

Shared Genetic Architectures between Coronary Artery Disease and Type 2 Diabetes Mellitus in East Asian and European Populations

Abstract
:1. Introduction
2. Materials and Methods
2.1. Individual Data for CAD and T2DM of Chinese Cohort
2.2. GWAS Summary Statistics for CAD and T2DM of European and Japanese Populations
2.3. Global and Local Genetic Correlation Analysis
2.4. Single-Variant Pleiotropic Analysis (ASSET)
2.5. Definition of Common Pleiotropic Loci and Functional Annotation
2.6. Multivariate Gene-Based Association Analysis
2.7. Multiethnic Fine-Mapping of Shared Loci/Genes
2.8. Functional Enrichment Analysis in Datasets of Different Populations
3. Results
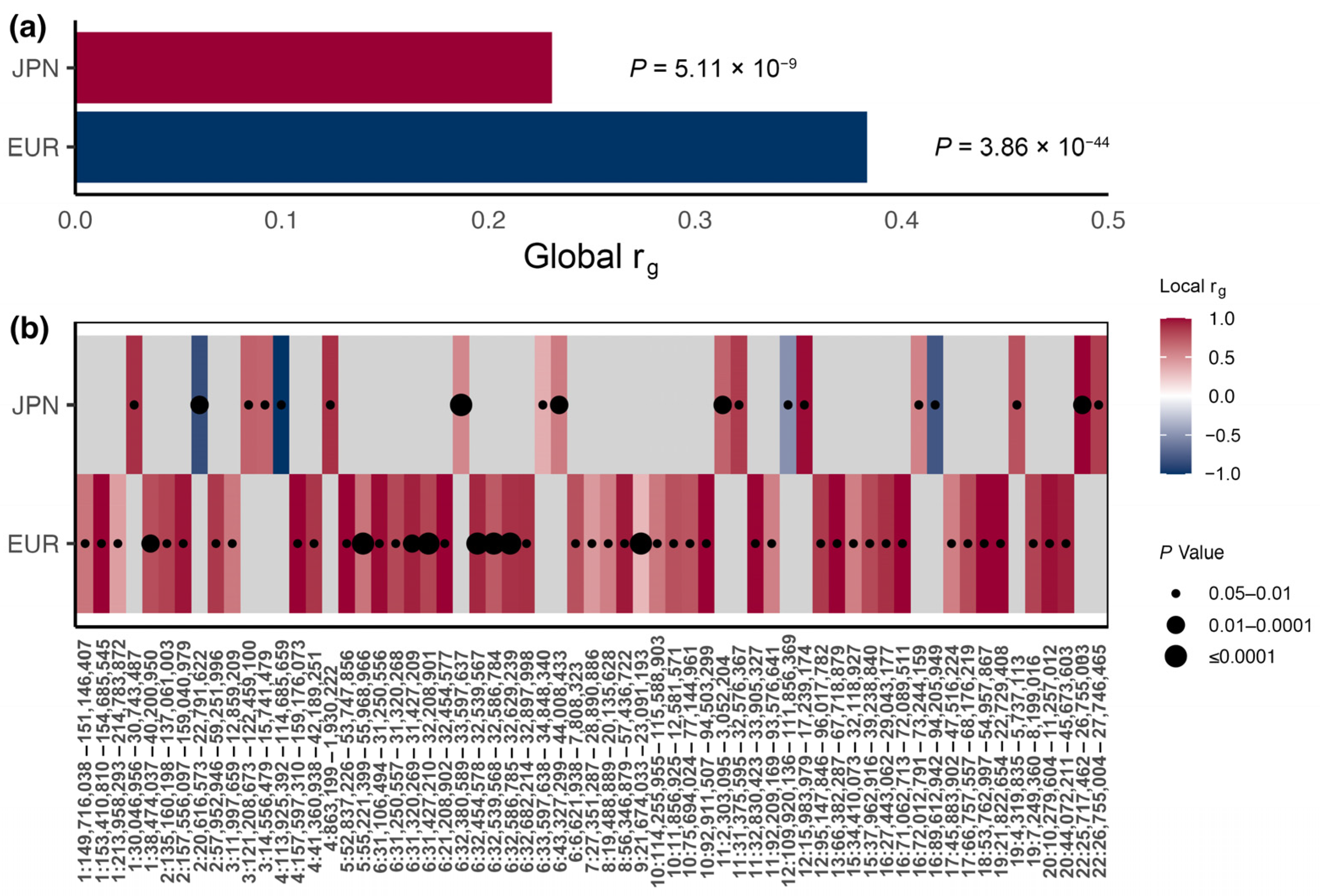
3.1. Global and Local Genetic Correlations
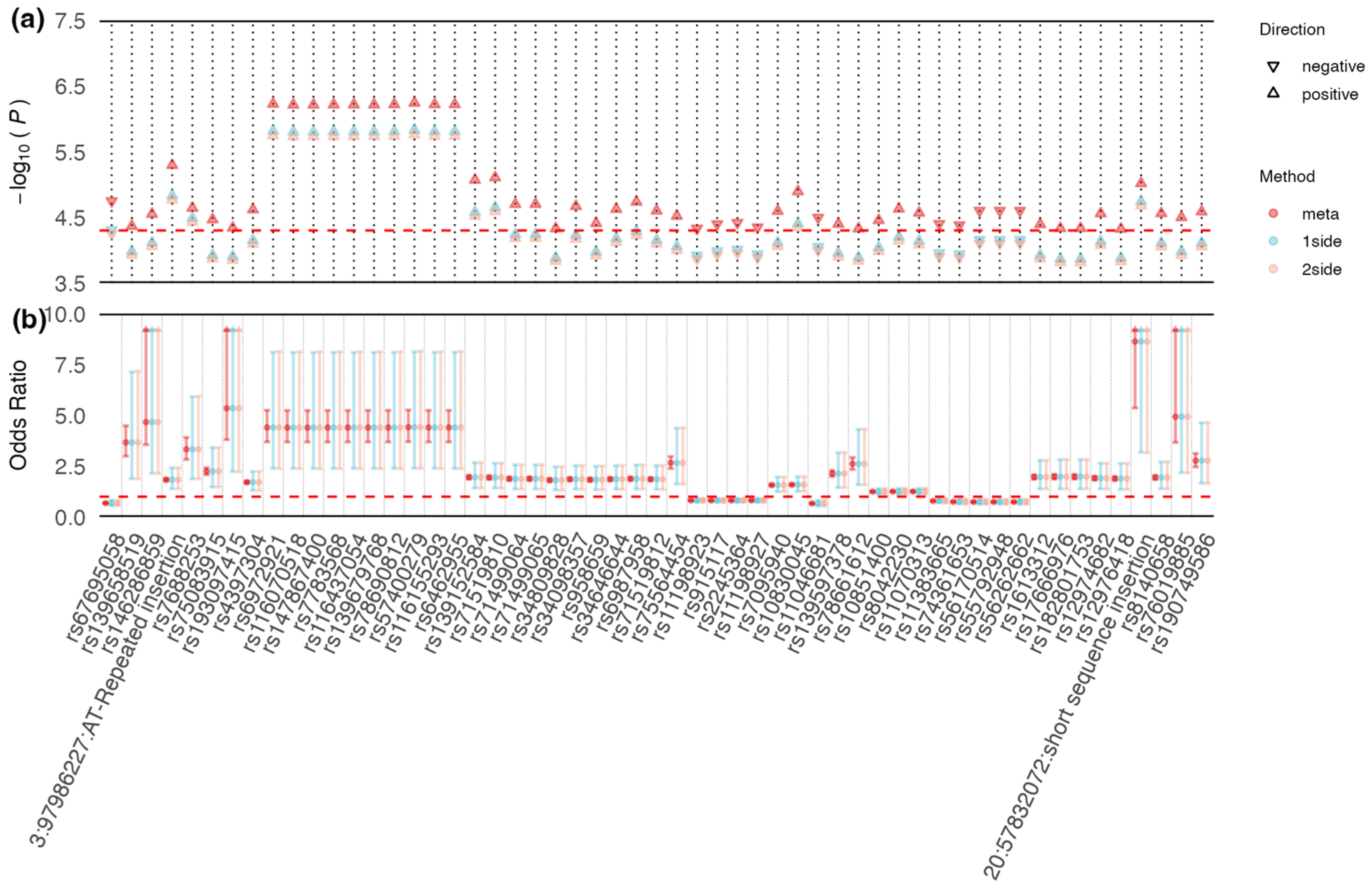
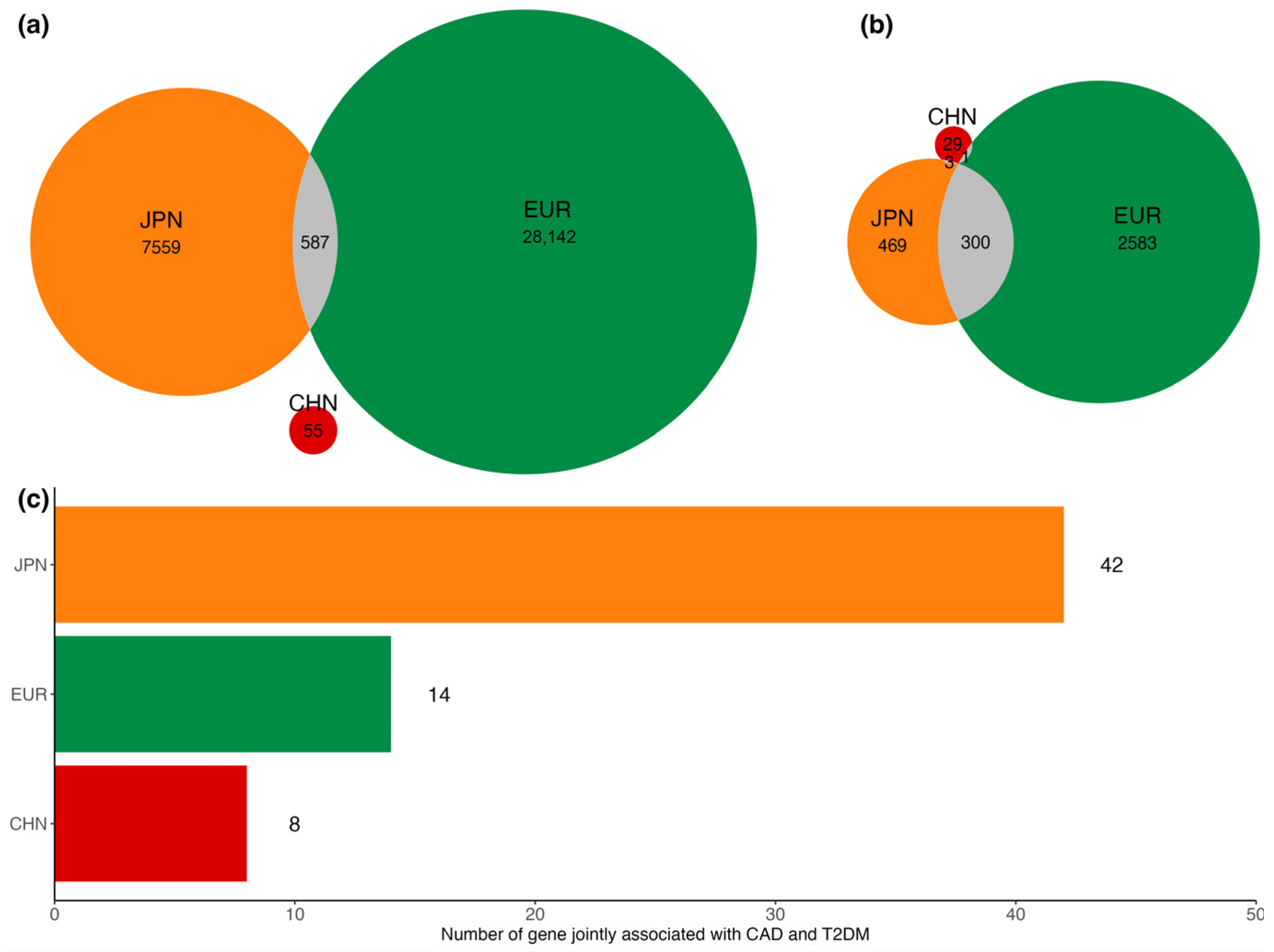
3.2. Shared Signals between CAD and T2DM in East Asian and European Populations
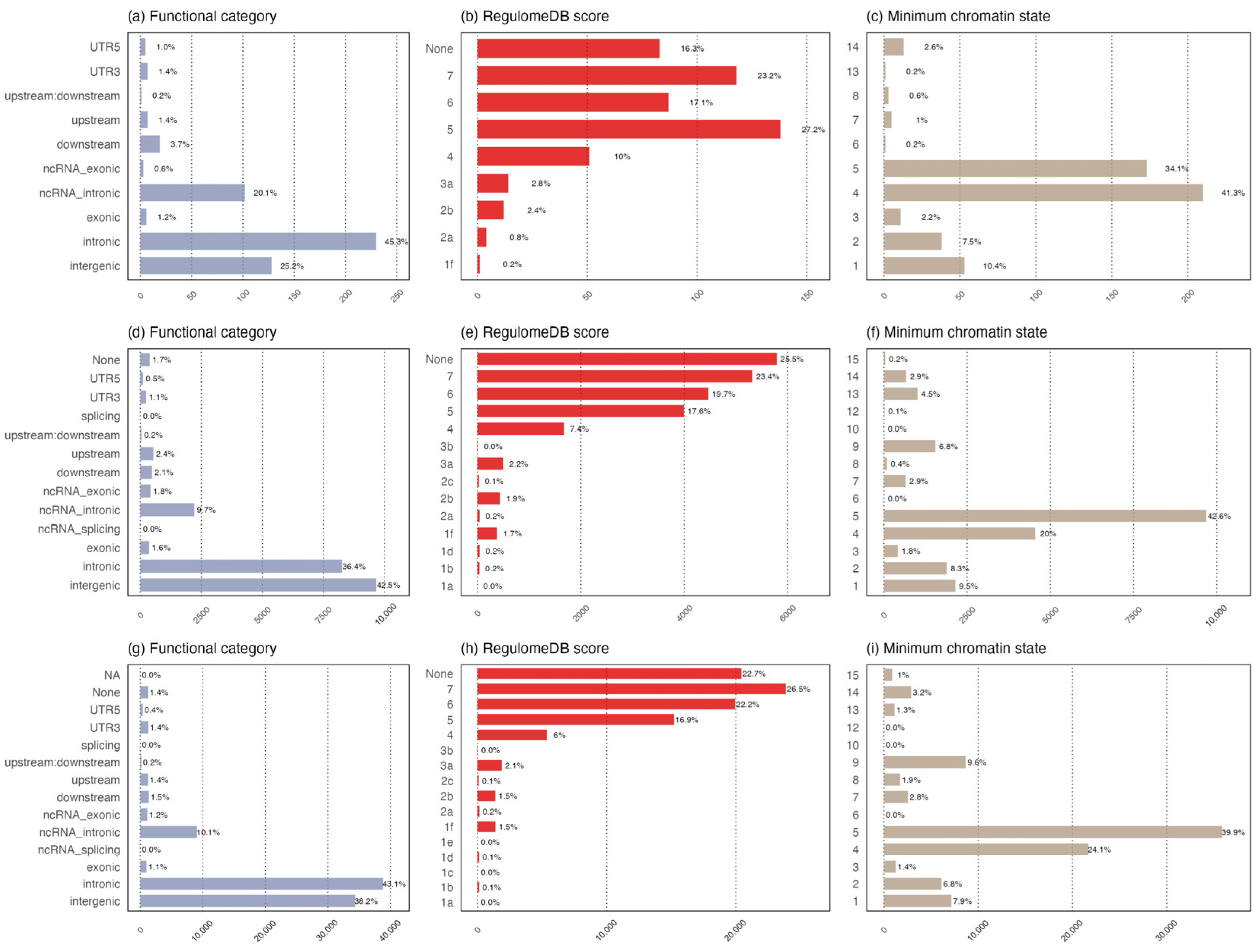
3.3. Functional Annotation of Shared Common Loci
3.4. Gene-Based Association Analysis for Shared Noncommon Signals
3.5. Fine Mapping of Shared Loci/Genes between CAD and T2DM
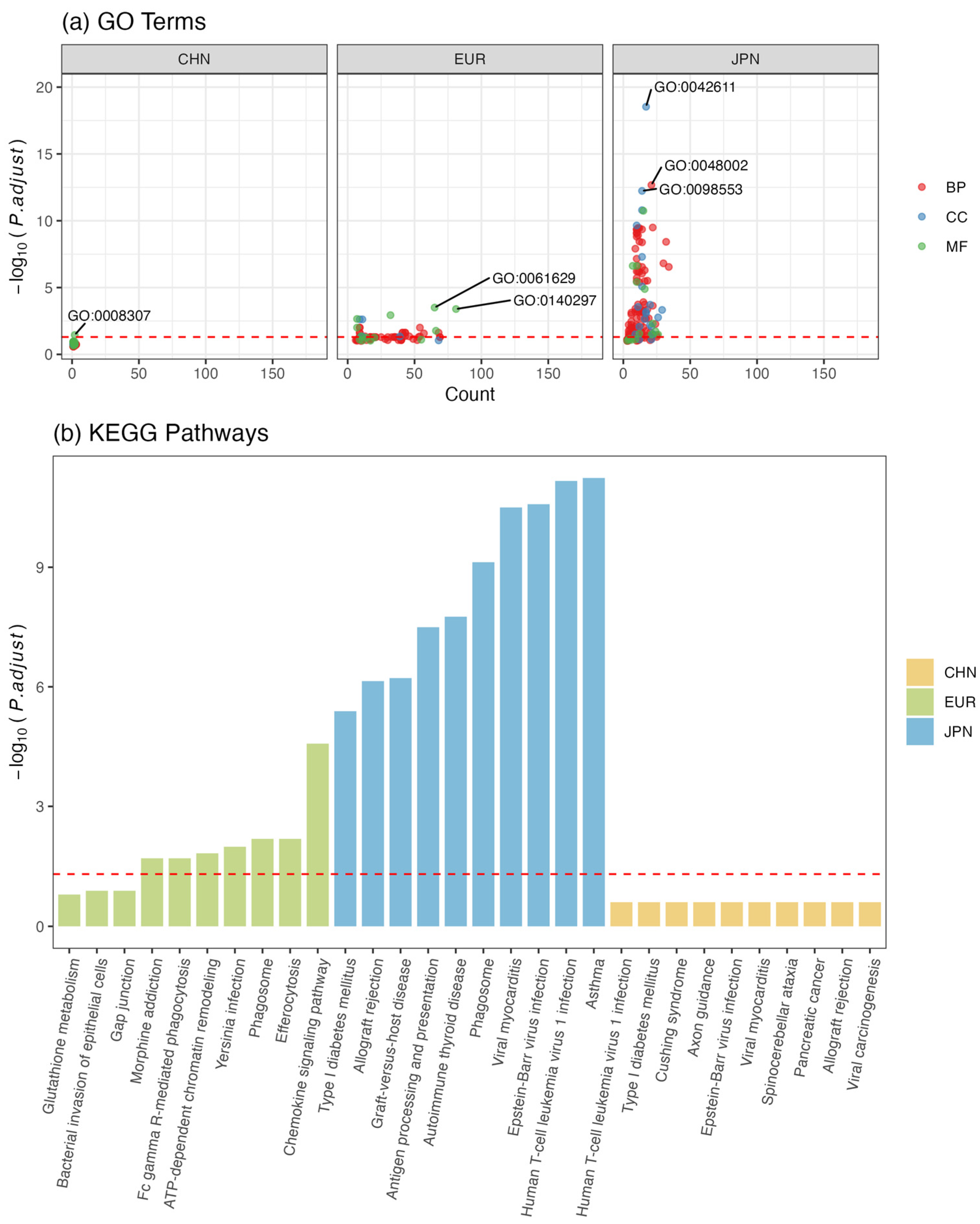
3.6. Functional Enrichment Analysis of Shared Loci/Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- GBD 2019 Diseases and Injuries Collaborators. Global burden of 369 diseases and injuries in 204 countries and territories, 1990–2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet 2020, 396, 1204–1222. [Google Scholar] [CrossRef] [PubMed]
- Einarson, T.R.; Acs, A.; Ludwig, C.; Panton, U.H. Prevalence of cardiovascular disease in type 2 diabetes: A systematic literature review of scientific evidence from across the world in 2007–2017. Cardiovasc. Diabetol. 2018, 17, 83. [Google Scholar] [CrossRef] [PubMed]
- Sarwar, N.; Gao, P.; Seshasai, S.R.; Gobin, R.; Kaptoge, S.; Di Angelantonio, E.; Ingelsson, E.; Lawlor, D.A.; Selvin, E.; Stampfer, M.; et al. Diabetes mellitus, fasting blood glucose concentration, and risk of vascular disease: A collaborative meta-analysis of 102 prospective studies. Lancet 2010, 375, 2215–2222. [Google Scholar] [PubMed]
