
5.1. Function of APOA5
5.2. Epigenetic Regulation and Therapeutic Potential of APOA5
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Berberich, A.J.; Hegele, R.A. A Modern Approach to Dyslipidemia. Endocr. Rev. 2022, 43, 611–653. [Google Scholar] [CrossRef] [PubMed]
- Kopin, L.; Lowenstein, C. Dyslipidemia. Ann. Intern. Med. 2017, 167, ITC81–ITC96. [Google Scholar] [CrossRef] [PubMed]
- Arvanitis, M.; Lowenstein, C.J. Dyslipidemia. Ann. Intern. Med. 2023, 176, ITC81–ITC96. [Google Scholar] [CrossRef] [PubMed]
- Nussbaumerova, B.; Rosolova, H. Obesity and Dyslipidemia. Curr. Atheroscler. Rep. 2023, 25, 947–955. [Google Scholar] [CrossRef] [PubMed]
- Bahiru, E.; Hsiao, R.; Phillipson, D.; Watson, K.E. Mechanisms and Treatment of Dyslipidemia in Diabetes. Curr. Cardiol. Rep. 2021, 23, 26. [Google Scholar] [CrossRef] [PubMed]
- Machado, M.V.; Diehl, A.M. Pathogenesis of Nonalcoholic Steatohepatitis. Gastroenterology 2016, 150, 1769–1777. [Google Scholar] [CrossRef] [PubMed]
- Katsiki, N.; Mikhailidis, D.P.; Mantzoros, C.S. Non-alcoholic fatty liver disease and dyslipidemia: An update. Metabolism 2016, 65, 1109–1123. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.L.; McNabb-Baltar, J. Hypertriglyceridemia and acute pancreatitis. Pancreatology 2020, 20, 795–800. [Google Scholar] [CrossRef] [PubMed]
- Baass, A.; Paquette, M.; Bernard, S.; Hegele, R.A. Familial chylomicronemia syndrome: An under-recognized cause of severe hypertriglyceridaemia. J. Intern. Med. 2020, 287, 340–348. [Google Scholar] [CrossRef]
- Parhofer, K.G.; Laufs, U. Lipid Profile and Lipoprotein(a) Testing. Dtsch. Arztebl. Int. 2023, 120, 582–588. [Google Scholar] [CrossRef]
- Basavaraju, P.; Balasubramani, R.; Kathiresan, D.S.; Devaraj, I.; Babu, K.; Alagarsamy, V.; Puthamohan, V.M. Genetic Regulatory Networks of Apolipoproteins and Associated Medical Risks. Front. Cardiovasc. Med. 2021, 8, 788852. [Google Scholar] [CrossRef]
- Lai, C.-Q.; Parnell, L.D.; Ordovas, J.M. The APOA1/C3/A4/A5 gene cluster, lipid metabolism and cardiovascular disease risk. Curr. Opin. Lipidol. 2005, 16, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Zhang, Z.; Yao, C.; Zhao, S. Emerging evidences for the opposite role of apolipoprotein C3 and apolipoprotein A5 in lipid metabolism and coronary artery disease. Lipids Health Dis. 2019, 18, 220. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.-B.; Zhou, Y.-F.; Yao, J.-L.; Sun, S.-J.; Rui, Q.; Yang, X.-J.; Li, X.-B. Apolipoprotein A1 polymorphisms and risk of coronary artery disease: A meta-analysis. Arch. Med. Sci. AMS 2017, 13, 813–819. [Google Scholar] [CrossRef] [PubMed]
- de Luis, D.A.; Izaola, O.; Primo, D.; Aller, R. Role of rs670 variant of APOA1 gene on lipid profile, insulin resistance and adipokine levels in obese subjects after weight loss with a dietary intervention. Diabetes Res. Clin. Pract. 2018, 142, 139–145. [Google Scholar] [CrossRef] [PubMed]
- de Luis, D.; Izaola, O.; Primo, D.; Aller, R. Role of rs670 variant of APOA1 gene on metabolic response after a high fat vs. a low fat hypocaloric diets in obese human subjects. J. Diabetes Complicat. 2019, 33, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Tall, A.R.; Thomas, D.G.; Gonzalez-Cabodevilla, A.G.; Goldberg, I.J. Addressing dyslipidemic risk beyond LDL-cholesterol. J. Clin. Investig. 2022, 132, e148559. [Google Scholar] [CrossRef] [PubMed]
- Au, A.; Griffiths, L.R.; Irene, L.; Kooi, C.W.; Wei, L.K. The impact of APOA5, APOB, APOC3 and ABCA1 gene polymorphisms on ischemic stroke: Evidence from a meta-analysis. Atherosclerosis 2017, 265, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Azúa-López, Z.R.d.; Pezzotti, M.R.; González-Díaz, Á.; Meilhac, O.; Ureña, J.; Amaya-Villar, R.; Castellano, A.; Varela, L.M. HDL anti-inflammatory function is impaired and associated with high SAA1 and low APOA4 levels in aneurysmal subarachnoid hemorrhage. J. Cereb. Blood Flow. Metab. Off. J. Int. Soc. Cereb. Blood Flow. Metab. 2023, 43, 1919–1930. [Google Scholar] [CrossRef]
