
Cerebral Microbleeds Associate with Brain Endothelial Cell Activation-Dysfunction and Blood–Brain Barrier Dysfunction/Disruption with Increased Risk of Hemorrhagic and Ischemic Stroke


Abstract
:
1. Introduction
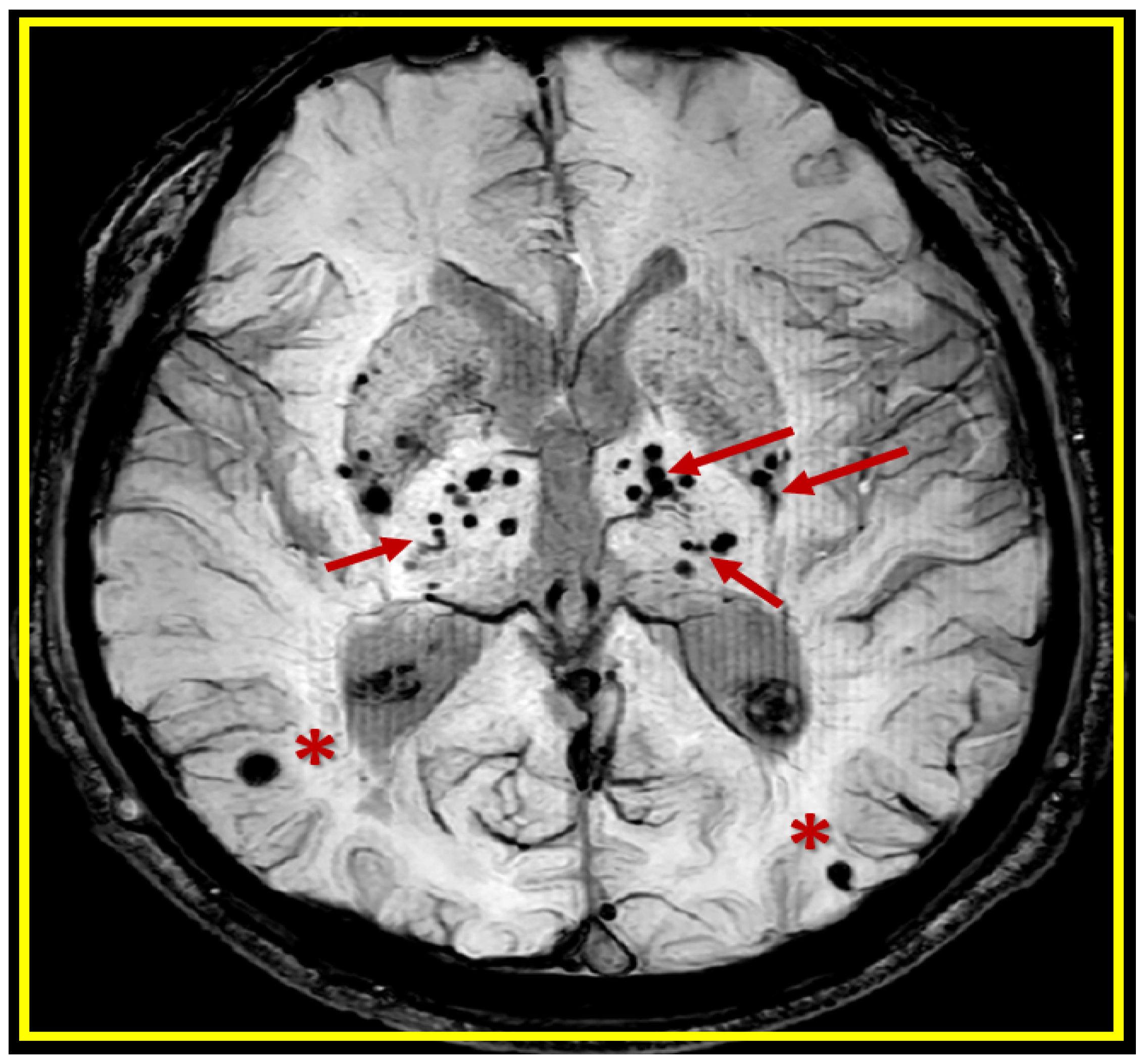
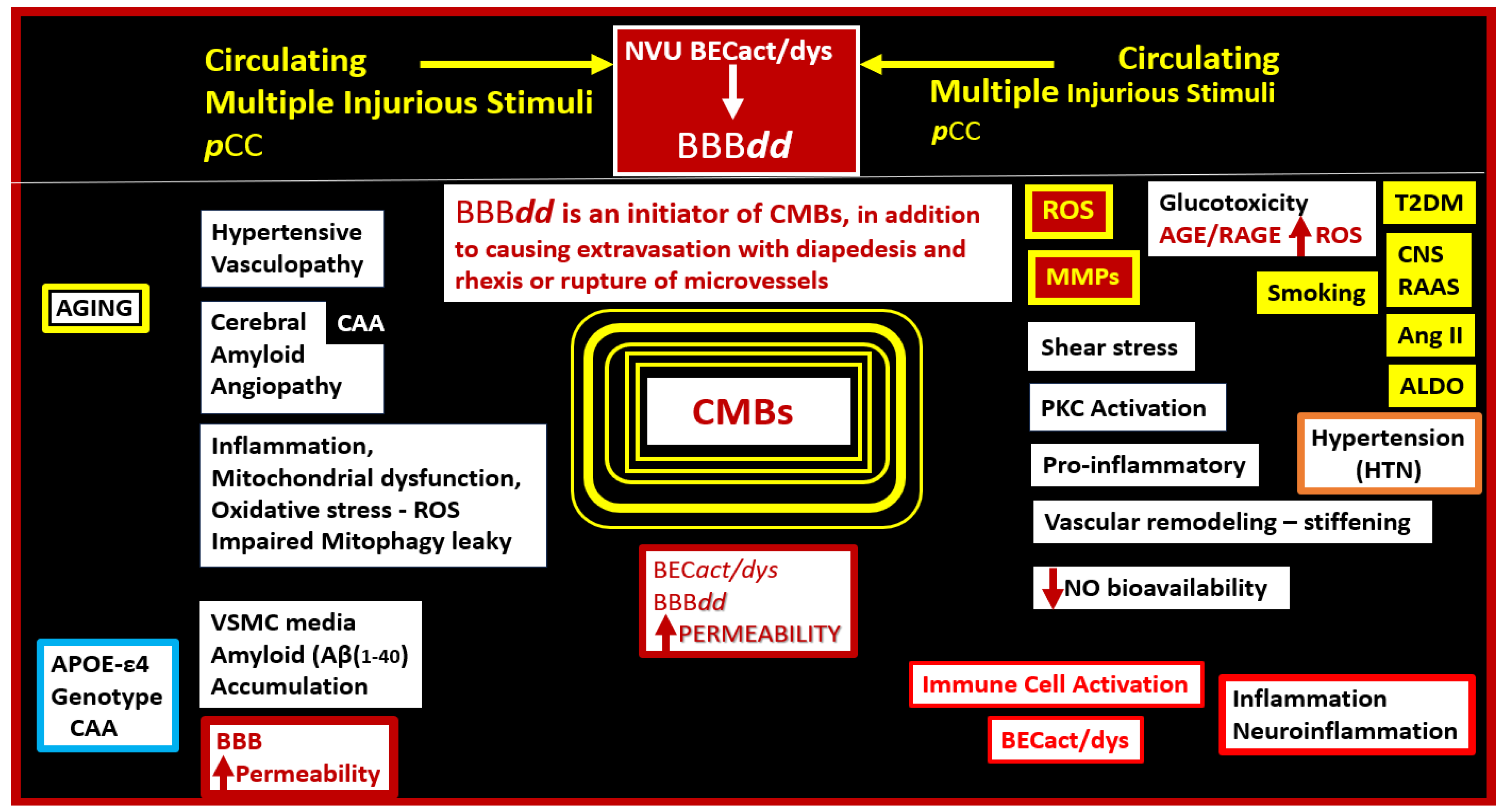
2. A Possible Sequence of Events in the Development of Cerebral Microvessel Bleeds (CMBs)
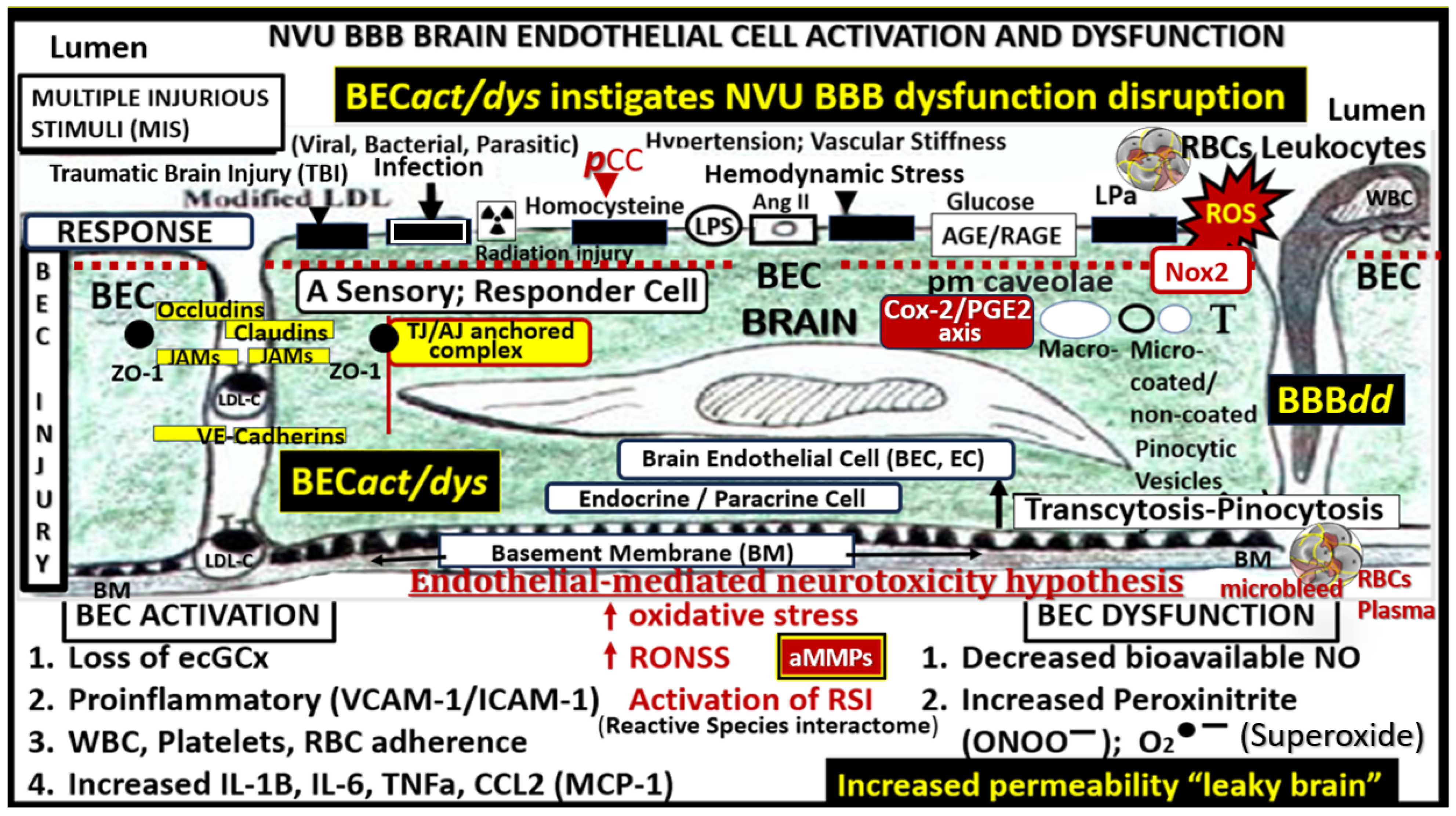
2.1. Brain Endothelial Cell Activation and Dysfunction (BECact/dys)
2.2. Blood–Brain Barrier Dysfunction and/or Disruption (BBBdd) with Increased Permeability
2.3. Hypertensive (HTN) Vasculopathy
2.4. Cerebral Amyloid Angiopathy (CAA)
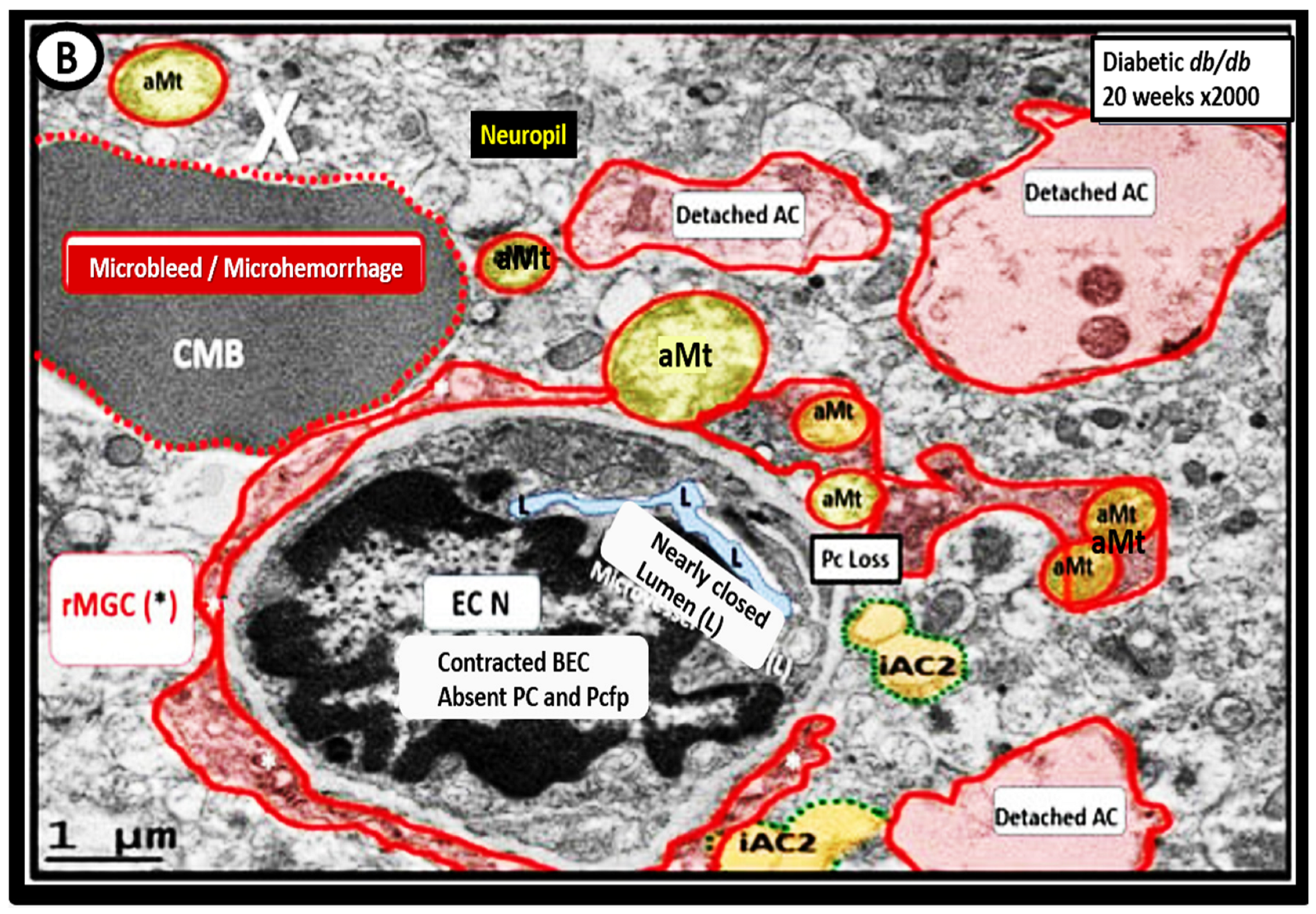
3. Transmission Electron Microscopy (TEM) Imaging of BECact/dys), BBBdd with CMBs
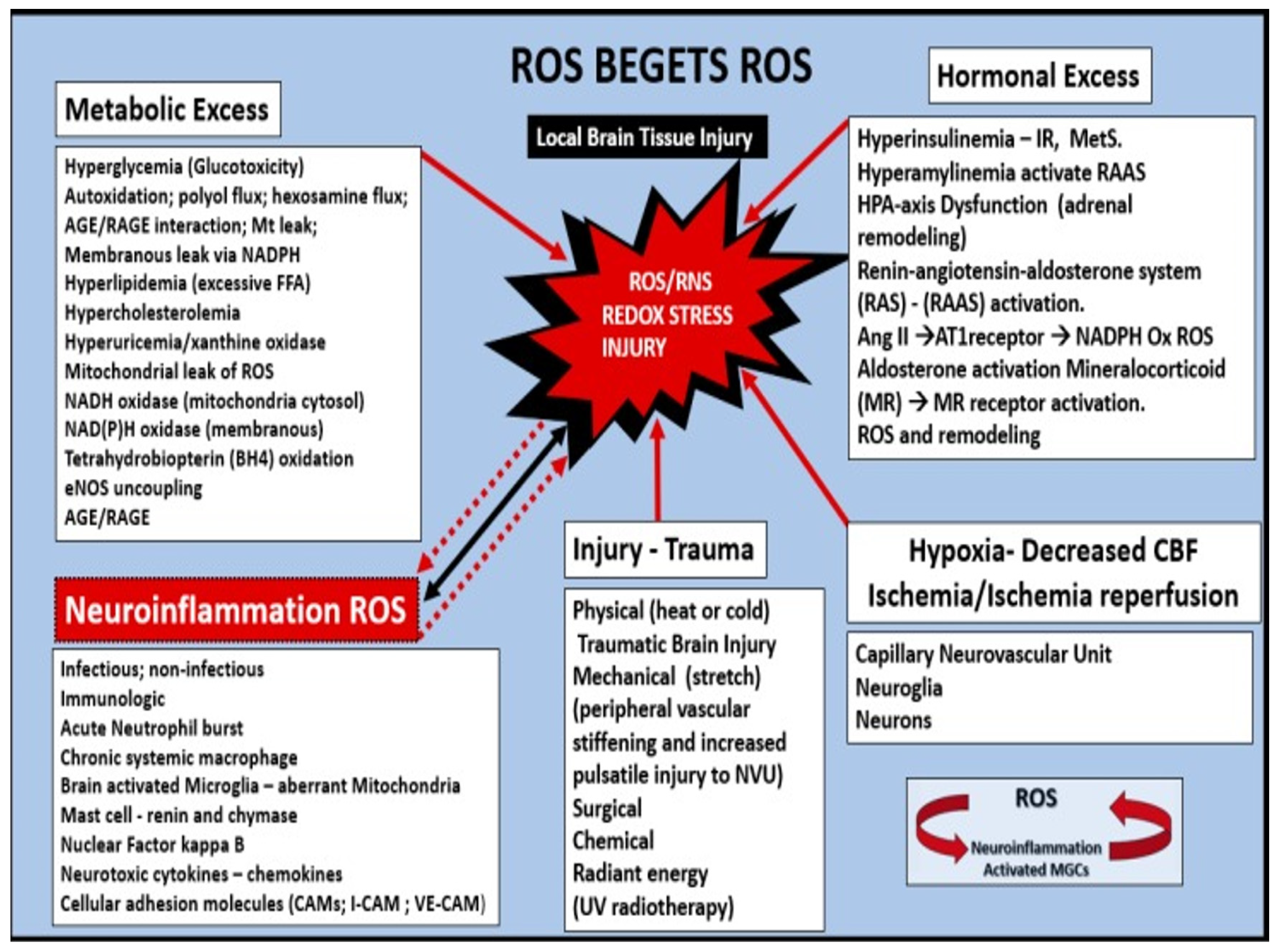
Oxidative—Redox Stress: Implications in the Development of CAA, LOAD, SVD, and CMBs
4. Atrial Fibrillation (AF) Association with Cerebral Microbleeds and Stroke
5. Apolipoprotein E Association with CMBs
6. Conclusions
7. Future Directions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Wardlaw, J.M.; Smith, E.E.; Biessels, G.J.; Cordonnier, C.; Fazekas, F.; Frayne, R.; Lindley, R.I.; O’Brien, J.T.; Barkhof, F.; Benavente, O.R.; et al. Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol. 2013, 12, 822–838. [Google Scholar] [CrossRef]
- Greenberg, S.M.; Vernooij, M.W.; Cordonnier, C.; Viswanathan, A.; Salman, R.A.; Warach, S.; Launer, L.J.; Van Buchem, M.A.; Breteler, M.M. Cerebral microbleeds: A guide to detection and interpretation. Lancet Neurol. 2009, 8, 165–174. [Google Scholar] [CrossRef]
- Lee, J.; Sohn, E.H.; Oh, E.; Lee, A.Y. Characteristics of Cerebral Microbleeds. Dement. Neurocogn. Disord. 2018, 17, 73–82. [Google Scholar] [CrossRef]
- Shao, P.; Xu, H.; Sheng, X.; Qin, R.; Ma, J.; Luo, Y.; Lee, A.; Shi, L.; Huang, L.; Cheng, Y.; et al. Lobar Cerebral Microbleeds Are Associated with Cognitive Decline in Patients with Type 2 Diabetes Mellitus. Front. Neurol. 2022, 13, 843260. [Google Scholar] [CrossRef]
- Yakushiji, Y.; Hara, H. Cerebral microbleeds: Clinical features and management. Rinsho Shinkeigaku 2012, 52, 1106–1109. [Google Scholar] [CrossRef]
- Elmståhl, S.; Ellström, K.; Siennicki-Lantz, A.; Abul-Kasim, K. Association between cerebral microbleeds and hypertension in the Swedish general population “Good Aging in Skåne” study. J. Clin. Hypertens. 2019, 21, 1099–1107. [Google Scholar] [CrossRef]
- Akoudad, S.; Portegies, M.L.; Koudstaal, P.J.; Hofman, A.; van der Lugt, A.; Ikram, M.A.; Vernooij, M.W. Cerebral Microbleeds Are Associated with an Increased Risk of Stroke. The Rotterdam Study. Circulation 2015, 132, 509–516. [Google Scholar] [CrossRef]
- Yakushiji, Y.; Werring, D.J. Cerebrovascular disease: Lobar cerebral microbleeds signal early cognitive impairment. Nat. Rev. Neurol. 2016, 12, 680–682. [Google Scholar] [CrossRef]
- Pantoni, L. Cerebral small vessel disease: From pathogenesis and clinical characteristics to therapeutic challenges. Lancet Neurol. 2010, 9, 689–701. [Google Scholar] [CrossRef]
- Marini, S.; Anderson, C.D.; Rosand, J. Genetics of Cerebral Small Vessel Disease. Stroke 2020, 51, 12–20. [Google Scholar] [CrossRef]
- Gao, Y.; Li, D.; Lin, J.; Thomas, A.M.; Miao, J.; Chen, D.; Li, S.; Chu, C. Cerebral small vessel disease: Pathological mechanisms and potential therapeutic targets. Front. Aging Neurosci. 2022, 14, 961661. [Google Scholar] [CrossRef]
- Zhao, L.; Lee, A.; Fan, Y.-H.; Mok, V.C.; Shi, L. Magnetic resonance imaging manifestations of cerebral small vessel disease: Automated quantification and clinical application. Chin. Med. J. 2021, 134, 151–160. [Google Scholar] [CrossRef]
- Shulyatnikova, T.; Hayden, M.R. Why Are Perivascular Spaces Important? Medicina 2023, 59, 917. [Google Scholar] [CrossRef]
- Stokum, J.A.; Cannarsa, G.J.; Wessell, A.P.; Shea, P.; Wenger, N.; Simard, J.M. When the Blood Hits Your Brain: The Neurotoxicity of Extravasated Blood. Int. J. Mol. Sci. 2021, 22, 5132. [Google Scholar] [CrossRef]
- Hayden, M.R. A Closer Look at the Perivascular Unit in the Development of Enlarged Perivascular Spaces in Obesity, Metabolic Syndrome, and Type 2 Diabetes Mellitus. Biomedicines 2024, 12, 96. [Google Scholar] [CrossRef]