- Hughes, T.M.; Althouse, A.D.; Niemczyk, N.A.; Hawkins, M.S.; Kuipers, A.L.; Sutton-Tyrrell, K. Effects of weight loss and insulin reduction on arterial stiffness in the SAVE trial. Cardiovasc. Diabetol. 2012, 11, 114. [Google Scholar] [CrossRef]
- Andreozzi, F.; Gastaldelli, A.; Mannino, G.C.; Sciacqua, A.; Succurro, E.; Arturi, F.; Folli, F.; Perticone, F. Increased carotid intima-media thickness in the physiologic range is associated with impaired postprandial glucose metabolism, insulin resistance and beta cell dysfunction. Atherosclerosis 2013, 229, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.B.; Mitchell, G.F.; Gill, D.; Burgess, S.; Rahman, M.; Hanff, T.C.; Ramachandran, V.S.; Mutalik, K.M.; Townsend, R.R.; Chirinos, J.A. Arterial Stiffness and Diabetes Risk in Framingham Heart Study and UK Biobank. Circ. Res. 2022, 131, 545–554. [Google Scholar] [CrossRef]
- Izzo, R.; de Simone, G.; Trimarco, V.; Gerdts, E.; Giudice, R.; Vaccaro, O.; De Luca, N.; Trimarco, B. Hypertensive target organ damage predicts incident diabetes mellitus. Eur. Heart J. 2013, 34, 3419–3426. [Google Scholar] [CrossRef] [PubMed]
- La Sala, L.; Prattichizzo, F.; Ceriello, A. The link between diabetes and atherosclerosis. Eur. J. Prev. Cardiol. 2019, 26 (Suppl. 2), 15–24. [Google Scholar] [CrossRef] [PubMed]
- Drobni, Z.D.; Kolossvary, M.; Karady, J.; Jermendy, A.L.; Tarnoki, A.D.; Tarnoki, D.L.; Simon, J.; Szilveszter, B.; Littvay, L.; Voros, S.; et al. Heritability of Coronary Artery Disease: Insights from a Classical Twin Study. Circ. Cardiovasc. Imaging 2022, 15, e013348. [Google Scholar] [CrossRef]
- Willemsen, G.; Ward, K.J.; Bell, C.G.; Christensen, K.; Bowden, J.; Dalgård, C.; Harris, J.R.; Kaprio, J.; Lyle, R.; Magnusson, P.K.; et al. The Concordance and Heritability of Type 2 Diabetes in 34,166 Twin Pairs from International Twin Registers: The Discordant Twin (DISCOTWIN) Consortium. Twin Res. Hum. Genet. 2015, 18, 762–771. [Google Scholar] [CrossRef]