- Peters, K.E.; Xu, J.; Bringans, S.D.; Davis, W.A.; Davis, T.M.E.; Hansen, M.K.; Lipscombe, R.J. PromarkerD Predicts Renal Function Decline in Type 2 Diabetes in the Canagliflozin Cardiovascular Assessment Study (CANVAS). J. Clin. Med. 2020, 9, 3212. [Google Scholar] [CrossRef]
- Abdullah, M.M.H.; Vazquez-Vidal, I.; Baer, D.J.; House, J.D.; Jones, P.J.H.; Desmarchelier, C. Common Genetic Variations Involved in the Inter-Individual Variability of Circulating Cholesterol Concentrations in Response to Diets: A Narrative Review of Recent Evidence. Nutrients 2021, 13, 695. [Google Scholar] [CrossRef]
- Lin, Y.-C.; Nunez, V.; Johns, R.; Shiao, S.P.K. APOA5 Gene Polymorphisms and Cardiovascular Diseases: Metaprediction in Global Populations. Nurs. Res. 2017, 66, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Aung, L.H.H.; Yin, R.-X.; Wu, D.-F.; Wang, W.; Liu, C.-W.; Pan, S.-L. Association of the variants in the BUD13-ZNF259 genes and the risk of hyperlipidaemia. J. Cell. Mol. Med. 2014, 18, 1417–1428. [Google Scholar] [CrossRef] [PubMed]
- Säll, J.; Pettersson, A.M.L.; Björk, C.; Henriksson, E.; Wasserstrom, S.; Linder, W.; Zhou, Y.; Hansson, O.; Andersson, D.P.; Ekelund, M.; et al. Salt-inducible kinase 2 and -3 are downregulated in adipose tissue from obese or insulin-resistant individuals: Implications for insulin signalling and glucose uptake in human adipocytes. Diabetologia 2017, 60, 314–323. [Google Scholar] [CrossRef]
- Masjoudi, S.; Sedaghati-Khayat, B.; Givi, N.J.; Bonab, L.N.H.; Azizi, F.; Daneshpour, M.S. Kernel machine SNP set analysis finds the association of BUD13, ZPR1, and APOA5 variants with metabolic syndrome in Tehran Cardio-metabolic Genetics Study. Sci. Rep. 2021, 11, 10305. [Google Scholar] [CrossRef]
- Song, D.; Yin, L.; Wang, C.; Wen, X. Adenovirus-mediated expression of SIK1 improves hepatic glucose and lipid metabolism in type 2 diabetes mellitus rats. PLoS ONE 2019, 14, e0210930. [Google Scholar] [CrossRef] [PubMed]
- Fitz-James, M.H.; Cavalli, G. Molecular mechanisms of transgenerational epigenetic inheritance. Nat. Rev. Genet. 2022, 23, 325–341. [Google Scholar] [CrossRef]
- Nacev, B.A.; Jones, K.B.; Intlekofer, A.M.; Yu, J.S.E.; Allis, C.D.; Tap, W.D.; Ladanyi, M.; Nielsen, T.O. The epigenomics of sarcoma. Nat. Rev. Cancer 2020, 20, 608–623. [Google Scholar] [CrossRef]
- Rhee, E.-J.; Byrne, C.D.; Sung, K.-C. The HDL cholesterol/apolipoprotein A-I ratio: An indicator of cardiovascular disease. Curr. Opin. Endocrinol. Diabetes Obes. 2017, 24, 148–153. [Google Scholar] [CrossRef]
- Kluck, G.E.G.; Yoo, J.-A.; Sakarya, E.H.; Trigatti, B.L. Good Cholesterol Gone Bad? HDL and COVID-19. Int. J. Mol. Sci. 2021, 22, 10182. [Google Scholar] [CrossRef]
- Pownall, H.J.; Rosales, C.; Gillard, B.K.; Gotto, A.M. High-density lipoproteins, reverse cholesterol transport and atherogenesis. Nat. Rev. Cardiol. 2021, 18, 712–723. [Google Scholar] [CrossRef] [PubMed]
- Escolà-Gil, J.C.; Rotllan, N.; Julve, J.; Blanco-Vaca, F. Reverse Cholesterol Transport Dysfunction Is a Feature of Familial Hypercholesterolemia. Curr. Atheroscler. Rep. 2021, 23, 29. [Google Scholar] [CrossRef] [PubMed]
- Jomard, A.; Osto, E. High Density Lipoproteins: Metabolism, Function, and Therapeutic Potential. Front. Cardiovasc. Med. 2020, 7, 39. [Google Scholar] [CrossRef] [PubMed]
- Márquez, A.B.; Nazir, S.; van der Vorst, E.P.C. High-Density Lipoprotein Modifications: A Pathological Consequence or Cause of Disease Progression? Biomedicines 2020, 8, 549. [Google Scholar] [CrossRef]