- Hayden, M.R. Brain Injury: Response to Injury Wound-Healing Mechanisms and Enlarged Perivascular Spaces in Obesity, Metabolic Syndrome, and Type 2 Diabetes Mellitus. Medicina 2023, 59, 1337. [Google Scholar] [CrossRef]
- Wang, H.-L.; Zhang, C.-L.; Qiu, Y.-M.; Chen, A.-Q.; Li, Y.-N.; Hu, B. Dysfunction of the Blood-brain Barrier in Cerebral Microbleeds: From Bedside to Bench. Aging Dis. 2021, 12, 1898–1919. [Google Scholar] [CrossRef]
- Cheng, Z.; Dai, L.; Wu, Y.; Cao, Y.; Chai, X.; Wang, P.; Liu, C.; Ni, M.; Gao, F.; Wang, Q.; et al. Correlation of blood–brain barrier leakage with cerebral small vessel disease including cerebral microbleeds in Alzheimer’s disease. Front. Neurol. 2023, 14, 1077860. [Google Scholar] [CrossRef]
- Li, Y.; Li, M.; Zuo, L.; Li, X.; Hou, Y.; Hu, W. Cerebral Microbleeds Are Associated with Widespread Blood-Brain Barrier Leakage. Eur. Neurol. 2023, 86, 395–403. [Google Scholar] [CrossRef]
- Lee, J.M.; Zhai, G.; Liu, Q.; Gonzales, E.R.; Yin, K.; Yan, P.; Hsu, C.Y.; Vo, K.D.; Lin, W. Vascular permeability precedes spontaneous intracerebral hemorrhage in stroke-prone spontaneously hypertensive rats. Stroke 2007, 38, 3289–3291. [Google Scholar] [CrossRef]
- Freeze, W.M.; Jacobs, H.I.L.; Schreuder, F.H.B.M.; van Oostenbrugge, R.J.; Backes, W.H.; Verhey, F.R.; Klijn, C.J.M. Blood-Brain Barrier Dysfunction in Small Vessel Disease Related Intracerebral Hemorrhage. Front. Neurol. 2018, 9, 926. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.; Ambler, G.; Lee, K.-J.; Lim, J.-S.; Shiozawa, M.; Koga, M.; Li, L.; Lovelock, C.; Chabriat, H.; Hennerici, M.; et al. Cerebral microbleeds and stroke risk after ischaemic stroke or transient ischaemic attack: A pooled analysis of individual patient data from cohort studies. Lancet Neurol. 2019, 18, 653–665. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, T.; Ueba, T.; Kajiwara, M.; Fujisawa, I.; Miyamatsu, N.; Yamashita, K. Cerebral microbleeds predict first-ever symptomatic cerebrovascular events. Clin. Neurol. Neurosurg. 2009, 111, 825–828. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.-B.; Kang, D.-W.; Cho, A.-H.; Lee, E.-M.; Choi, C.G.; Kwon, S.U.; Kim, J.S. Initial microbleeds at MR imaging can predict recurrent intracerebral hemorrhage. J. Neurol. 2007, 254, 508–512. [Google Scholar] [CrossRef] [PubMed]
- Senior, K. Microbleeds may predict cerebral bleeding after stroke. Lancet 2002, 359, 769. [Google Scholar] [CrossRef] [PubMed]
- Akoudad, S.; Wolters, F.J.; Viswanathan, A.; de Bruijn, R.F.; van der Lugt, A.; Hofman, A.; Koudstaal, P.J.; Ikram, M.A.; Vernooij, M.W. Association of Cerebral Microbleeds with Cognitive Decline and Dementia. JAMA Neurol. 2016, 73, 934–943. [Google Scholar] [CrossRef] [PubMed]
- Thompson, C.S.; Hakim, A.M. Living beyond our physiological means: Small vessel disease of the brain is an expression of a systemic failure in arteriolar function: A unifying hypothesis. Stroke 2009, 40, e322–e330. [Google Scholar] [CrossRef] [PubMed]
- Charidimou, A.; Werring, D.J. Cerebral microbleeds: Detection, mechanisms and clinical challenges. Futur. Neurol. 2011, 6, 587–611. [Google Scholar] [CrossRef]
- Ungvari, Z.; Tarantini, S.; Kirkpatrick, A.C.; Csiszar, A.; Prodan, C.I. Cerebral microhemorrhages: Mechanisms, consequences, and prevention. Am. J. Physiol. Heart Circ. Physiol. 2017, 312, H1128–H1143. [Google Scholar] [CrossRef]
- Fulop, G.A.; Tarantini, S.; Yabluchanskiy, A.; Molnar, A.; Prodan, C.I.; Kiss, T.; Csipo, T.; Lipecz, A.; Balasubramanian, P.; Farkas, E.; et al. Role of age-related alterations of the cerebral venous circulation in the pathogenesis of vascular cognitive impairment. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H1124–H1140. [Google Scholar] [CrossRef]
- Stolarz, A.J.; Mu, S.; Zhang, H.; Fouda, A.Y.; Rusch, N.J.; Ding, Z. Opinion: Endothelial Cells—Macrophage-Like Gatekeepers? Front. Immunol. 2022, 13, 902945. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.K. Linking endothelial dysfunction with endothelial cell activation. J. Clin. Investig. 2013, 123, 540–541. [Google Scholar] [CrossRef] [PubMed]
- Pober, J.S. Cytokine-mediated activation of vascular endothelium. Am. J. Pathol. 1988, 133, 426–433. [Google Scholar] [PubMed]
