
DREAM On, DREAM Off: A Review of the Estrogen Paradox in Luminal A Breast Cancers


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Estrogen-Induced Regression in ER+ Cancers
2.1. Estrogen-Induced Apoptosis in Luminal B Cancers
2.2. Evidence for a Non-Apoptotic Pathway of Estrogen-Induced Regression
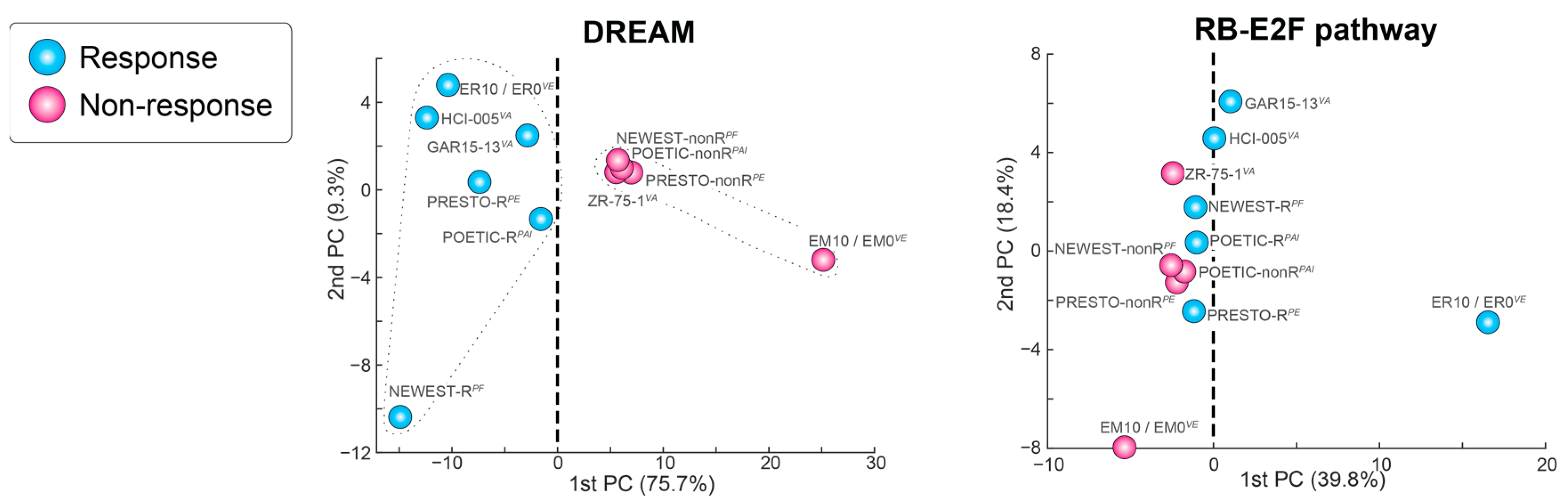
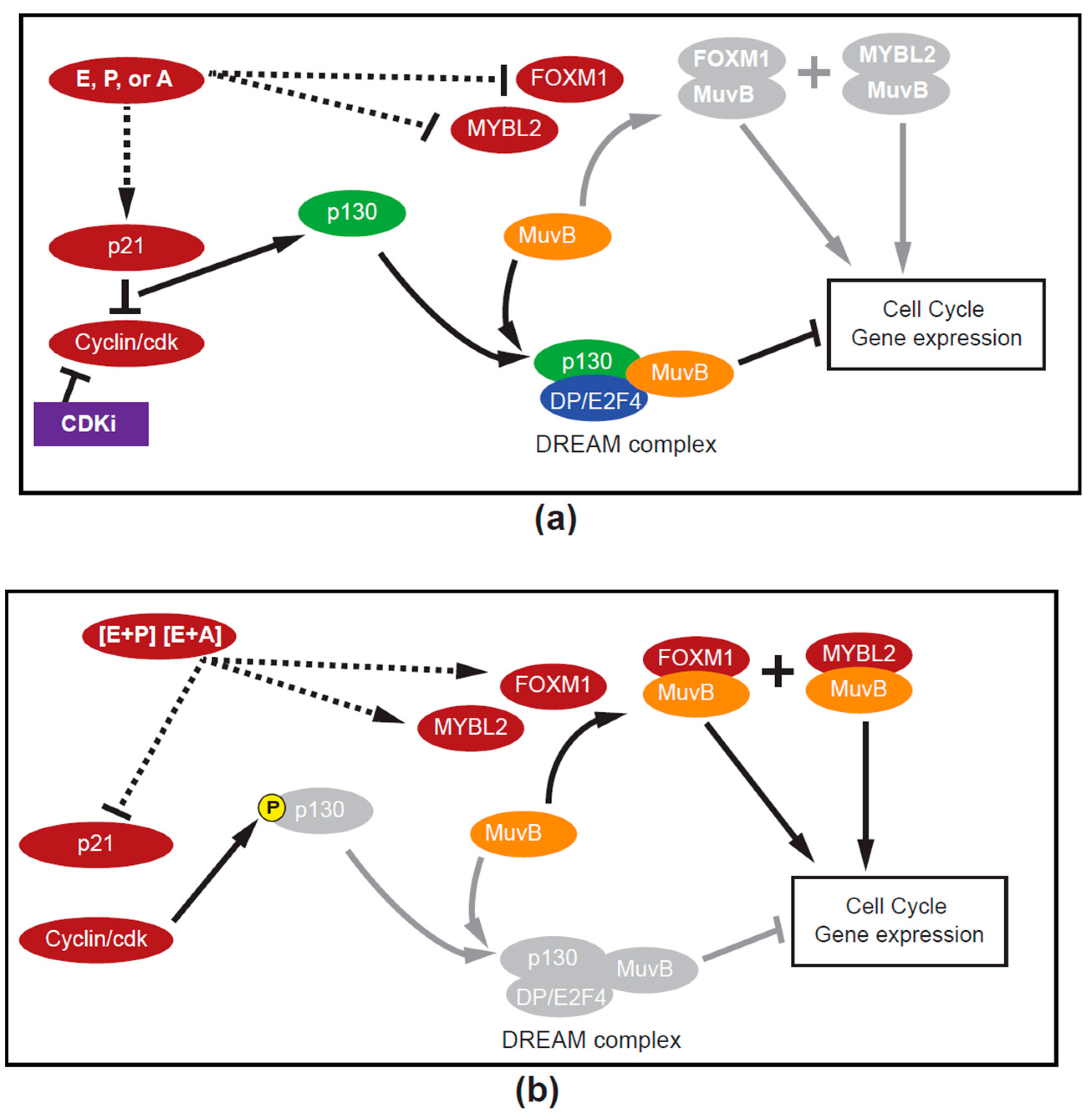
2.3. How Can Estrogen Promote and Restrict the Growth of Luminal A Cancers? DREAM
2.4. Why Does Estrogen Inhibition Require a 5-Year Gap? Estrogen in Context
3. A Hypothesis for the Estrogen Paradox in Luminal A Cancers
3.1. DREAM ON, DREAM OFF
3.2. One Hormone vs. Two Hormones
3.3. Estrogen and Anti-Estrogens

4. Conclusions
5. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
| Log2fold change in mRNA of ten RB target genes after estrogen treatment *. | ||||||||||
| PRESTO Group | CCNE2 | RIBC2 | PEG10 | PRIM1 | CDKN2C | NRM | SESTD1 | SLC1A4 | HIRIP3 | LRRCC1 |
| Non-Responders | 0.0485 | −0.0073 | 0.2826 | 0.2401 | 0.0778 | −0.0744 | −0.1907 | 0.0925 | −0.0721 | 0.1740 |
| Responders | −1.1384 | −1.4648 | −0.5749 | −0.0495 | 0.0671 | −0.0601 | −0.3463 | −0.0002 | −0.4182 | −0.4683 |
| * None of the fold changes shown had a significant Padj value. | ||||||||||
Appendix B


References
- Haddow, A.; Watkinson, J.M.; Paterson, E.; Koller, P.C. Influence of Synthetic Oestrogens on Advanced Malignant Disease. Br. Med. J. 1944, 2, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Coelingh Bennink, H.J.T.; Verhoeven, C.; Dutman, A.E.; Thijssen, J. The use of high-dose estrogens for the treatment of breast cancer. Maturitas 2017, 95, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Kautz, H. Androgens and estrogens in the treatment of disseminated mammary carcinoma. J. Am. Med. Assoc. 1960, 172, 135–147. [Google Scholar]
- Chlebowski, R.T.; Anderson, G.L.; Aragaki, A.K.; Manson, J.E.; Stefanick, M.L.; Pan, K.; Barrington, W.; Kuller, L.H.; Simon, M.S.; Lane, D.; et al. Association of Menopausal Hormone Therapy With Breast Cancer Incidence and Mortality During Long-term Follow-up of the Women’s Health Initiative Randomized Clinical Trials. JAMA 2020, 324, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Burstein, H.J. Systemic Therapy for Estrogen Receptor-Positive, HER2-Negative Breast Cancer. N. Engl. J. Med. 2020, 383, 2557–2570. [Google Scholar] [CrossRef] [PubMed]
- Shete, N.; Calabrese, J.; Tonetti, D.A. Revisiting Estrogen for the Treatment of Endocrine-Resistant Breast Cancer: Novel Therapeutic Approaches. Cancers 2023, 15, 3647. [Google Scholar] [CrossRef] [PubMed]
- Traphagen, N.A.; Hosford, S.R.; Jiang, A.; Marotti, J.D.; Brauer, B.L.; Demidenko, E.; Miller, T.W. High estrogen receptor alpha activation confers resistance to estrogen deprivation and is required for therapeutic response to estrogen in breast cancer. Oncogene 2021, 40, 3408–3421. [Google Scholar] [CrossRef] [PubMed]