- Genest, J.; Schwertani, A.; Choi, H.Y. Membrane microdomains and the regulation of HDL biogenesis. Curr. Opin. Lipidol. 2018, 29, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Kontush, A. HDL and Reverse Remnant-Cholesterol Transport (RRT): Relevance to Cardiovascular Disease. Trends Mol. Med. 2020, 26, 1086–1100. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Song, Z.; Mao, B.; Xu, G. Apolipoprotein A1-Related Proteins and Reverse Cholesterol Transport in Antiatherosclerosis Therapy: Recent Progress and Future Perspectives. Cardiovasc. Ther. 2022, 2022, 4610834. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. ApoA1 and ApoA1-specific self-antibodies in cardiovascular disease. Lab. Investig. J. Tech. Methods Pathol. 2016, 96, 708–718. [Google Scholar] [CrossRef] [PubMed]
- Tsompanidi, E.M.; Brinkmeier, M.S.; Fotiadou, E.H.; Giakoumi, S.M.; Kypreos, K.E. HDL biogenesis and functions: Role of HDL quality and quantity in atherosclerosis. Atherosclerosis 2010, 208, 3–9. [Google Scholar] [CrossRef]
- Singh, V.; Kaur, R.; Kumari, P.; Pasricha, C.; Singh, R. ICAM-1 and VCAM-1: Gatekeepers in various inflammatory and cardiovascular disorders. Clin. Chim. Acta Int. J. Clin. Chem. 2023, 548, 117487. [Google Scholar] [CrossRef]
- Li, J.; Wang, W.; Han, L.; Feng, M.; Lu, H.; Yang, L.; Hu, X.; Shi, S.; Jiang, S.; Wang, Q.; et al. Human apolipoprotein A-I exerts a prophylactic effect on high-fat diet-induced atherosclerosis via inflammation inhibition in a rabbit model. Acta Biochim. Biophys. Sin. 2017, 49, 149–158. [Google Scholar] [CrossRef]
- Iqbal, A.J.; Barrett, T.J.; Taylor, L.; McNeill, E.; Manmadhan, A.; Recio, C.; Carmineri, A.; Brodermann, M.H.; White, G.E.; Cooper, D.; et al. Acute exposure to apolipoprotein A1 inhibits macrophage chemotaxis in vitro and monocyte recruitment in vivo. eLife 2016, 5, e15190. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Li, L.; Xie, W.; Wu, J.-F.; Yao, F.; Tan, Y.-L.; Xia, X.-D.; Liu, X.-Y.; Liu, D.; Lan, G.; et al. Apolipoprotein A-1 binding protein promotes macrophage cholesterol efflux by facilitating apolipoprotein A-1 binding to ABCA1 and preventing ABCA1 degradation. Atherosclerosis 2016, 248, 149–159. [Google Scholar] [CrossRef]
- Halley, P.; Kadakkuzha, B.M.; Faghihi, M.A.; Magistri, M.; Zeier, Z.; Khorkova, O.; Coito, C.; Hsiao, J.; Lawrence, M.; Wahlestedt, C. Regulation of the apolipoprotein gene cluster by a long noncoding RNA. Cell Rep. 2014, 6, 222–230. [Google Scholar] [CrossRef]
- Ramasamy, T.; Ruttala, H.B.; Munusamy, S.; Chakraborty, N.; Kim, J.O. Nano drug delivery systems for antisense oligonucleotides (ASO) therapeutics. J. Control. Release Off. J. Control. Release Soc. 2022, 352, 861–878. [Google Scholar] [CrossRef]
- Kianmehr, A.; Qujeq, D.; Bagheri, A.; Mahrooz, A. Oxidized LDL-regulated microRNAs for evaluating vascular endothelial function: Molecular mechanisms and potential biomarker roles in atherosclerosis. Crit. Rev. Clin. Lab. Sci. 2022, 59, 40–53. [Google Scholar] [CrossRef]
- Wang, J.; Cai, Y.; Lu, H.; Zhang, F.; Zheng, J. LncRNA APOA1-AS facilitates proliferation and migration and represses apoptosis of VSMCs through TAF15-mediated SMAD3 mRNA stabilization. Cell Cycle 2021, 20, 1642–1652. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Wang, Q.; Yang, X.; Ren, Y.; Jiao, S.; Zhu, Q.; Guo, D.; Xia, K.; Wang, Y.; Li, C.; et al. Qishen granule attenuates cardiac fibrosis by regulating TGF-β/Smad3 and GSK-3β pathway. Phytomed. Int. J. Phytother. Phytopharm. 2019, 62, 152949. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Zhu, M.; Ju, J.; Yang, H. Effects of Dietary Cholesterol Regulation on Spermatogenesis of Gobiocypris rarus Rare Minnow. Int. J. Mol. Sci. 2023, 24, 7492. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, M.; Zhu, Z.; Yang, H.; Wei, W.; Li, B. Bisphenol A regulates apolipoprotein A1 expression through estrogen receptors and DNA methlylation and leads to cholesterol disorder in rare minnow testis. Aquat. Toxicol. 2021, 241, 105999. [Google Scholar] [CrossRef]