- Mahajan, A.; Spracklen, C.N.; Zhang, W.; Ng, M.C.Y.; Petty, L.E.; Kitajima, H.; Yu, G.Z.; Rüeger, S.; Speidel, L.; Kim, Y.J.; et al. Multi-ancestry genetic study of type 2 diabetes highlights the power of diverse populations for discovery and translation. Nat. Genet. 2022, 54, 560–572. [Google Scholar] [CrossRef] [PubMed]
- Aragam, K.G.; Jiang, T.; Goel, A.; Kanoni, S.; Wolford, B.N.; Atri, D.S.; Weeks, E.M.; Wang, M.; Hindy, G.; Zhou, W.; et al. Discovery and systematic characterization of risk variants and genes for coronary artery disease in over a million participants. Nat. Genet. 2022, 54, 1803–1815. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Stringer, S.; Frei, O.; Umićević Mirkov, M.; de Leeuw, C.; Polderman, T.J.C.; van der Sluis, S.; Andreassen, O.A.; Neale, B.M.; Posthuma, D. A global overview of pleiotropy and genetic architecture in complex traits. Nat. Genet. 2019, 51, 1339–1348. [Google Scholar] [CrossRef] [PubMed]
- Weissbrod, O.; Flint, J.; Rosset, S. Estimating SNP-Based Heritability and Genetic Correlation in Case-Control Studies Directly and with Summary Statistics. Am. J. Hum. Genet. 2018, 103, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Bulik-Sullivan, B.; Finucane, H.K.; Anttila, V.; Gusev, A.; Day, F.R.; Loh, P.R.; Duncan, L.; Perry, J.R.; Patterson, N.; Robinson, E.B.; et al. An atlas of genetic correlations across human diseases and traits. Nat. Genet. 2015, 47, 1236–1241. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, S.; Rajaraman, P.; Jacobs, K.B.; Wheeler, W.A.; Melin, B.S.; Hartge, P.; Yeager, M.; Chung, C.C.; Chanock, S.J.; Chatterjee, N. A subset-based approach improves power and interpretation for the combined analysis of genetic association studies of heterogeneous traits. Am. J. Hum. Genet. 2012, 90, 821–835. