
Rasagiline Exerts Neuroprotection towards Oxygen–Glucose-Deprivation/Reoxygenation-Induced GAPDH-Mediated Cell Death by Activating Akt/Nrf2 Signaling

 ,
, 

Abstract

1. Introduction
2. Materials and Methods
2.1. Materials
2.2. PC12 Cell Cultures
2.3. Ischemic Insult Protocol
2.4. Measurement of Mitochondrial Reactive Oxygen Species (ROS) Levels
2.5. Nuclear Protein Extraction
2.6. Western Blotting
2.7. Knockdown of Nrf 2 Gene Expression Using siRNA
2.8. RNA Extraction and RT-PCR
2.9. Immunofluorescence and Confocal Microscopy
2.10. Statistical Analysis
3. Results
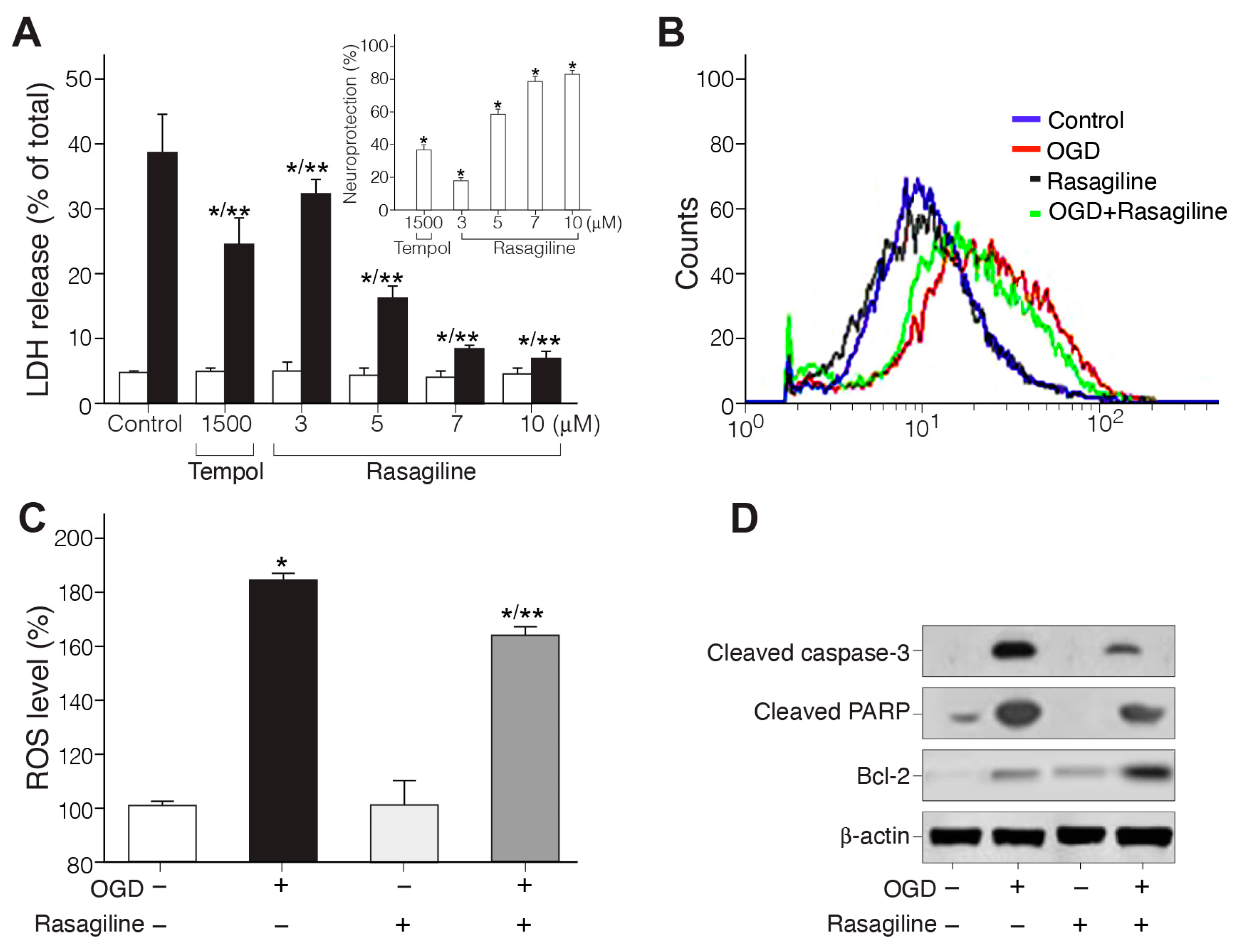
3.1. Rasagiline Conferred Neuroprotection towards Ischemic Insult
3.1.1. Rasagiline Inhibited Ischemic Insult-Induced Necrotic Cell Death
3.1.2. Rasagiline Inhibited Ischemic Insult-Induced ROS Production
3.1.3. Rasagiline Inhibited Ischemic Insult-Induced Apoptotic Cell Death