- Yang, Y.-M.; Huang, A.; Kaley, G.; Sun, D.; Chang, F.; Flavahan, S.; Flavahan, N.A.; Verkaik, M.; Juni, R.P.; van Loon, E.P.M.; et al. eNOS uncoupling and endothelial dysfunction in aged vessels. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1829–H1836. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.R.; Tyagi, S.C. Intimal redox stress: Accelerated atherosclerosis in metabolic syndrome and type 2 diabetes mellitus. Atheroscleropathy. Cardiovasc. Diabetol. 2002, 1, 3. [Google Scholar] [CrossRef] [PubMed]
- Owens, T.; Bechmann, I.; Engelhardt, B. Perivascular spaces and the two steps to neuroinflammation. J. Neuropathol. Exp. Neurol. 2008, 67, 1113–1121. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.R. The Brain Endothelial Cell Glycocalyx Plays a Crucial Role in the Development of Enlarged Perivascular Spaces in Obesity, Metabolic Syndrome, and Type 2 Diabetes Mellitus. Life 2023, 13, 1955. [Google Scholar] [CrossRef] [PubMed]
- Noguchi-Shinohara, M.; Komatsu, J.; Samuraki, M.; Matsunari, I.; Ikeda, T.; Sakai, K.; Hamaguchi, T.; Ono, K.; Nakamura, H.; Yamada, M. Cerebral amyloid angiopathy-related microbleeds and cerebrospinal fluid biomarkers in alzheimer’s disease. J. Alzheimer’s Dis. 2017, 55, 905–913. [Google Scholar] [CrossRef]
- Petrea, R.E.; O’donnell, A.; Beiser, A.S.; Habes, M.; Aparicio, H.; DeCarli, C.; Seshadri, S.; Romero, J.R. Mid to late life hypertension trends and cerebral small vessel disease in the framingham heart study. Hypertension 2020, 76, 707–714. [Google Scholar] [CrossRef]
- Toth, P.; Tarantini, S.; Springo, Z.; Tucsek, Z.; Gautam, T.; Giles, C.B.; Wren, J.D.; Koller, A.; Sonntag, W.E.; Csiszar, A.; et al. Aging exacerbates hypertension-induced cerebral microhemorrhages in mice: Role of resveratrol treatment in vasoprotection. Aging Cell 2015, 14, 400–408. [Google Scholar] [CrossRef]
- Nyúl-Tóth, Á.; Tarantini, S.; Kiss, T.; Toth, P.; Galvan, V.; Tarantini, A.; Yabluchanskiy, A.; Csiszar, A.; Ungvari, Z. Increases in hypertension-induced cerebral microhemorrhages exacerbate gait dysfunction in a mouse model of alzheimer’s disease. GeroScience 2020, 42, 1685–1698. [Google Scholar] [CrossRef] [PubMed]
- Poels, M.; Ikram, M.; van der Lugt, A.; Hofman, A.; Niessen, W.; Krestin, G.; Breteler, M.; Vernooij, M. Cerebral microbleeds are associated with worse cognitive function: The rotterdam scan study. Neurology 2012, 78, 326–333. [Google Scholar] [CrossRef]
- Baggeroer, C.E.; Cambronero, F.E.; Savan, N.A.; Jefferson, A.L.; Santisteban, M.M. Basic Mechanisms of Brain Injury and Cognitive Decline in Hypertension. Hypertension 2024, 81, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Katsi, V.; Marketou, M.; Maragkoudakis, S.; Didagelos, M.; Charalambous, G.; Parthenakis, F.; Tsioufis, C.; Tousoulis, D. Blood–brain barrier dysfunction: The undervalued frontier of hypertension. J. Hum. Hypertens. 2020, 34, 682–691. [Google Scholar] [CrossRef]
- Biffi, A.; Greenberg, S.M. Cerebral amyloid angiopathy: A systematic review. J. Clin. Neurol. 2011, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Li, T.; Ma, X.; Liu, S.; Li, C.; Huang, Z.; Lin, Y.; Wu, R.; Wang, S.; Lu, D.; et al. Macrophage lineage cells-derived migrasomes activate complement-dependent blood-brain barrier damage in cerebral amyloid angiopathy mouse model. Nat. Commun. 2023, 14, 3945. [Google Scholar] [CrossRef] [PubMed]
- Pezzini, A.; Del Zotto, E.; Volonghi, I.; Giossi, A.; Costa, P.; Padovani, A. Cerebral amyloid angiopathy: A common cause of cerebral hemorrhage. Curr. Med. Chem. 2009, 16, 2498–2513. [Google Scholar] [CrossRef] [PubMed]
- Roher, A.E.; Lowenson, J.D.; Clarke, S.; Woods, A.S.; Cotter, R.J.; Gowing, E.; Ball, M.J. beta-Amyloid-(1-42) is a major component of cerebrovascular amyloid deposits: Implications for the pathology of Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 10836–10840. [Google Scholar] [CrossRef]
- Chow, V.W.; Mattson, M.P.; Wong, P.C.; Gleichmann, M. An overview of APP processing enzymes and products. Neuromolecular Med. 2010, 12, 1–12. [Google Scholar] [CrossRef]
- Mestre, H.; Kostrikov, S.; Mehta, R.I.; Nedergaard, M. Perivascular Spaces, Glymphatic Dysfunction, and Small Vessel Disease. Clin. Sci. 2017, 131, 2257–2274. [Google Scholar] [CrossRef]