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.R.; Gignoux, C.R.; Walters, R.K.; Wojcik, G.L.; Neale, B.M.; Gravel, S.; Daly, M.J.; Bustamante, C.D.; Kenny, E.E. Human Demographic History Impacts Genetic Risk Prediction across Diverse Populations. Am. J. Hum. Genet. 2017, 100, 635–649. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Nielsen, J.B.; Fritsche, L.G.; Dey, R.; Gabrielsen, M.E.; Wolford, B.N.; LeFaive, J.; VandeHaar, P.; Gagliano, S.A.; Gifford, A.; et al. Efficiently controlling for case-control imbalance and sample relatedness in large-scale genetic association studies. Nat. Genet. 2018, 50, 1335–1341. [Google Scholar] [CrossRef] [PubMed]
- Bycroft, C.; Freeman, C.; Petkova, D.; Band, G.; Elliott, L.T.; Sharp, K.; Motyer, A.; Vukcevic, D.; Delaneau, O.; O’Connell, J.; et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 2018, 562, 203–209. [Google Scholar] [CrossRef]
- Denny, J.C.; Bastarache, L.; Ritchie, M.D.; Carroll, R.J.; Zink, R.; Mosley, J.D.; Field, J.R.; Pulley, J.M.; Ramirez, A.H.; Bowton, E.; et al. Systematic comparison of phenome-wide association study of electronic medical record data and genome-wide association study data. Nat. Biotechnol. 2013, 31, 1102–1110. [Google Scholar] [CrossRef]
- Ishigaki, K.; Akiyama, M.; Kanai, M.; Takahashi, A.; Kawakami, E.; Sugishita, H.; Sakaue, S.; Matoba, N.; Low, S.K.; Okada, Y.; et al. Large-scale genome-wide association study in a Japanese population identifies novel susceptibility loci across different diseases. Nat. Genet. 2020, 52, 669–679. [Google Scholar] [CrossRef] [PubMed]
- Sakaue, S.; Kanai, M.; Tanigawa, Y.; Karjalainen, J.; Kurki, M.; Koshiba, S.; Narita, A.; Konuma, T.; Yamamoto, K.; Akiyama, M.; et al. A cross-population atlas of genetic associations for 220 human phenotypes. Nat. Genet. 2021, 53, 1415–1424. [Google Scholar] [CrossRef] [PubMed]
- Bulik-Sullivan, B.K.; Loh, P.R.; Finucane, H.K.; Ripke, S.; Yang, J.; Patterson, N.; Daly, M.J.; Price, A.L.; Neale, B.M. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 2015, 47, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Li, B.; Ou, D.; Erlendsdottir, M.; Powles, R.L.; Jiang, T.; Hu, Y.; Chang, D.; Jin, C.; Dai, W.; et al. A Powerful Approach to Estimating Annotation-Stratified Genetic Covariance via GWAS Summary Statistics. Am. J. Hum. Genet. 2017, 101, 939–964. [Google Scholar] [CrossRef] [PubMed]