- Gangabadage, C.S.; Zdunek, J.; Tessari, M.; Nilsson, S.; Olivecrona, G.; Wijmenga, S.S. Structure and dynamics of human apolipoprotein CIII. J. Biol. Chem. 2008, 283, 17416–17427. [Google Scholar] [CrossRef] [PubMed]
- D’Erasmo, L.; Di Costanzo, A.; Gallo, A.; Bruckert, E.; Arca, M. ApoCIII: A multifaceted protein in cardiometabolic disease. Metabolism 2020, 113, 154395. [Google Scholar] [CrossRef] [PubMed]
- Wolska, A.; Lo, L.; Sviridov, D.O.; Pourmousa, M.; Pryor, M.; Ghosh, S.S.; Kakkar, R.; Davidson, M.; Wilson, S.; Pastor, R.W.; et al. A dual apolipoprotein C-II mimetic-apolipoprotein C-III antagonist peptide lowers plasma triglycerides. Sci. Transl. Med. 2020, 12, 528. [Google Scholar] [CrossRef]
- Larsson, M.; Allan, C.M.; Jung, R.S.; Heizer, P.J.; Beigneux, A.P.; Young, S.G.; Fong, L.G. Apolipoprotein C-III inhibits triglyceride hydrolysis by GPIHBP1-bound LPL. J. Lipid Res. 2017, 58, 1893–1902. [Google Scholar] [CrossRef]
- Qin, W.; Sundaram, M.; Wang, Y.; Zhou, H.; Zhong, S.; Chang, C.-C.; Manhas, S.; Yao, E.F.; Parks, R.J.; McFie, P.J.; et al. Missense mutation in APOC3 within the C-terminal lipid binding domain of human ApoC-III results in impaired assembly and secretion of triacylglycerol-rich very low density lipoproteins: Evidence that ApoC-III plays a major role in the formation of lipid precursors within the microsomal lumen. J. Biol. Chem. 2011, 286, 27769–27780. [Google Scholar] [PubMed]
- Gordts, P.L.S.M.; Nock, R.; Son, N.-H.; Ramms, B.; Lew, I.; Gonzales, J.C.; Thacker, B.E.; Basu, D.; Lee, R.G.; Mullick, A.E.; et al. ApoC-III inhibits clearance of triglyceride-rich lipoproteins through LDL family receptors. J. Clin. Investig. 2016, 126, 2855–2866. [Google Scholar] [CrossRef]
- Zha, Y.; Lu, Y.; Zhang, T.; Yan, K.; Zhuang, W.; Liang, J.; Cheng, Y.; Wang, Y. CRISPR/Cas9-mediated knockout of APOC3 stabilizes plasma lipids and inhibits atherosclerosis in rabbits. Lipids Health Dis. 2021, 20, 180. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, A.; Aikawa, M.; Libby, P.; Alcaide, P.; Luscinskas, F.W.; Sacks, F.M. Apolipoprotein CIII in apolipoprotein B lipoproteins enhances the adhesion of human monocytic cells to endothelial cells. Circulation 2006, 113, 691–700. [Google Scholar] [CrossRef]
- Li, H.; Han, Y.; Qi, R.; Wang, Y.; Zhang, X.; Yu, M.; Tang, Y.; Wang, M.; Shu, Y.-N.; Huang, W.; et al. Aggravated restenosis and atherogenesis in ApoCIII transgenic mice but lack of protection in ApoCIII knockouts: The effect of authentic triglyceride-rich lipoproteins with and without ApoCIII. Cardiovasc. Res. 2015, 107, 579–589. [Google Scholar] [CrossRef]
- Yingchun, H.; Yahong, M.; Jiangping, W.; Xiaokui, H.; Xiaohong, Z. Increased inflammation, endoplasmic reticulum stress and oxidative stress in endothelial and macrophage cells exacerbate atherosclerosis in ApoCIII transgenic mice. Lipids Health Dis. 2018, 17, 220. [Google Scholar] [CrossRef]
- Qamar, A.; Khetarpal, S.A.; Khera, A.V.; Qasim, A.; Rader, D.J.; Reilly, M.P. Plasma apolipoprotein C-III levels, triglycerides, and coronary artery calcification in type 2 diabetics. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1880–1888. [Google Scholar] [CrossRef] [PubMed]
- Taskinen, M.-R.; Borén, J. Why Is Apolipoprotein CIII Emerging as a Novel Therapeutic Target to Reduce the Burden of Cardiovascular Disease? Curr. Atheroscler. Rep. 2016, 18, 59. [Google Scholar] [CrossRef] [PubMed]
- Reeskamp, L.F.; Tromp, T.R.; Stroes, E.S.G. The next generation of triglyceride-lowering drugs: Will reducing apolipoprotein C-III or angiopoietin like protein 3 reduce cardiovascular disease? Curr. Opin. Lipidol. 2020, 31, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.-L.; Cui, G.-L.; Huang, J.; Jiang, J.-G.; Wang, D.-W. An APOC3 3′UTR variant associated with plasma triglycerides levels and coronary heart disease by creating a functional miR-4271 binding site. Sci. Rep. 2016, 6, 32700. [Google Scholar] [CrossRef] [PubMed]