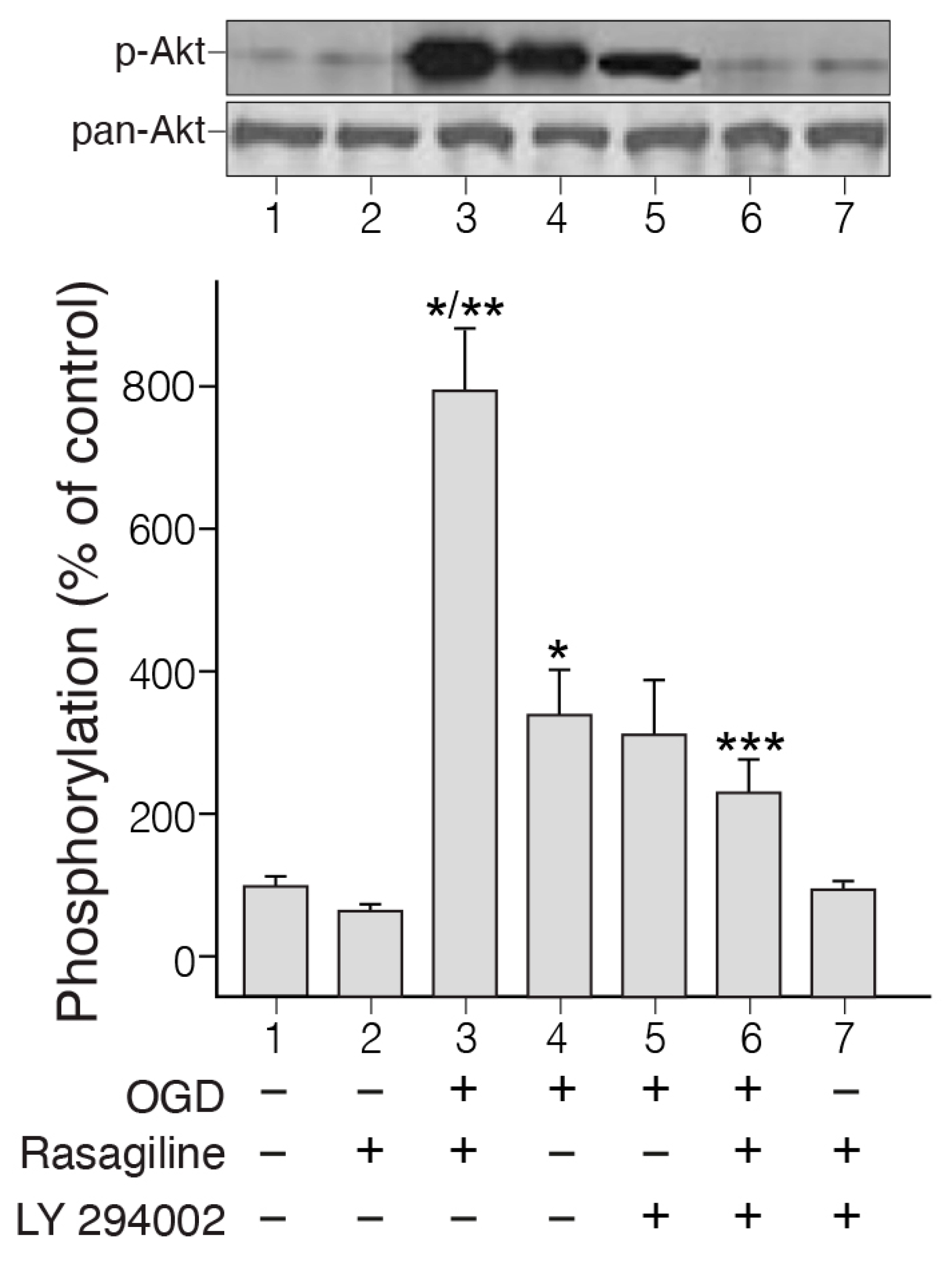
3.2. Rasagiline Potentiated PI3K/Akt Signaling in PC12 Cell Cultures Exposed to Ischemic Insult
3.3. Rasagiline Inhibited the Ischemic Insult-Induced GAPDH Nuclear Translocation
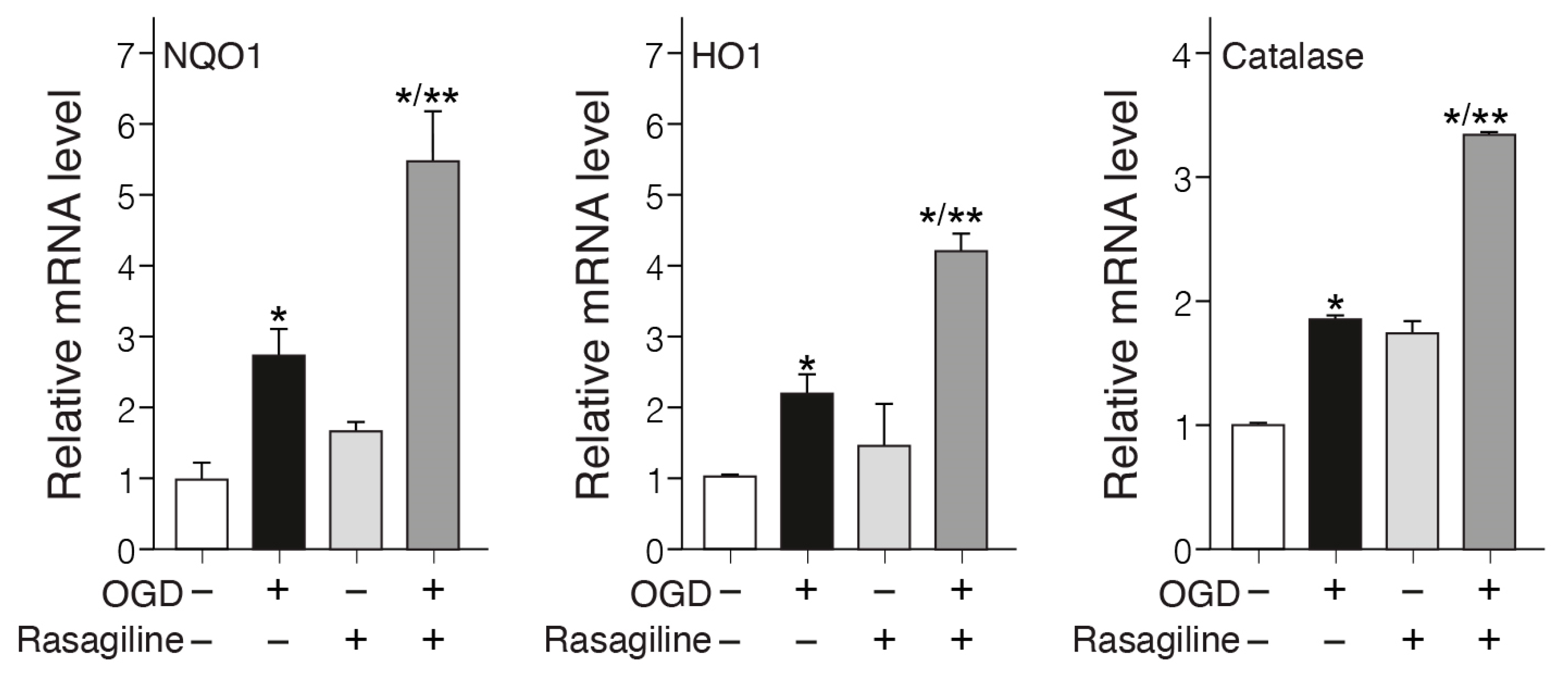
3.4. Rasagiline Increased Translocation of Nrf2 into the Nucleus and Transcription of ARE Phase II Antioxidant Enzyme Genes during the Ischemic Insult
3.5. Rasagiline Decreased the Expression Levels of α-Synuclein during Ischemic Insult
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kalia, L.V.; Lang, A.E. Parkinson’s Disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef] [PubMed]
- Masato, A.; Plotegher, N.; Boassa, D.; Bubacco, L. Impaired Dopamine Metabolism in Parkinson’s Disease Pathogenesis. Mol. Neurodegener. 2019, 14, 35. [Google Scholar] [CrossRef] [PubMed]