- Werme, J.; van der Sluis, S.; Posthuma, D.; de Leeuw, C.A. An integrated framework for local genetic correlation analysis. Nat. Genet. 2022, 54, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B (Methodol.) 2018, 57, 289–300. [Google Scholar] [CrossRef]
- Freedman, M.L.; Reich, D.; Penney, K.L.; McDonald, G.J.; Mignault, A.A.; Patterson, N.; Gabriel, S.B.; Topol, E.J.; Smoller, J.W.; Pato, C.N.; et al. Assessing the impact of population stratification on genetic association studies. Nat. Genet. 2004, 36, 388–393. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Taskesen, E.; van Bochoven, A.; Posthuma, D. Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 2017, 8, 1826. [Google Scholar] [CrossRef] [PubMed]
- Oscanoa, J.; Sivapalan, L.; Gadaleta, E.; Dayem Ullah, A.Z.; Lemoine, N.R.; Chelala, C. SNPnexus: A web server for functional annotation of human genome sequence variation (2020 update). Nucleic Acids Res. 2020, 48, W185–W192. [Google Scholar] [CrossRef]
- Luo, L.; Shen, J.; Zhang, H.; Chhibber, A.; Mehrotra, D.V.; Tang, Z.Z. Multi-trait analysis of rare-variant association summary statistics using MTAR. Nat. Commun. 2020, 11, 2850. [Google Scholar] [CrossRef]
- Kichaev, G.; Roytman, M.; Johnson, R.; Eskin, E.; Lindström, S.; Kraft, P.; Pasaniuc, B. Improved methods for multi-trait fine mapping of pleiotropic risk loci. Bioinformatics 2017, 33, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Boyle, E.I.; Weng, S.; Gollub, J.; Jin, H.; Botstein, D.; Cherry, J.M.; Sherlock, G. GO::TermFinder—Open source software for accessing Gene Ontology information and finding significantly enriched Gene Ontology terms associated with a list of genes. Bioinformatics 2004, 20, 3710–3715. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Tikoo, S.; Maity, S.; Sengupta, S.; Sengupta, S.; Kaur, A.; Bachhawat, A.K. Mammalian proapoptotic factor ChaC1 and its homologues function as γ-glutamyl cyclotransferases acting specifically on glutathione. EMBO Rep. 2012, 13, 1095–1101. [Google Scholar] [CrossRef] [PubMed]