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; Van De Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Neve, R.M.; Chin, K.; Fridlyand, J.; Yeh, J.; Baehner, F.L.; Fevr, T.; Clark, L.; Bayani, N.; Coppe, J.-P.; Tong, F.; et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 2006, 10, 515–527. [Google Scholar] [CrossRef]
- Kao, J.; Salari, K.; Bocanegra, M.; Choi, Y.-L.; Girard, L.; Gandhi, J.; Kwei, K.A.; Hernandez-Boussard, T.; Wang, P.; Gazdar, A.F.; et al. Molecular profiling of breast cancer cell lines defines relevant tumor models and provides a resource for cancer gene discovery. PLoS ONE 2009, 4, e6146. [Google Scholar] [CrossRef]
- Prat, A.; Karginova, O.; Parker, J.S.; Fan, C.; He, X.; Bixby, L.; Harrell, J.C.; Roman, E.; Adamo, B.; Troester, M.; et al. Characterization of cell lines derived from breast cancers and normal mammary tissues for the study of the intrinsic molecular subtypes. Breast Cancer Res. Treat. 2013, 142, 237–255. [Google Scholar] [CrossRef] [PubMed]
- Pommerenke, C.; Nagel, S.; Haake, J.; Koelz, A.L.; Christgen, M.; Steenpass, L.; Eberth, S. Molecular Characterization and Subtyping of Breast Cancer Cell Lines Provide Novel Insights into Cancer Relevant Genes. Cells 2024, 13, 301. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Shen, D.; Shao, J.; Crowder, R.; Liu, W.; Prat, A.; He, X.; Liu, S.; Hoog, J.; Lu, C.; et al. Endocrine-Therapy-Resistant ESR1 Variants Revealed by Genomic Characterization of Breast-Cancer-Derived Xenografts. Cell Rep. 2013, 4, 1116–1130. [Google Scholar] [CrossRef] [PubMed]
- Mori, H.; Saeki, K.; Chang, G.; Wang, J.; Wu, X.; Hsu, P.-Y.; Kanaya, N.; Wang, X.; Somlo, G.; Nakamura, M.; et al. Influence of estrogen treatment on esr1+ and esr1− cells in er+ breast cancer: Insights from single-cell analysis of patient-derived xenograft models. Cancers 2021, 13, 6375. [Google Scholar] [CrossRef] [PubMed]
- Ntai, I.; LeDuc, R.D.; Fellers, R.T.; Erdmann-Gilmore, P.; Davies, S.R.; Rumsey, J.; Early, B.P.; Thomas, P.M.; Li, S.; Compton, P.D.; et al. Integrated Bottom-Up and Top-Down Proteomics of Patient-Derived Breast Tumor Xenografts. Mol. Cell. Proteom. 2016, 15, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Yoshitake, R.; Mori, H.; Ha, D.; Wu, X.; Wang, J.; Wang, X.; Saeki, K.; Chang, G.; Shim, H.J.; Chan, Y.; et al. Molecular features of luminal breast cancer defined through spatial and single-cell transcriptomics. Clin. Transl. Med. 2024, 14, e1548. [Google Scholar] [CrossRef] [PubMed]
- Davies, C.; Pan, H.; Godwin, J.; Gray, R.; Arriagada, R.; Raina, V.; Abraham, M.; Medeiros Alencar, V.H.; Badran, A.; Bonfill, X.; et al. Long-term effects of continuing adjuvant tamoxifen to 10 years versus stopping at 5 years after diagnosis of oestrogen receptor-positive breast cancer: ATLAS, a randomised trial. Lancet 2013, 381, 805–816. [Google Scholar] [CrossRef] [PubMed]
- Zajchowski, D.A.; Sager, R.; Webster, L. Estrogen Inhibits the Growth of Estrogen Receptor-negative, but not Estrogen Receptor-positive, Human Mammary Epithelial Cells Expressing a Recombinant Estrogen Receptor. Cancer Res. 1993, 53, 5004–5011. [Google Scholar] [PubMed]