- Kostopoulou, F.; Malizos, K.N.; Papathanasiou, I.; Tsezou, A. MicroRNA-33a regulates cholesterol synthesis and cholesterol efflux-related genes in osteoarthritic chondrocytes. Arthritis Res. Ther. 2015, 17, 42. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhang, M.; Dai, Y.; Xu, Z. MicroRNA-424-5p regulates aortic smooth muscle cell function in atherosclerosis by blocking APOC3-mediated nuclear factor-κB signalling pathway. Exp. Physiol. 2020, 105, 1035–1049. [Google Scholar] [CrossRef]
- Cui, R.; Li, C.; Wang, J.; Dai, J. Induction of hepatic miR-34a by perfluorooctanoic acid regulates metabolism-related genes in mice. Environ. Pollut. 2019, 244, 270–278. [Google Scholar] [CrossRef]
- Cui, G.; Li, Z.; Li, R.; Huang, J.; Wang, H.; Zhang, L.; Ding, H.; Wang, D.W. A functional variant in APOA5/A4/C3/A1 gene cluster contributes to elevated triglycerides and severity of CAD by interfering with microRNA 3201 binding efficiency. J. Am. Coll. Cardiol. 2014, 64, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Caussy, C.; Charrière, S.; Marçais, C.; Di Filippo, M.; Sassolas, A.; Delay, M.; Euthine, V.; Jalabert, A.; Lefai, E.; Rome, S.; et al. An APOA5 3′ UTR variant associated with plasma triglycerides triggers APOA5 downregulation by creating a functional miR-485-5p binding site. Am. J. Hum. Genet. 2014, 94, 129–134. [Google Scholar] [CrossRef]
- Nordestgaard, B.G.; Nicholls, S.J.; Langsted, A.; Ray, K.K.; Tybjærg-Hansen, A. Advances in lipid-lowering therapy through gene-silencing technologies. Nat. Rev. Cardiol. 2018, 15, 261–272. [Google Scholar] [CrossRef]
- Alexander, V.J.; Xia, S.; Hurh, E.; Hughes, S.G.; O’Dea, L.; Geary, R.S.; Witztum, J.L.; Tsimikas, S. N-acetyl galactosamine-conjugated antisense drug to APOC3 mRNA, triglycerides and atherogenic lipoprotein levels. Eur. Heart J. 2019, 40, 2785–2796. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Hu, L.; Shen, T.; Yang, R.; Jiang, L. Recent Advances in Gene Therapy for Familial Hypercholesterolemia: An Update Review. J. Clin. Med. 2022, 11, 6773. [Google Scholar] [CrossRef] [PubMed]
- Shamsudeen, I.; Hegele, R.A. Safety and efficacy of therapies for chylomicronemia. Expert Rev. Clin. Pharmacol. 2022, 15, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Witztum, J.L.; Gaudet, D.; Freedman, S.D.; Alexander, V.J.; Digenio, A.; Williams, K.R.; Yang, Q.; Hughes, S.G.; Geary, R.S.; Arca, M.; et al. Volanesorsen and Triglyceride Levels in Familial Chylomicronemia Syndrome. N. Engl. J. Med. 2019, 381, 531–542. [Google Scholar] [CrossRef] [PubMed]
- Gouni-Berthold, I.; Alexander, V.J.; Yang, Q.; Hurh, E.; Steinhagen-Thiessen, E.; Moriarty, P.M.; Hughes, S.G.; Gaudet, D.; Hegele, R.A.; O’Dea, L.S.L.; et al. Efficacy and safety of volanesorsen in patients with multifactorial chylomicronaemia (COMPASS): A multicentre, double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Diabetes Endocrinol. 2021, 9, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Tardif, J.-C.; Karwatowska-Prokopczuk, E.; Amour, E.S.; Ballantyne, C.M.; Shapiro, M.D.; Moriarty, P.M.; Baum, S.J.; Hurh, E.; Bartlett, V.J.; Kingsbury, J.; et al. Apolipoprotein C-III reduction in subjects with moderate hypertriglyceridaemia and at high cardiovascular risk. Eur. Heart J. 2022, 43, 1401–1412. [Google Scholar] [CrossRef]
- Prohaska, T.A.; Alexander, V.J.; Karwatowska-Prokopczuk, E.; Tami, J.; Xia, S.; Witztum, J.L.; Tsimikas, S. APOC3 inhibition with volanesorsen reduces hepatic steatosis in patients with severe hypertriglyceridemia. J. Clin. Lipidol. 2023, 17, 406–411. [Google Scholar] [CrossRef]
- Li, J.; Zhu, X.; Yu, K.; Jiang, H.; Zhang, Y.; Deng, S.; Cheng, L.; Liu, X.; Zhong, J.; Zhang, X.; et al. Genome-Wide Analysis of DNA Methylation and Acute Coronary Syndrome. Circ. Res. 2017, 120, 1754–1767. [Google Scholar] [CrossRef] [PubMed]