- Mungrue, I.N.; Pagnon, J.; Kohannim, O.; Gargalovic, P.S.; Lusis, A.J. CHAC1/MGC4504 is a novel proapoptotic component of the unfolded protein response, downstream of the ATF4-ATF3-CHOP cascade. J. Immunol. 2009, 182, 466–476. [Google Scholar] [CrossRef]
- Mise, K.; Imamura, M.; Yamaguchi, S.; Teshigawara, S.; Tone, A.; Uchida, H.A.; Eguchi, J.; Nakatsuka, A.; Ogawa, D.; Yoshida, M.; et al. Identification of Novel Urinary Biomarkers for Predicting Renal Prognosis in Patients with Type 2 Diabetes by Glycan Profiling in a Multicenter Prospective Cohort Study: U-CARE Study 1. Diabetes Care 2018, 41, 1765–1775. [Google Scholar] [CrossRef] [PubMed]
- Bruni, A.; Pepper, A.R.; Pawlick, R.L.; Gala-Lopez, B.; Gamble, A.F.; Kin, T.; Seeberger, K.; Korbutt, G.S.; Bornstein, S.R.; Linkermann, A.; et al. Ferroptosis-inducing agents compromise in vitro human islet viability and function. Cell Death Dis. 2018, 9, 595. [Google Scholar] [CrossRef]
- Li, Z.; Nguyen, L.O.; Organ, C.L.; Lefer, D.J.; Mungrue, I.N. Genetic inhibition of the UPR gene Chac1 preserves cardiac function in a murine model of pressure overload induced heart failure. J. Mol. Cell. Cardiol. 2017, 112, 160. [Google Scholar] [CrossRef]
- Hou, H.T.; Chen, H.X.; Wang, X.L.; Yuan, C.; Yang, Q.; Liu, Z.G.; He, G.W. Genetic characterisation of 22q11.2 variations and prevalence in patients with congenital heart disease. Arch. Dis. Child. 2020, 105, 367–374. [Google Scholar] [CrossRef]
- Jin, W.; Mulas, F.; Gaertner, B.; Sui, Y.; Wang, J.; Matta, I.; Zeng, C.; Vinckier, N.; Wang, A.; Nguyen-Ngoc, K.V.; et al. A Network of microRNAs Acts to Promote Cell Cycle Exit and Differentiation of Human Pancreatic Endocrine Cells. iScience 2019, 21, 681–694. [Google Scholar] [CrossRef]