- Zhao, H.; Yu, J.; Peltier, C.P.; Davie, J.R. Elevated expression of the estrogen receptor prevents the down-regulation of p21Waf1/Cip1 in hormone dependent breast cancer cells. J. Cell. Biochem. 2004, 93, 619–628. [Google Scholar] [CrossRef]
- Lazennec, G.; Alcorn, J.L.; Katzenellenbogen, B.S. Adenovirus-mediated delivery of a dominant negative estrogen receptor gene abrogates estrogen-stimulated gene expression and breast cancer cell proliferation. Mol. Endocrinol. 1999, 13, 969–980. [Google Scholar] [CrossRef]
- Haddon, L. Increased Estrogen Receptor Expression Leads to a Novel DNA Binding Signature Which Differentiates Luminal A and Luminal B Breast Cancers. Ph.D. Thesis, University of Alberta, Edmonton, AB, Canada, 2018. [Google Scholar] [CrossRef]
- Harvey, J.M.; Clark, G.M.; Osborne, C.K.; Allred, D.C. Estrogen receptor status by immunohistochemistry is superior to the ligand-binding assay for predicting response to adjuvant endocrine therapy in breast cancer. J. Clin. Oncol. 1999, 17, 1474–1481. [Google Scholar] [CrossRef] [PubMed]
- Brooks, S.C.; Locke, E.R.; Soule, H.D. Estrogen receptor in a human cell line (MCF-7) from breast carcinoma. J. Biol. Chem. 1973, 248, 6251–6253. [Google Scholar] [CrossRef] [PubMed]
- Longacre, T.A.; Bartow, S.A. A correlative morphologic study of human breast and endometrium in the menstrual cycle. Am. J. Surg. Pathol. 1986, 10, 382–393. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, R.; Khan, S.A.; Badve, S. Morphological changes in breast tissue with menstrual cycle. Mod. Pathol. 2002, 15, 1348–1356. [Google Scholar] [CrossRef] [PubMed]
- Diep, C.H.; Daniel, A.R.; Mauro, L.J.; Knutson, T.P.; A Lange, C. Progesterone action in breast, uterine, and ovarian cancers. J. Mol. Endocrinol. 2015, 54, R31–R53. [Google Scholar] [CrossRef] [PubMed]
- Hugh, J.C.; Haddon, L.S.; Githaka, J.M.; Bigras, G.; Hu, X.; Madden, B.; Hanson, J.; Gabos, Z.; Giannakopoulos, N.V.; Huang, F.; et al. DREAM, a possible answer to the estrogen paradox of the Women’s Health Initiative Trial. Heliyon 2022, 8, e08666. [Google Scholar] [CrossRef] [PubMed]
- Ellis, M.J.; Gao, F.; Dehdashti, F.; Jeffe, D.B.; Marcom, P.K.; Carey, L.A.; Dickler, M.N.; Silverman, P.; Fleming, G.F.; Kommareddy, A.; et al. Lower-dose vs high-dose oral estradiol therapy of hormone receptor-positive, aromatase inhibitor-resistant advanced breast cancer: A phase 2 randomized study. JAMA 2009, 302, 774–780. [Google Scholar] [CrossRef] [PubMed]
- Ariazi, E.A.; Cunliffe, H.E.; Lewis-Wambi, J.S.; Slifker, M.J.; Willis, A.L.; Ramos, P.; Tapia, C.; Kim, H.R.; Yerrum, S.; Sharma, C.G.N.; et al. Estrogen induces apoptosis in estrogen deprivation-resistant breast cancer through stress responses as identified by global gene expression across time. Proc. Natl. Acad. Sci. USA 2011, 108, 18879–18886. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, T.O.; Leung, S.C.Y.; Rimm, D.L.; Dodson, A.; Acs, B.; Badve, S.; Denkert, C.; Ellis, M.J.; Fineberg, S.; Flowers, M.; et al. Assessment of Ki67 in Breast Cancer: Updated Recommendations From the International Ki67 in Breast Cancer Working Group. JNCI J. Natl. Cancer Inst. 2021, 113, 808–819. [Google Scholar] [CrossRef]