- Keable, A.; Fenna, K.; Yuen, H.M.; Johnston, D.A.; Smyth, N.R.; Smith, C.; Salman, R.A.; Samarasekera, N.; Nicoll, J.A.; Attems, J.; et al. Deposition of amyloid β in the walls of human leptomeningeal arteries in relation to perivascular drainage pathways in cerebral amyloid angiopathy. Biochim. Biophys. Acta 2016, 1862, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, S.M.; Bacskai, B.J.; Hernandez-Guillamon, M.; Pruzin, J.; Sperling, R.; van Veluw, S.J. Cerebral amyloid angiopathy and Alzheimer disease—One peptide, two pathways. Nat. Rev. Neurol. 2020, 16, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Hsu, M.-J.; Hsu, C.Y.; Chen, B.-C.; Chen, M.-C.; Ou, G.; Lin, C.-H. Apoptosis signal-regulating kinase 1 in amyloid β peptide-induced cerebral endothelial cell apoptosis. J. Neurosci. 2007, 27, 5719–5729. [Google Scholar] [CrossRef]
- Parodi-Rullán, R.; Sone, J.Y.; Fossati, S. Endothelial Mitochondrial Dysfunction in Cerebral Amyloid Angiopathy and Alzheimer’s Disease. J. Alzheimer’s Dis. 2019, 72, 1019–1039. [Google Scholar] [CrossRef] [PubMed]
- Teng, T.; Ridgley, D.M.; Tsoy, A.; Sun, G.Y.; Askarova, S.; Lee, J.C. Azelnidipine Attenuates the Oxidative and NFκB Pathways in Amyloid-β-Stimulated Cerebral Endothelial Cells. ACS Chem. Neurosci. 2019, 10, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Salameh, T.S.; Shah, G.N.; Price, T.O.; Hayden, M.R.; Banks, W.A. Blood–Brain Barrier Disruption and Neurovascular Unit Dysfunction in Diabetic Mice: Protection with the Mitochondrial Carbonic Anhydrase Inhibitor Topiramate. J. Pharmacol. Exp. Ther. 2016, 359, 452–459. [Google Scholar] [CrossRef]
- Du, H.; Li, P.; Wang, J.; Qing, X.; Li, W. The interaction of amyloid β and the receptor for advanced glycation endproducts induces matrix metalloproteinase-2 expression in brain endothelial cells. Cell. Mol. Neurobiol. 2012, 32, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Yin, K.; Hsin, I.; Chen, S.; Fryer, J.D.; Holtzman, D.M.; Hsu, C.Y.; Xu, J. Matrix metalloproteinase–9 and spontaneous hemorrhage in an animal model of cerebral amyloid angiopathy. Ann. Neurol. 2003, 54, 379–382. [Google Scholar] [CrossRef]
- Vukic, V.; Callaghan, D.; Walker, D.; Lue, L.-F.; Liu, Q.Y.; Couraud, P.-O.; Romero, I.A.; Weksler, B.; Stanimirovic, D.B.; Zhang, W. Expression of inflammatory genes induced by beta-amyloid peptides in human brain endothelial cells and in Alzheimer’s brain is mediated by the JNK-AP1 signaling pathway. Neurobiol. Dis. 2009, 34, 95–106. [Google Scholar] [CrossRef]
- Arvanitakis, Z.; Leurgans, S.E.; Wang, Z.; Wilson, R.S.; Bennett, D.A.; Schneider, J.A. Cerebral amyloid angiopathy pathology and cognitive domains in older persons. Ann. Neurol. 2011, 69, 320–327. [Google Scholar] [CrossRef]
- Jellinger, K.A. Alzheimer disease and cerebrovascular pathology: An update. J. Neural Transm. 2002, 109, 813–836. [Google Scholar] [CrossRef]
- Ellis, R.J.; Olichney, J.M.; Thal, L.J.; Mirra, S.S.; Morris, J.C.; Beekly, D.; Heyman, A. Cerebral amyloid angiopathy in the brains of patients with Alzheimer’s disease: The CERAD experience, Part XV. Neurology 1996, 46, 1592–1596. [Google Scholar] [CrossRef] [PubMed]
- Reddy, S.T.; Savitz, S.I. Hypertension-Related Cerebral Microbleeds. Case Rep. Neurol. 2020, 12, 266–269. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wang, Z.; Zheng, A.; Yuan, R.; Shu, Y.; Zhang, S.; Lei, P.; Wu, B.; Liu, M. Blood pressure and outcomes in patients with different etiologies of intracerebral hemorrhage: A multicenter cohort study. J. Am. Heart Assoc. 2020, 9, e016766. [Google Scholar] [CrossRef] [PubMed]
- Stanisavljevic, A.; Schrader, J.M.; Zhu, X.; Mattar, J.M.; Hanks, A.; Xu, F.; Majchrzak, M.; Robinson, J.K.; Van Nostrand, W.E. Impact of non-pharmacological chronic hypertension on a transgenic rat model of cerebral amyloid angiopathy. Front. Neurosci. 2022, 16, 811371. [Google Scholar] [CrossRef] [PubMed]
- Jolink, W.M.; van Veluw, S.J.; Zwanenburg, J.J.; Rozemuller, A.J.; van Hecke, W.; Frosch, M.P.; Bacskai, B.J.; Rinkel, G.J.; Greenberg, S.M.; Klijn, C.J. Histopathology of cerebral microin-farcts and microbleeds in spontaneous intracerebral hemorrhage. Transl. Stroke Res. 2022, 14, 174–184. [Google Scholar] [CrossRef]