- O’Connor, M.J.; Schroeder, P.; Huerta-Chagoya, A.; Cortés-Sánchez, P.; Bonàs-Guarch, S.; Guindo-Martínez, M.; Cole, J.B.; Kaur, V.; Torrents, D.; Veerapen, K.; et al. Recessive Genome-Wide Meta-analysis Illuminates Genetic Architecture of Type 2 Diabetes. Diabetes 2022, 71, 554–565. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Xu, H.; Xu, H.; Geng, Q.; Mak, W.H.; Ling, F.; Su, Z.; Yang, F.; Zhang, T.; Chen, J.; et al. New genetic variants associated with major adverse cardiovascular events in patients with acute coronary syndromes and treated with clopidogrel and aspirin. Pharmacogenom. J. 2021, 21, 664–672. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Horibe, H.; Oguri, M.; Sakuma, J.; Takeuchi, I.; Yasukochi, Y.; Kato, K.; Sawabe, M. Identification of novel hyper- or hypomethylated CpG sites and genes associated with atherosclerotic plaque using an epigenome-wide association study. Int. J. Mol. Med. 2018, 41, 2724–2732. [Google Scholar] [CrossRef] [PubMed]
- Pugazhenthi, S.; Qin, L.; Reddy, P.H. Common neurodegenerative pathways in obesity, diabetes, and Alzheimer’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1037–1045. [Google Scholar] [CrossRef] [PubMed]
- Nordestgaard, L.T.; Christoffersen, M.; Frikke-Schmidt, R. Shared Risk Factors between Dementia and Atherosclerotic Cardiovascular Disease. Int. J. Mol. Sci. 2022, 23, 9777. [Google Scholar] [CrossRef] [PubMed]
- Strawbridge, R.J.; van Zuydam, N.R. Shared Genetic Contribution of Type 2 Diabetes and Cardiovascular Disease: Implications for Prognosis and Treatment. Curr. Diab. Rep. 2018, 18, 59. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, F.; Akiyama, M.; Matoba, N.; Katsuya, T.; Nakatochi, M.; Tabara, Y.; Narita, A.; Saw, W.Y.; Moon, S.; Spracklen, C.N.; et al. Interethnic analyses of blood pressure loci in populations of East Asian and European descent. Nat. Commun. 2018, 9, 5052. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.; Zhao, L.; Manuel, G.; Taylor, H.; Liu, D. Approaches to therapeutic angiogenesis for ischemic heart disease. J. Mol. Med. 2019, 97, 141–151. [Google Scholar] [CrossRef]