- Ghose, S.; Ghosh, S.; Tanwar, V.S.; Tolani, P.; Kutum, R.; Sharma, A.; Bhardwaj, N.; Shamsudheen, K.V.; Verma, A.; Jayarajan, R.; et al. Investigating Coronary Artery Disease methylome through targeted bisulfite sequencing. Gene 2019, 721, 144107. [Google Scholar] [CrossRef]
- Pu, Z.; Wang, W.; Xie, H.; Wang, W. Apolipoprotein C3 (ApoC3) facilitates NLRP3 mediated pyroptosis of macrophages through mitochondrial damage by accelerating of the interaction between SCIMP and SYK pathway in acute lung injury. Int. Immunopharmacol. 2024, 128, 111537. [Google Scholar] [CrossRef]
- Wang, F.; Kohan, A.B.; Lo, C.-M.; Liu, M.; Howles, P.; Tso, P. Apolipoprotein A-IV: A protein intimately involved in metabolism. J. Lipid Res. 2015, 56, 1403–1418. [Google Scholar] [CrossRef]
- Mokhtar, F.B.A.; Plat, J.; Mensink, R.P. Genetic variation and intestinal cholesterol absorption in humans: A systematic review and a gene network analysis. Prog. Lipid Res. 2022, 86, 101164. [Google Scholar] [CrossRef]
- Deng, X.; Morris, J.; Dressmen, J.; Tubb, M.R.; Tso, P.; Jerome, W.G.; Davidson, W.S.; Thompson, T.B. The structure of dimeric apolipoprotein A-IV and its mechanism of self-association. Structure 2012, 20, 767–779. [Google Scholar] [CrossRef] [PubMed]
- Hockey, K.J.; Anderson, R.A.; Cook, V.R.; Hantgan, R.R.; Weinberg, R.B. Effect of the apolipoprotein A-IV Q360H polymorphism on postprandial plasma triglyceride clearance. J. Lipid Res. 2001, 42, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Duverger, N.; Tremp, G.; Caillaud, J.M.; Emmanuel, F.; Castro, G.; Fruchart, J.C.; Steinmetz, A.; Denèfle, P. Protection against atherogenesis in mice mediated by human apolipoprotein A-IV. Science 1996, 273, 966–968. [Google Scholar] [CrossRef]
- Cohen, R.D.; Castellani, L.W.; Qiao, J.H.; Van Lenten, B.J.; Lusis, A.J.; Reue, K. Reduced aortic lesions and elevated high density lipoprotein levels in transgenic mice overexpressing mouse apolipoprotein A-IV. J. Clin. Investig. 1997, 99, 1906–1916. [Google Scholar] [CrossRef] [PubMed]
- Ostos, M.A.; Conconi, M.; Vergnes, L.; Baroukh, N.; Ribalta, J.; Girona, J.; Caillaud, J.M.; Ochoa, A.; Zakin, M.M. Antioxidative and antiatherosclerotic effects of human apolipoprotein A-IV in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1023–1028. [Google Scholar] [CrossRef]
- Cheng, C.; Liu, X.-H.; He, J.; Gao, J.; Zhou, J.-T.; Fan, J.-N.; Jin, X.; Zhang, J.; Chang, L.; Xiong, Z.; et al. Apolipoprotein A4 Restricts Diet-Induced Hepatic Steatosis via SREBF1-Mediated Lipogenesis and Enhances IRS-PI3K-Akt Signaling. Mol. Nutr. Food Res. 2022, 66, e2101034. [Google Scholar] [CrossRef]
- Li, W.-H.; Zhang, L.; Li, Y.-Y.; Wang, X.-Y.; Li, J.-L.; Zhao, S.-N.; Ni, M.-Q.; Li, Q.; Sun, H. Apolipoprotein A-IV Has Bi-Functional Actions in Alcoholic Hepatitis by Regulating Hepatocyte Injury and Immune Cell Infiltration. Int. J. Mol. Sci. 2022, 24, 670. [Google Scholar] [CrossRef]
- Li, X.; Liu, X.; Zhang, Y.; Cheng, C.; Fan, J.; Zhou, J.; Garstka, M.A.; Li, Z. Hepatoprotective effect of apolipoprotein A4 against carbon tetrachloride induced acute liver injury through mediating hepatic antioxidant and inflammation response in mice. Biochem. Biophys. Res. Commun. 2021, 534, 659–665. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, Z.; Wei, Y.; Li, X.; Li, S. Apolipoprotein A4 regulates the immune response in carbon tetrachloride-induced chronic liver injury in mice. Int. Immunopharmacol. 2021, 90, 107222. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Li, X.; Xie, L.; Li, S.; Liu, J.; Jia, L.; Dong, X.; Ren, X.; Xiao, J.; Yang, C.; et al. A long non-coding RNA, APOA4-AS, regulates APOA4 expression depending on HuR in mice. Nucleic Acids Res. 2016, 44, 6423–6433. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xiang, D.; Mei, S.; Jin, Y.; Sun, D.; Chen, C.; Hu, D.; Li, S.; Li, H.; Wang, Y.; et al. The novel long noncoding RNA Lnc19959.2 modulates triglyceride metabolism-associated genes through the interaction with Purb and hnRNPA2B1. Mol. Metab. 2020, 37, 100996. [Google Scholar] [CrossRef] [PubMed]