- Fazekas, F.; Kleinert, R.; Roob, G.; Kleinert, G.; Kapeller, P.; Schmidt, R.; Hartung, H.P. Histopathologic analysis of foci of signal loss on gradient-echo T2*-weighted MR images in patients with spontaneous intracerebral hemorrhage: Evidence of microangiopathy-related microbleeds. AJNR Am. J. Neuroradiol. 1999, 20, 637–642. [Google Scholar] [PubMed]
- Zhu, Y.; Liu, L.; Zhong, L.; Cheng, Y.; Zhang, S.; Wu, B.; Wang, D.; Xu, M. The association between hypertensive angiopathy and cerebral amyloid angiopathy in primary intracerebral hemorrhage. Front. Neurol. 2023, 14, 1257896. [Google Scholar] [CrossRef]
- Hayden, M.R. Type 2 Diabetes Mellitus Increases the Risk of Late-Onset Alzheimer’s Disease: Ultrastructural Remodeling of the Neurovascular Unit and Diabetic Gliopathy. Brain Sci. 2019, 9, 262. [Google Scholar] [CrossRef]
- Teng, Z.; Feng, J.; Liu, R.; Dong, Y.; Chen, H.; Xu, J.; Jiang, X.; Li, R.; Lv, P. Cerebral Small Vessel Disease is Associated with Mild Cognitive Impairment in Type 2 Diabetes Mellitus. Diabetes Metab. Syndr. Obes. Targets Ther. 2022, 15, 1985–1994. [Google Scholar] [CrossRef]
- Srikanth, V.; Sinclair, A.J.; Hill-Briggs, F.; Moran, C.; Biessels, G.J. Type 2 diabetes and cognitive dysfunction—Towards effective management of both comorbidities. Lancet Diabetes Endocrinol. 2020, 8, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Biessels, G.J.; Despa, F. Cognitive decline and dementia in diabetes mellitus: Mechanisms and clinical implications. Nat. Rev. Endocrinol. 2018, 14, 591–604. [Google Scholar] [CrossRef] [PubMed]
- Kullmann, S.; Kleinridders, A.; Small, D.M.; Fritsche, A.; Häring, H.-U.; Preissl, H.; Heni, M. Central nervous pathways of insulin action in the control of metabolism and food intake. Lancet Diabetes Endocrinol. 2020, 8, 524–534. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-C.; Lu, B.-Z.; Shu, Y.-C.; Sun, Y.-T. Spatiotemporal dynamics of cerebral vascular permeability in Type 2 diabetes-related cerebral microangiopathy. Front. Endocrinol. 2021, 12, 805637. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, O.K.L.; Backhouse, E.V.; Janssen, E.; Jochems, A.C.C.; Maher, C.; Ritakari, T.E.; Stevenson, A.J.; Xia, L.; Deary, I.J.; Wardlaw, J.M. Cognitive impairment in sporadic cerebral small vessel disease: A systematic review and meta-analysis. Alzheimer’s Dement. 2021, 17, 665–685. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Zhang, L.; Ding, G.; Davoodi-Bojd, E.; Li, Q.; Li, L.; Sadry, N.; Nedergaard, M.; Chopp, M.; Zhang, Z. Impairment of the glymphatic system after diabetes. J. Cereb. Blood Flow Metab. 2017, 37, 1326–1337. [Google Scholar] [CrossRef] [PubMed]
- Munis, O.B. Association of Type 2 Diabetes Mellitus With Perivascular Spaces and Cerebral Amyloid Angiopathy in Alzheimer’s Disease: Insights From MRI Imaging. Dement. Neurocogn. Disord. 2023, 22, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Cukierman, T.; Gerstein, H.C.; Williamson, J.D. Cognitive decline and dementia in diabetes—Systematic overview of prospective observational studies. Diabetologia 2005, 48, 2460–2469. [Google Scholar] [CrossRef] [PubMed]
- Mitaki, S.; Takayoshi, H.; Nakagawa, T.; Nagai, A.; Oguro, H.; Yamaguchi, S. Metabolic syndrome is associated with incidence of deep cerebral microbleeds. PLoS ONE 2018, 13, e0194182. [Google Scholar] [CrossRef]
- Diabetes Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Study Research Group; Jacobson, A.M.; Musen, G.; Ryan, C.M.; Silvers, N.; Cleary, P.; Waberski, B.; Burwood, A.; Weinger, K.; Bayless, M.; et al. Long-Term Effect of Diabetes and Its Treatment on Cognitive Function. N. Engl. J. Med. 2007, 356, 1842–1852. [Google Scholar] [CrossRef]
- van Sloten, T.T.; Sedaghat, S.; Carnethon, M.R.; Launer, L.J.; Stehouwer, C.D.A. Cerebral microvascular complications of type 2 diabetes: Stroke, cognitive dysfunction, and depression. Lancet Diabetes Endocrinol. 2020, 8, 325–336. [Google Scholar] [CrossRef] [PubMed]
- McCrimmon, R.J.; Ryan, C.M.; Frier, B.M. Diabetes and cognitive dysfunction. Lancet 2012, 379, 2291–2299. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.R.; Grant, D.G.; Aroor, A.R.; DeMarco, V.G. Ultrastructural Remodeling of the Neurovascular Unit in the Female Diabetic db/db Model—Part I: Astrocyte. Neuroglia 2018, 1, 220–244. [Google Scholar] [CrossRef]