- Raoufinia, R.; Rahimi, H.R.; Saburi, E.; Moghbeli, M. Advances and challenges of the cell-based therapies among diabetic patients. J. Transl. Med. 2024, 22, 435. [Google Scholar] [CrossRef]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef]
- Boyle, A.P.; Hong, E.L.; Hariharan, M.; Cheng, Y.; Schaub, M.A.; Kasowski, M.; Karczewski, K.J.; Park, J.; Hitz, B.C.; Weng, S.; et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 2012, 22, 1790–1797. [Google Scholar] [CrossRef] [PubMed]
- Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J.; Ziller, M.J.; et al. Integrative analysis of 111 reference human epigenomes. Nature 2015, 518, 317–330. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CHR | Gene | Number of SNPs 1 | MTAR-O | cMTAR | iMTAR | cctP |
|---|---|---|---|---|---|---|
| 4 | TBC1D9 | 705 | ||||
| 7 | AC072061.2 | 269 | ||||
| 8 | MYOM2 | 1469 | ||||
| 10 | DOCK1 | 3541 | ||||
| 15 | CHAC1 | 20 | ||||
| 16 | ADAT1 | 166 | ||||
| 22 | TANGO2 | 375 | ||||
| 22 | ZNF74 | 6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Zhou, Z.; Ma, Y.; Ding, K.; Xiao, H.; Chen, D.; Liu, N. Shared Genetic Architectures between Coronary Artery Disease and Type 2 Diabetes Mellitus in East Asian and European Populations. Biomedicines 2024, 12, 1243. https://doi.org/10.3390/biomedicines12061243
Li X, Zhou Z, Ma Y, Ding K, Xiao H, Chen D, Liu N. Shared Genetic Architectures between Coronary Artery Disease and Type 2 Diabetes Mellitus in East Asian and European Populations. Biomedicines. 2024; 12(6):1243. https://doi.org/10.3390/biomedicines12061243
Chicago/Turabian StyleLi, Xiaoyi, Zechen Zhou, Yujia Ma, Kexin Ding, Han Xiao, Dafang Chen, and Na Liu. 2024. "Shared Genetic Architectures between Coronary Artery Disease and Type 2 Diabetes Mellitus in East Asian and European Populations" Biomedicines 12, no. 6: 1243. https://doi.org/10.3390/biomedicines12061243