- Rimm, D.L.; Leung, S.C.Y.; McShane, L.M.; Bai, Y.; Bane, A.L.; Bartlett, J.M.S.; Bayani, J.; Chang, M.C.; Dean, M.; Denkert, C.; et al. An international multicenter study to evaluate reproducibility of automated scoring for assessment of Ki67 in breast cancer. Mod. Pathol. 2019, 32, 59–69. [Google Scholar] [CrossRef]
- Acs, B.; Leung, S.C.; Kidwell, K.M.; Arun, I.; Augulis, R.; Badve, S.S.; Bai, Y.; Bane, A.L.; Bartlett, J.M.; Bayani, J.; et al. Systematically higher Ki67 scores on core biopsy samples compared to corresponding resection specimen in breast cancer: A multi-operator and multi-institutional study. Mod. Pathol. 2022, 35, 1362–1369. [Google Scholar] [CrossRef] [PubMed]
- Romero, Q.; Bendahl, P.-O.; Klintman, M.; Loman, N.; Ingvar, C.; Rydén, L.; Rose, C.; Grabau, D.; Borgquist, S. Ki67 proliferation in core biopsies versus surgical samples—A model for neo-adjuvant breast cancer studies. BMC Cancer 2011, 11, 341. [Google Scholar] [CrossRef]
- Robertson, S.; Rönnlund, C.; de Boniface, J.; Hartman, J. Re-testing of predictive biomarkers on surgical breast cancer specimens is clinically relevant. Breast Cancer Res. Treat. 2019, 174, 795–805. [Google Scholar] [CrossRef] [PubMed]
- Zare, A.; Postovit, L.-M.; Githaka, J.M. Robust inflammatory breast cancer gene signature using nonparametric random forest analysis. Breast Cancer Res. 2021, 23, 92. [Google Scholar] [CrossRef] [PubMed]
- Walston, H.; Iness, A.N.; Litovchick, L. DREAM On: Cell Cycle Control in Development and Disease. Annu. Rev. Genet. 2021, 55, 309–329. [Google Scholar] [CrossRef] [PubMed]
- Uxa, S.; Bernhart, S.H.; Mages, C.F.S.; Fischer, M.; Kohler, R.; Hoffmann, S.; Stadler, P.F.; Engeland, K.; A Müller, G. DREAM and RB cooperate to induce gene repression and cell-cycle arrest in response to p53 activation. Nucleic Acids Res. 2019, 47, 9087–9103. [Google Scholar] [CrossRef]
- Finn, R.S.; Liu, Y.; Zhu, Z.; Martin, M.; Rugo, H.S.; Diéras, V.; Im, S.-A.; Gelmon, K.A.; Harbeck, N.; Lu, D.R.; et al. Biomarker Analyses of Response to Cyclin-Dependent Kinase 4/6 Inhibition and Endocrine Therapy in Women with Treatment-Naïve Metastatic Breast Cancer. Clin. Cancer Res. 2020, 26, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.; Thijssen, B.; McDermott, U.; Garnett, M.; A Wessels, L.F.; Bernards, R. Targeting the RB-E2F pathway in breast cancer. Oncogene 2016, 35, 4829–4835. [Google Scholar] [CrossRef] [PubMed]
- Mauro, L.J.; I Seibel, M.; Diep, C.H.; Spartz, A.; Kerkvliet, C.P.; Singhal, H.; Swisher, E.M.; E Schwartz, L.; Drapkin, R.; Saini, S.; et al. Progesterone Receptors Promote Quiescence and Ovarian Cancer Cell Phenotypes via DREAM in p53-Mutant Fallopian Tube Models. J. Clin. Endocrinol. Metab. 2021, 106, 1929–1955. [Google Scholar] [CrossRef]
- Nyquist, M.D.; Coleman, I.M.; Lucas, J.M.; Li, D.; Hanratty, B.; Meade, H.; Mostaghel, E.A.; Plymate, S.R.; Corey, E.; Haffner, M.C.; et al. Supraphysiological Androgens Promote the Tumor Suppressive Activity of the Androgen Receptor through cMYC Repression and Recruitment of the DREAM Complex. Cancer Res. 2023, 83, 2938–2951. [Google Scholar] [CrossRef]