- Hayden, M.R.; Grant, D.G.; Aroor, A.R.; DeMarco, V.G. Empagliflozin Ameliorates Type 2 Diabetes-Induced Ultrastructural Remodeling of the Neurovascular Unit and Neuroglia in the Female db/db Mouse. Brain Sci. 2019, 9, 57. [Google Scholar] [CrossRef] [PubMed]
- Han, B.H.; Zhou, M.-L.; Johnson, A.W.; Singh, I.; Liao, F.; Vellimana, A.K.; Nelson, J.W.; Milner, E.; Cirrito, J.R.; Basak, J.; et al. Contribution of reactive oxygen species to cerebral amyloid angiopathy, vasomotor dysfunction, and microhemorrhage in aged Tg2576 mice. Proc. Natl. Acad. Sci. USA 2015, 112, E881–E890. [Google Scholar] [CrossRef] [PubMed]
- Markesbery, W.R. Oxidative stress hypothesis in Alzheimer’s disease. Free Radic. Biol. Med. 1997, 23, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Vargas-Soria, M.; Ramos-Rodriguez, J.J.; del Marco, A.; Hierro-Bujalance, C.; Carranza-Naval, M.J.; Calvo-Rodriguez, M.; van Veluw, S.J.; Stitt, A.W.; Simó, R.; Bacskai, B.J.; et al. Accelerated amyloid angiopathy and related vascular alterations in a mixed murine model of Alzheimer’s disease and type two diabetes. Fluids Barriers CNS 2022, 19, 88. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.J.; Zhang, X.; Chen, W.W. Role of oxidative stress in Alzheimer’s disease. Biomed. Rep. 2016, 4, 519–522. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, C.; Moreira, P.I. Oxidative Stress: A Major Player in Cerebrovascular Alterations Associated to Neurodegenerative Events. Front. Physiol. 2018, 9, 806. [Google Scholar] [CrossRef]
- Jomova, K.; Vondrakova, D.; Lawson, M.; Valko, M. Metals, oxidative stress and neurodegenerative disorders. Mol. Cell. Biochem. 2010, 345, 91–104. [Google Scholar] [CrossRef]
- Beaman, C.; Kozii, K.; Hilal, S.; Liu, M.; Spagnolo-Allende, A.J.; Polanco-Serra, G.; Chen, C.; Cheng, C.-Y.; Zambrano, D.; Arikan, B.; et al. Cerebral Microbleeds, Cerebral Amyloid Angiopathy, and Their Relationships to Quantitative Markers of Neurodegeneration. Neurology 2022, 98, E1605–E1616. [Google Scholar] [CrossRef]
- Zhao, L.; Arbel-Ornath, M.; Wang, X.; Betensky, R.A.; Greenberg, S.M.; Frosch, M.P.; Bacskai, B.J. Matrix metalloproteinase 9–mediated intracerebral hemorrhage induced by cerebral amyloid angiopathy. Neurobiol. Aging 2015, 36, 2963–2971. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, G.A. Matrix metalloproteinases and their multiple roles in neurodegenerative diseases. Lancet Neurol. 2009, 8, 205–216. [Google Scholar] [CrossRef]
- Selim, M.; Diener, H.C. Atrial Fibrillation and Microbleeds. Stroke 2017, 48, 2660–2664. [Google Scholar] [CrossRef]
- Wolf, P.A.; Dawber, T.R.; Thomas, H.E., Jr.; Kannel, W.B. Epidemiologic assessment of chronic atrial fibrillation and risk of stroke: The fiamingham Study. Neurology 1978, 28, 973–977. [Google Scholar] [CrossRef]
- Bokura, H.; Saika, R.; Yamaguchi, T.; Nagai, A.; Oguro, H.; Kobayashi, S.; Yamaguchi, S. Microbleeds are associated with subsequent hemorrhagic and ischemic stroke in healthy elderly individuals. Stroke 2011, 42, 1867–1871. [Google Scholar] [CrossRef] [PubMed]
- Thijs, V.; Lemmens, R.; Schoofs, C.; Görner, A.; Van Damme, P.; Schrooten, M.; Demaerel, P. Microbleeds and the risk of recurrent stroke. Stroke 2010, 41, 2005–2009. [Google Scholar] [CrossRef]
- Hirata, Y.; Kato, N.; Muraga, K.; Shindo, A.; Nakamura, N.; Matsuura, K.; Ii, Y.; Shiga, M.; Tabei, K.-I.; Satoh, M.; et al. Cerebral Microbleeds With Atrial Fibrillation After Ablation Therapy. Front. Cell. Neurosci. 2022, 16, 818288. [Google Scholar] [CrossRef] [PubMed]
- Bunch, T.J. Atrial fibrillation and dementia. Circulation 2020, 142, 618–620. [Google Scholar] [CrossRef]
- Junejo, R.T.; Lip, G.Y.H.; Fisher, J.P. Cerebrovascular Dysfunction in Atrial Fibrillation. Front. Physiol. 2020, 11, 1066. [Google Scholar] [CrossRef]
- Soo, Y.; Zietz, A.; Yiu, B.; Mok, V.C.T.; Polymeris, A.A.; Seiffge, D.; Ambler, G.; Wilson, D.; Leung, T.W.H.; Tsang, S.F.; et al. Impact of Cerebral Microbleeds in Stroke Patients with Atrial Fibrillation. Ann. Neurol. 2023, 94, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Fisher, M. MRI screening for chronic anticoagulation in atrial fibrillation. Front. Neurol. 2013, 4, 137. [Google Scholar] [CrossRef]