- Gong, T.; Zhang, P.; Ruan, S.-F.; Xiao, Z.; Chen, W.; Lin, M.; Zhong, Q.; Luo, R.; Xu, Q.; Peng, J.; et al. APOA4 as a novel predictor of prognosis in Stevens-Johnson syndrome/toxic epidermal necrolysis: A proteomics analysis from two prospective cohorts. J. Am. Acad. Dermatol. 2023, 89, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Clarke, R.; Von Ende, A.; Schmidt, L.E.; Yin, X.; Hill, M.; Hughes, A.D.; Pechlaner, R.; Willeit, J.; Kiechl, S.; Watkins, H.; et al. Apolipoprotein Proteomics for Residual Lipid-Related Risk in Coronary Heart Disease. Circ. Res. 2023, 132, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Eslam, M.; Valenti, L.; Romeo, S. Genetics and epigenetics of NAFLD and NASH: Clinical impact. J. Hepatol. 2018, 68, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Cespiati, A.; Youngson, N.A.; Tourna, A.; Valenti, L. Genetics and Epigenetics in the Clinic: Precision Medicine in the Management of Fatty Liver Disease. Curr. Pharm. Des. 2020, 26, 998–1009. [Google Scholar] [CrossRef] [PubMed]
- Tryndyak, V.P.; Willett, R.A.; Avigan, M.I.; Sanyal, A.J.; Beland, F.A.; Rusyn, I.; Pogribny, I.P. Non-alcoholic fatty liver disease-associated DNA methylation and gene expression alterations in the livers of Collaborative Cross mice fed an obesogenic high-fat and high-sucrose diet. Epigenetics 2022, 17, 1462–1476. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, S.K.; Heeren, J.; Olivecrona, G.; Merkel, M. Apolipoprotein A-V; a potent triglyceride reducer. Atherosclerosis 2011, 219, 15–21. [Google Scholar] [CrossRef]
- Forte, T.M.; Ryan, R.O. Apolipoprotein A5: Extracellular and Intracellular Roles in Triglyceride Metabolism. Curr. Drug Targets 2015, 16, 1274–1280. [Google Scholar] [CrossRef]
- Zhang, L.S.; Sato, H.; Yang, Q.; Ryan, R.O.; Wang, D.Q.H.; Howles, P.N.; Tso, P. Apolipoprotein A-V is present in bile and its secretion increases with lipid absorption in Sprague-Dawley rats. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G918–G925. [Google Scholar] [CrossRef] [PubMed]
- Treviño-Villarreal, J.H.; Reynolds, J.S.; Bartelt, A.; Langston, P.K.; MacArthur, M.R.; Arduini, A.; Tosti, V.; Veronese, N.; Bertozzi, B.; Brace, L.E.; et al. Dietary protein restriction reduces circulating VLDL triglyceride levels via CREBH-APOA5-dependent and -independent mechanisms. JCI Insight 2018, 3, e99470. [Google Scholar] [CrossRef] [PubMed]
- Ress, C.; Moschen, A.R.; Sausgruber, N.; Tschoner, A.; Graziadei, I.; Weiss, H.; Schgoer, W.; Ebenbichler, C.F.; Konrad, R.J.; Patsch, J.R.; et al. The role of apolipoprotein A5 in non-alcoholic fatty liver disease. Gut 2011, 60, 985–991. [Google Scholar] [CrossRef] [PubMed]
- Camporez, J.P.G.; Kanda, S.; Petersen, M.C.; Jornayvaz, F.R.; Samuel, V.T.; Bhanot, S.; Petersen, K.F.; Jurczak, M.J.; Shulman, G.I. ApoA5 knockdown improves whole-body insulin sensitivity in high-fat-fed mice by reducing ectopic lipid content. J. Lipid Res. 2015, 56, 526–536. [Google Scholar] [CrossRef] [PubMed]
- Merkel, M.; Loeffler, B.; Kluger, M.; Fabig, N.; Geppert, G.; Pennacchio, L.A.; Laatsch, A.; Heeren, J. Apolipoprotein AV accelerates plasma hydrolysis of triglyceride-rich lipoproteins by interaction with proteoglycan-bound lipoprotein lipase. J. Biol. Chem. 2005, 280, 21553–21560. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.-Y.; Yu, B.-L.; Xie, Y.-F.; Zhao, S.-P.; Wu, C.-L. Apolipoprotein A5 regulates intracellular triglyceride metabolism in adipocytes. Mol. Med. Report. 2017, 16, 6771–6779. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.Q.; Pottanat, T.G.; Zhen, E.Y.; Siegel, R.W.; Ehsani, M.; Qian, Y.-W.; Konrad, R.J. ApoA5 lowers triglyceride levels via suppression of ANGPTL3/8-mediated LPL inhibition. J. Lipid Res. 2021, 62, 100068. [Google Scholar] [CrossRef] [PubMed]