- Mauro, L.J.; Seibel, M.I.; Diep, C.H.; Spartz, A.; Perez Kerkvliet, C.; Singhal, H.; Lange, C.A. Data from: Progesterone receptors promote quiescence and ovarian cancer cell phenotypes via regulation of dream in p53-mutant fallopian tube models. J. Clin. Endocrinol. Metab. 2021, 106, 1929–1955. [Google Scholar] [CrossRef] [PubMed]
- Méndez-García, L.A.; Nava-Castro, K.E.; Ochoa-Mercado, T.d.L.; Palacios-Arreola, M.I.; Ruiz-Manzano, R.A.; Segovia-Mendoza, M.; Solleiro-Villavicencio, H.; Cázarez-Martínez, C.; Morales-Montor, J. Breast Cancer Metastasis: Are Cytokines Important Players During Its Development and Progression? J. Interf. Cytokine Res. 2019, 39, 39–55. [Google Scholar] [CrossRef] [PubMed]
- Maroni, P.; Bendinelli, P.; Ferraretto, A.; Lombardi, G. Interleukin 11 (IL-11): Role(s) in Breast Cancer Bone Metastases. Biomedicines 2021, 9, 659. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yin, L.; Shen, S.; Hou, Y. Inflammation and cancer: Paradoxical roles in tumorigenesis and implications in immunotherapies. Genes Dis. 2023, 10, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Beral, V.; Million Women Study Collaborators. Breast cancer and hormone-replacement therapy in the Million Women Study. Lancet 2003, 362, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Clemmesen, J. Carcinoma of the breast; results from statistical research. Br. J. Radiol. 1948, 21, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Collaborative Group on Hormonal Factors in Breast Cancer. Menarche, menopause, and breast cancer risk: Individual participant meta-analysis, including 118 964 women with breast cancer from 117 epidemiological studies. Lancet Oncol. 2012, 13, 1141–1151. [Google Scholar] [CrossRef] [PubMed]
- Gleason, M.X.; Mdzinarishvili, T.; Sherman, S. Breast cancer incidence in black and white women stratified by estrogen and progesterone receptor statuses. PLoS ONE 2012, 7, e49359. [Google Scholar] [CrossRef] [PubMed]
- Lasley, B.L.; Crawford, S.; McConnell, D.S. Adrenal androgens and the menopausal transition. Obstet. Gynecol. Clin. N. Am. 2011, 38, 467–475. [Google Scholar] [CrossRef]
- Aribas, E.; van Lennep, J.E.R.; De Rijke, Y.B.; Laven, J.S.E.; Ikram, M.A.; Peeters, R.P.; Kavousi, M. Sex steroids and sex steroid-binding globulin levels amongst middle-aged and elderly men and women from general population. Eur. J. Clin. Investig. 2022, 52, e13866. [Google Scholar] [CrossRef]
- Lasley, B.L.; Crawford, S.L.; Laughlin, G.A.; Santoro, N.; McConnell, D.S.; Crandall, C.; Greendale, G.A.; Polotsky, A.J.; Vuga, M. Circulating dehydroepiandrosterone sulfate levels in women who underwent bilateral salpingo-oophorectomy during the menopausal transition. Menopause 2011, 18, 494–498. [Google Scholar] [CrossRef]
- Trabert, B.; Bauer, D.C.; Buist, D.S.M.; Cauley, J.A.; Falk, R.T.; Geczik, A.M.; Gierach, G.L.; Hada, M.; Hue, T.F.; Lacey, J.V.; et al. Association of Circulating Progesterone With Breast Cancer Risk Among Postmenopausal Women. JAMA Netw. Open 2020, 3, e203645. [Google Scholar] [CrossRef]