- Lip, G.Y.H. Atrial fibrillation in 2011: Stroke prevention in AF. Nat. Rev. Cardiol. 2011, 9, 71–73. [Google Scholar] [CrossRef]
- De Meyer, S.F.; Stoll, G.; Wagner, D.D.; Kleinschnitz, C. von Willebrand factor: An emerging target in stroke therapy. Stroke 2012, 43, 599–606. [Google Scholar] [CrossRef]
- Stirbys, P. Review And Insights Into The Bleeding Mechanism Incited By Antithrombotic Therapy: Mechanistic Nuances Of Dual Pro-Hemorrhagic Substrate Incorporating Drug-Induced Microvascular Leakage. J. Atr. Fibrillation 2015, 8, 1254. [Google Scholar] [CrossRef] [PubMed]
- De Caterina, R.; Husted, S.; Wallentin, L.; Andreotti, F.; Arnesen, H.; Bachmann, F.; Baigent, C.; Huber, K.; Jespersen, J.; Kristensen, S.D.; et al. General mechanisms of coagulation and targets of anticoagulants (Section I). Position Paper of the ESC Working Group on Thrombosis—Task Force on Anticoagulants in Heart Disease. Thromb. Haemost. 2013, 109, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Mahley, R.W. Apolipoprotein E: Structure and Function in Lipid Metabolism, Neurobiology, and Alzheimer’s Diseases. Neurobiol. Dis. 2014, 72 Pt A, 3–12. [Google Scholar] [CrossRef]
- Ingala, S.; Mazzai, L.; Sudre, C.H.; Salvadó, G.; Brugulat-Serrat, A.; Wottschel, V.; Falcon, C.; Operto, G.; Tijms, B.; Gispert, J.D.; et al. The relation between APOE genotype and cerebral microbleeds in cognitively unimpaired middle- and old-aged individuals. Neurobiol. Aging 2020, 95, 104–114. [Google Scholar] [CrossRef]
- Li, J.; Shen, D.; Zhou, Y.; Jin, Y.; Jin, L.; Ye, X.; Tong, L.; Gao, F. Underlying microangiopathy and functional outcome of simultaneous multiple intracerebral hemorrhage. Front. Aging Neurosci. 2022, 14, 1000573. [Google Scholar] [CrossRef]
- Wang, S.; Lv, Y.; Zheng, X.; Qiu, J.; Chen, H.-S. The impact of cerebral microbleeds on intracerebral hemorrhage and poor functional outcome of acute ischemic stroke patients treated with intravenous thrombolysis: A systematic review and meta-analysis. J. Neurol. 2017, 264, 1309–1319. [Google Scholar] [CrossRef]
- Chacon-Portillo, M.A.; Llinas, R.H.; Marsh, E.B. Cerebral microbleeds shouldn’t dictate treatment of acute stroke: A retrospective cohort study evaluating risk of intracerebral hemorrhage. BMC Neurol. 2018, 18, 33. [Google Scholar] [CrossRef] [PubMed]
- Montagne, A.; Zhao, Z.; Zlokovic, B.V. Alzheimer’s disease: A matter of blood–brain barrier dysfunction? J. Exp. Med. 2017, 214, 3151–3169. [Google Scholar] [CrossRef] [PubMed]
- Poels, M.M.; Vernooij, M.W.; Ikram, M.A.; Hofman, A.; Krestin, G.P.; van der Lugt, A.; Breteler, M.M. Prevalence and risk factors of cerebral microbleeds: An update of the Rotterdam scan study. Stroke 2010, 41 (Suppl. S10), S103–S106. [Google Scholar] [CrossRef] [PubMed]
- Nahirney, P.C.; Reeson, P.; Brown, C.E. Ultrastructural analysis of blood–brain barrier breakdown in the peri-infarct zone in young adult and aged mice. J. Cereb. Blood Flow Metab. 2016, 36, 413–425. [Google Scholar] [CrossRef]
- Bai, T.; Yu, S.; Feng, J. Advances in the Role of Endothelial Cells in Cerebral Small Vessel Disease. Front. Neurol. 2022, 13, 861714. [Google Scholar] [CrossRef] [PubMed]
- Offenbacher, H.; Fazekas, F.; Schmidt, R.; Koch, M.; Fazekas, G.; Kapeller, P. MR of cerebral abnormalities concomitant with primary intrac-erebral hematomas. AJNR Am. J. Neuroradiol. 1996, 17, 573–578. [Google Scholar]
- Cordonnier, C.; Salman, R.A.-S.; Wardlaw, J. Spontaneous brain microbleeds: Systematic review, subgroup analyses and standards for study design and reporting. Brain 2007, 130, 1988–2003. [Google Scholar] [CrossRef]
- Shoamanesh, A.; Kwok, C.; Benavente, O. Cerebral microbleeds: Histopathological correlation of neuroimaging. Cerebrovasc. Dis. 2011, 32, 528–534. [Google Scholar] [CrossRef]
- Vernooij, M.W.; Ikram, M.A.; Tanghe, H.L.; Vincent, A.J.; Hofman, A.; Krestin, G.P.; Niessen, W.J.; Breteler, M.M.; van der Lugt, A. Incidental findings on brain MRI in the general population. New Engl. J. Med. 2007, 357, 1821–1828. [Google Scholar] [CrossRef]
- van Veluw, S.J.; Zwanenburg, J.J.; Engelen-Lee, J.; Spliet, W.G.; Hendrikse, J.; Luijten, P.R.; Biessels, G.J. In vivo detection of cerebral cortical microinfarcts with high-resolution 7T MRI. J. Cereb. Blood Flow Metab. 2013, 33, 322–329. [Google Scholar] [CrossRef]