- Shu, X.; Nelbach, L.; Weinstein, M.M.; Burgess, B.L.; Beckstead, J.A.; Young, S.G.; Ryan, R.O.; Forte, T.M. Intravenous injection of apolipoprotein A-V reconstituted high-density lipoprotein decreases hypertriglyceridemia in apoav−/− mice and requires glycosylphosphatidylinositol-anchored high-density lipoprotein-binding protein 1. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2504–2509. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, S.K.; Lookene, A.; Beckstead, J.A.; Gliemann, J.; Ryan, R.O.; Olivecrona, G. Apolipoprotein A-V interaction with members of the low density lipoprotein receptor gene family. Biochemistry 2007, 46, 3896–3904. [Google Scholar] [CrossRef]
- Gonzales, J.C.; Gordts, P.L.S.M.; Foley, E.M.; Esko, J.D. Apolipoproteins E and AV mediate lipoprotein clearance by hepatic proteoglycans. J. Clin. Investig. 2013, 123, 2742–2751. [Google Scholar] [CrossRef]
- Oliva, I.; Guardiola, M.; Vallvé, J.-C.; Ibarretxe, D.; Plana, N.; Masana, L.; Monk, D.; Ribalta, J. APOA5 genetic and epigenetic variability jointly regulate circulating triacylglycerol levels. Clin. Sci. 2016, 130, 2053–2059. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhou, Y.; Zhu, L.; Liu, G.; Wang, X.; Wang, X.; Wang, J.; You, L.; Ji, C.; Guo, X.; et al. Genome-wide analysis reveals that altered methylation in specific CpG loci is associated with childhood obesity. J. Cell. Biochem. 2018, 119, 7490–7497. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Author | Gene | MicroRNA | Study Design | Major Outcomes |
|---|---|---|---|---|
| Fotini Kostopoulou et al. [65] | APOA1 | MicroRNA-33a | The treatment of human normal chondrocytes with miR-33a | Reduced APOA1 mRNA expression levels; induction of cholesterol metabolism disorders and osteoarthritic phenotype in normal chondrocytes |
| Li et al. [66] | APOC3 | MicroRNA-424-5p | Aortic smooth muscle cells were treated with miR-424-5p mimic | Silence of APOC3; the proliferation, migration, and inflammation of aortic smooth muscle cells are inhibited, and apoptosis is promoted |
| Hu et al. [64] | APOC3 | MicroRNA-4271 | Investigating the effect of APOC3 variants on microRNA binding | MicroRNA-4271 binds to APOC3 and inhibits its transcription to reduce CHD risk |
| Cui et al. [67] | APOA4 | MicroRNA-34a | Investigating gene expression levels in the livers of miR-34a−/−mice after perfluorooctanoic acid (PFOA) exposure | Under PFOA treatment, PPAR significantly up-regulates APOA4 expression, while microRNA-34a only plays a moderate role |
| Cui et al. [68] | APOA5 | MicroRNA-3201 | Investigating the effect of APOA5 variants on microRNA binding | The rs2266788 C allele interferes microRNA-3201 binding to APOA5, resulting in increased APOA5 expression levels and risk of CAD |
| Cyrielle Caussy et al. [69] | APOA5 | MicroRNA-485-5p | Investigating the effect of APOA5 variants on microRNA binding | The rs2266788 C allele mediates microRNA-485-5p binding to APOA5, resulting in downregulation of APOA5 and hypertriglyceridemic effect |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, Q.; Wang, J.; Wang, L.; Ding, H. APOA1/C3/A4/A5 Gene Cluster at 11q23.3 and Lipid Metabolism Disorders: From Epigenetic Mechanisms to Clinical Practices. Biomedicines 2024, 12, 1224. https://doi.org/10.3390/biomedicines12061224
Xiao Q, Wang J, Wang L, Ding H. APOA1/C3/A4/A5 Gene Cluster at 11q23.3 and Lipid Metabolism Disorders: From Epigenetic Mechanisms to Clinical Practices. Biomedicines. 2024; 12(6):1224. https://doi.org/10.3390/biomedicines12061224
Chicago/Turabian StyleXiao, Qianqian, Jing Wang, Luyun Wang, and Hu Ding. 2024. "APOA1/C3/A4/A5 Gene Cluster at 11q23.3 and Lipid Metabolism Disorders: From Epigenetic Mechanisms to Clinical Practices" Biomedicines 12, no. 6: 1224. https://doi.org/10.3390/biomedicines12061224