- Ross-Innes, C.S.; Stark, R.; Teschendorff, A.E.; Holmes, K.A.; Ali, H.R.; Dunning, M.J.; Brown, G.D.; Gojis, O.; Ellis, I.O.; Green, A.R.; et al. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 2012, 481, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Jenster, G.; Epner, D.E. Androgen Induction of Cyclin-Dependent Kinase Inhibitor p21 Gene: Role of Androgen Receptor and Transcription Factor Sp1 Complex. Mol. Endocrinol. 2000, 14, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Owen, G.I.; Richer, J.K.; Tung, L.; Takimoto, G.; Horwitz, K.B. Progesterone regulates transcription of the p21(WAF1) cyclin-dependent kinase inhibitor gene through Sp1 and CBP/p300. J. Biol. Chem. 1998, 273, 10696–10701. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, H.; Russell, I.A.; Stark, R.; Rueda, O.M.; Hickey, T.E.; Tarulli, G.A.; Serandour, A.A.A.; Birrell, S.N.; Bruna, A.; Saadi, A.; et al. Progesterone receptor modulates ERα action in breast cancer. Nature 2015, 523, 313–317. [Google Scholar] [CrossRef]
- Poortman, J.; Prenen, J.A.; Schwarz, F.; Thijssen, J.H. Interaction of delta-5-androstene-3beta, 17beta-diol with estradiol and dihydrotestosterone receptors in human myometrial and mammary cancer tissue. J. Clin. Endocrinol. Metab. 1975, 40, 373–379. [Google Scholar] [CrossRef]
- Honma, N.; Saji, S.; Hirose, M.; Horiguchi, S.-I.; Kuroi, K.; Hayashi, S.-I.; Utsumi, T.; Harada, N. Sex steroid hormones in pairs of tumor and serum from breast cancer patients and pathobiological role of androstene-3β, 17β-diol. Cancer Sci. 2011, 102, 1848–1854. [Google Scholar] [CrossRef]
- Chen, J.; Wang, W.-Q.; Lin, S.-X. Interaction of Androst-5-ene-3β,17β-diol and 5α-androstane-3β,17β-diol with estrogen and androgen receptors: A combined binding and cell study. J. Steroid Biochem. Mol. Biol. 2013, 137, 316–321. [Google Scholar] [CrossRef]
- Hackenberg, R.; Turgetto, I.; Filmer, A.; Schulz, K.D. Estrogen and androgen receptor mediated stimulation and inhibition of proliferation by androst-5-ene-3β,17β-diol in human mammary cancer cells. J. Steroid Biochem. Mol. Biol. 1993, 46, 597–603. [Google Scholar] [CrossRef]
- Boccuzzi, G.; Brignardello, E.; Di Monaco, M.; Gatto, V.; Leonardi, L.; Pizzini, A.; Gallo, M. 5-En-androstene-3β,17β-diol inhibits the growth of MCF-7 breast cancer cells when oestrogen receptors are blocked by oestradiol. Br. J. Cancer 1994, 70, 1035–1039. [Google Scholar] [CrossRef] [PubMed]
- Peters, A.A.; Buchanan, G.; Ricciardelli, C.; Bianco-Miotto, T.; Centenera, M.M.; Harris, J.M.; Jindal, S.; Segara, D.; Jia, L.; Moore, N.L.; et al. Androgen receptor inhibits estrogen receptor-α activity and is prognostic in breast cancer. Cancer Res. 2009, 69, 6131–6140. [Google Scholar] [CrossRef] [PubMed]
- Need, E.F.; Selth, L.A.; Harris, T.J.; Birrell, S.N.; Tilley, W.D.; Buchanan, G. Research resource: Interplay between the genomic and transcriptional networks of androgen receptor and estrogen receptor α in luminal breast cancer cells. Mol. Endocrinol. 2012, 26, 1941–1952. [Google Scholar] [CrossRef] [PubMed]