- Ghaznawi, R.; Zwartbol, M.; de Bresser, J.; Kuijf, H.; Vincken, K.; Rissanen, I.; Geerlings, M.; Hendrikse, J.; UCC-SMART-Study Group. Microinfarcts in the Deep Gray Matter on 7T MRI: Risk Factors, MRI Correlates, and Relation to Cognitive Functioning—The SMART-MR Study. Am. J. Neuroradiol. 2022, 43, 829–836. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.E.; Greenberg, S.M. Clinical diagnosis of cerebral amyloid angiopathy: Validation of the Boston Criteria. Curr. Atheroscler. Rep. 2003, 5, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Kling, M.A.; Trojanowski, J.Q.; Wolk, D.A.; Lee, V.M.; Arnold, S.E. Vascular Disease and Dementias: Paradigm Shifts to Drive Research in New Directions. Alzheimer’s Dement. 2013, 9, 76–92. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A. The enigma of vascular cognitive disorder and vascular dementia. Acta Neuropathol. 2007, 113, 349–388. [Google Scholar] [CrossRef] [PubMed]
- Attems, J.; Jellinger, K.A. The overlap between vascular disease and Alzheimer’s disease—Lessons from pathology. BMC Med. 2014, 12, 206. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, B.V. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat. Rev. Neurosci. 2011, 12, 723–738. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Factor | Cerebral Amyloid Arteriopathy (CAA) | Hypertension (HTN) |
|---|---|---|
| Definition | CAA is a condition where primarily amyloid beta Aβ (1–40) protein builds up in the walls of arteries and arterioles, primarily to the media VSMCs, ECM, and adventitia in the CNS, increasing the risk of cerebral microbleeds (CMBs). | Hypertension refers to high blood pressure, which can lead to changes in small blood vessels (microvessels) in the brain, potentially causing microbleeds (CMBs). |
| Molecular | Cerebral amyloid beta (Aβ1–40) accumulation in microvessels resulting in loss of integrity to the vessel wall with extrusion of blood contents by rhexis (rupture) and/or diapedesis. | Peripheral and CNS RAAS activation, SNS, increased AngII and ALDO. Multifactorial genetic and environmental causes lead to dysfunction and damage to intimal BECs, media VSMCs with loss of integrity, and extrusion of luminal blood contents by rhexis (rupture) and/or diapedesis. |
| Location | Lobar (cortex, gray–white matter junction, subcortical white matter, and leptomeninges) location. | Deep (infratentorial, basal ganglia, lacunal, internal and external capsule, thalamus, and brainstem). |
| Mechanisms—Pathogenesis | Vascular rupture due to endothelial and VSMC remodeling with loss of vascular integrity and rupture due to Aβ (1–40) direct effect on VSMC and ECM. BECact/dys and BBBdd. | Vascular stiffness, increased pulse pressure, vascular hyalinosis, atherosclerosis, arteriolosclerosis, oxidative stress, and inflammation. BECact/dys and BBBdd. |
| Associated conditions | Often seen in conjunction with Alzheimer’s disease and older age ≥ 60. | Age of onset may vary but usually increases with aging and can lead to various cardiovascular conditions such as heart disease and stroke. |
| Risk factors | Aging, genetics (e.g., presence of ApoE-ε4 allele genotype), and history of Alzheimer’s disease | Obesity, sedentary lifestyle, smoking, excessive alcohol consumption, and family history of hypertension. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hayden, M.R. Cerebral Microbleeds Associate with Brain Endothelial Cell Activation-Dysfunction and Blood–Brain Barrier Dysfunction/Disruption with Increased Risk of Hemorrhagic and Ischemic Stroke. Biomedicines 2024, 12, 1463. https://doi.org/10.3390/biomedicines12071463
Hayden MR. Cerebral Microbleeds Associate with Brain Endothelial Cell Activation-Dysfunction and Blood–Brain Barrier Dysfunction/Disruption with Increased Risk of Hemorrhagic and Ischemic Stroke. Biomedicines. 2024; 12(7):1463. https://doi.org/10.3390/biomedicines12071463
Chicago/Turabian StyleHayden, Melvin R. 2024. "Cerebral Microbleeds Associate with Brain Endothelial Cell Activation-Dysfunction and Blood–Brain Barrier Dysfunction/Disruption with Increased Risk of Hemorrhagic and Ischemic Stroke" Biomedicines 12, no. 7: 1463. https://doi.org/10.3390/biomedicines12071463