- Hickey, T.E.; Selth, L.A.; Chia, K.M.; Laven-Law, G.; Milioli, H.H.; Roden, D.; Jindal, S.; Hui, M.; Finlay-Schultz, J.; Ebrahimie, E.; et al. The androgen receptor is a tumor suppressor in estrogen receptor–positive breast cancer. Nat. Med. 2021, 27, 310–320. [Google Scholar] [CrossRef] [PubMed]
- Ingle, J.N.; Ahmann, D.L.; Green, S.J.; Edmonson, J.H.; Bisel, H.F.; Kvols, L.K.; Nichols, W.C.; Creagan, E.T.; Hahn, R.G.; Rubin, J.; et al. Randomized clinical trial of diethylstilbestrol versus tamoxifen in postmenopausal women with advanced breast cancer. N. Engl. J. Med. 1981, 304, 16–21. [Google Scholar] [CrossRef]
- Peethambaram, P.P.; Ingle, J.N.; Suman, V.J.; Hartmann, L.C.; Loprinzi, C.L. Randomized trial of diethylstilbestrol vs. tamoxifen in postmenopausal women with metastatic breast cancer. An updated analysis. Breast Cancer Res. Treat. 1999, 54, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Key, T.; Appleby, P.; Barnes, I.; Reeves, G. Endogenous Hormones and Breast Cancer Collaborative Group. Endogenous sex hormones and breast cancer in postmenopausal women: Reanalysis of nine prospective studies. J. Natl. Cancer Inst. 2002, 94, 606–616. [Google Scholar] [CrossRef] [PubMed]
- Lønning, P.E.; Haynes, B.P.; Straume, A.H.; Dunbier, A.; Helle, H.; Knappskog, S.; Dowsett, M. Recent data on intratumor estrogens in breast cancer. Steroids 2011, 76, 786–791. [Google Scholar] [CrossRef] [PubMed]
- Geisler, J.; Suzuki, T.; Helle, H.; Miki, Y.; Nagasaki, S.; Duong, N.K.; Ekse, D.; Aas, T.; Evans, D.B.; Lønning, P.E.; et al. Breast cancer aromatase expression evaluated by the novel antibody 677: Correlations to intra-tumor estrogen levels and hormone receptor status. J. Steroid Biochem. Mol. Biol. 2010, 118, 237–241. [Google Scholar] [CrossRef]
- Miller, W.R.; Stuart, M.; Sahmoud, T.; Dixon, J.M. Anastrozole (‘Arimidex’) blocks oestrogen synthesis both peripherally and within the breast in postmenopausal women with large operable breast cancer. Br. J. Cancer 2002, 87, 950–955. [Google Scholar] [CrossRef]
- Macedo, L.F.; Guo, Z.; Tilghman, S.L.; Sabnis, G.J.; Qiu, Y.; Brodie, A. Role of Androgens on MCF-7 Breast Cancer Cell Growth and on the Inhibitory Effect of Letrozole. Cancer Res. 2006, 66, 7775–7782. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; López-Knowles, E.; Cheang, M.C.U.; Morden, J.; Ribas, R.; Sidhu, K.; Evans, D.; Martins, V.; Dodson, A.; Skene, A.; et al. Impact of aromatase inhibitor treatment on global gene expression and its association with antiproliferative response in ER+ breast cancer in postmenopausal patients. Breast Cancer Res. 2019, 22, 2. [Google Scholar] [CrossRef] [PubMed]
- Cuzick, J.; Sestak, I.; Forbes, J.F.; Dowsett, M.; Knox, J.; Cawthorn, S.; Saunders, C.; Roche, N.; E Mansel, R.; von Minckwitz, G.; et al. Anastrozole for prevention of breast cancer in high-risk postmenopausal women (IBIS-II): An international, double-blind, randomised placebo-controlled trial. Lancet 2014, 383, 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- Goss, P.E.; Ingle, J.N.; Alés-Martínez, J.E.; Cheung, A.M.; Chlebowski, R.T.; Wactawski-Wende, J.; McTiernan, A.; Robbins, J.; Johnson, K.C.; Martin, L.W.; et al. Exemestane for Breast-Cancer Prevention in Postmenopausal Women. N. Engl. J. Med. 2011, 364, 2381–2391. [Google Scholar] [CrossRef] [PubMed]
- Visvanathan, K.; Fabian, C.J.; Bantug, E.; Brewster, A.M.; Davidson, N.E.; DeCensi, A.; Floyd, J.D.; Garber, J.E.; Hofstatter, E.W.; Khan, S.A.; et al. Use of Endocrine Therapy for Breast Cancer Risk Reduction: ASCO Clinical Practice Guideline Update. J. Clin. Oncol. 2019, 37, 3152–3165. [Google Scholar] [CrossRef] [PubMed]
- Patani, N.; Dunbier, A.K.; Anderson, H.; Ghazoui, Z.; Ribas, R.; Anderson, E.; Gao, Q.; A’Hern, R.; Mackay, A.; Lindemann, J.; et al. Differences in the Transcriptional Response to Fulvestrant and Estrogen Deprivation in ER-Positive Breast Cancer. Clin. Cancer Res. 2014, 20, 3962–3973. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xu, B.; Wang, W.; Zhai, X.; Chen, X. Efficacy and safety of fulvestrant in postmenopausal patients with hormone receptor-positive advanced breast cancer: A systematic literature review and meta-analysis. Breast Cancer Res. Treat. 2018, 171, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Mehta, R.S.; Barlow, W.E.; Albain, K.S.; Vandenberg, T.A.; Dakhil, S.R.; Tirumali, N.R.; Lew, D.L.; Hayes, D.F.; Gralow, J.R.; Linden, H.M.; et al. Overall Survival with Fulvestrant plus Anastrozole in Metastatic Breast Cancer. N. Engl. J. Med. 2019, 380, 1226–1234. [Google Scholar] [CrossRef] [PubMed]
- Carroll, J.S.; Prall, O.W.J.; Musgrove, E.A.; Sutherland, R.L. A Pure Estrogen Antagonist Inhibits Cyclin E-Cdk2 Activity in MCF-7 Breast Cancer Cells and Induces Accumulation of p130-E2F4 Complexes Characteristic of Quiescence. J. Biol. Chem. 2000, 275, 38221–38229. [Google Scholar] [CrossRef]
- Magge, T.; Rajendran, S.; Brufsky, A.M.; Foldi, J. CDK4/6 inhibitors: The Devil is in the Detail. Curr. Oncol. Rep. 2024. Online ahead of Print. [Google Scholar] [CrossRef]
- Witkiewicz, A.K.; Schultz, E.; Wang, J.; Hamilton, D.; Levine, E.; O’connor, T.; Knudsen, E.S. Determinants of response to CDK4/6 inhibitors in the real-world setting. npj Precis. Oncol. 2023, 7, 90. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hugh, J.C.; Haddon, L.S.J.; Githaka, J.M. DREAM On, DREAM Off: A Review of the Estrogen Paradox in Luminal A Breast Cancers. Biomedicines 2024, 12, 1300. https://doi.org/10.3390/biomedicines12061300
Hugh JC, Haddon LSJ, Githaka JM. DREAM On, DREAM Off: A Review of the Estrogen Paradox in Luminal A Breast Cancers. Biomedicines. 2024; 12(6):1300. https://doi.org/10.3390/biomedicines12061300
Chicago/Turabian StyleHugh, Judith C., Lacey S. J. Haddon, and John Maringa Githaka. 2024. "DREAM On, DREAM Off: A Review of the Estrogen Paradox in Luminal A Breast Cancers" Biomedicines 12, no. 6: 1300. https://doi.org/10.3390/biomedicines12061300
APA StyleHugh, J. C., Haddon, L. S. J., & Githaka, J. M. (2024). DREAM On, DREAM Off: A Review of the Estrogen Paradox in Luminal A Breast Cancers. Biomedicines, 12(6), 1300. https://doi.org/10.3390/biomedicines12061300