- Maetzler, W.; Liepelt, I.; Berg, D. Progression of Parkinson’s Disease in the Clinical Phase: Potential Markers. Lancet Neurol. 2009, 8, 1158–1171. [Google Scholar] [CrossRef] [PubMed]
- Pang, S.Y.Y.; Ho, P.W.L.; Liu, H.F.; Leung, C.T.; Li, L.; Chang, E.E.S.; Ramsden, D.B.; Ho, S.L. The Interplay of Aging, Genetics and Environmental Factors in the Pathogenesis of Parkinson’s Disease. Transl. Neurodegener. 2019, 8, 23. [Google Scholar] [CrossRef]
- Dionísio, P.A.; Amaral, J.D.; Rodrigues, C.M.P. Oxidative Stress and Regulated Cell Death in Parkinson’s Disease. Ageing Res. Rev. 2021, 67, 101263. [Google Scholar] [CrossRef]
- Jimenez-Moreno, N.; Lane, J.D. Autophagy and Redox Homeostasis in Parkinson’s: A Crucial Balancing Act. Oxid. Med. Cell. Longev. 2020, 2020, 8865611. [Google Scholar] [CrossRef]
- Trist, B.G.; Hare, D.J.; Double, K.L. Oxidative Stress in the Aging Substantia Nigra and the Etiology of Parkinson’s Disease. Aging Cell 2019, 18, e13031. [Google Scholar] [CrossRef]
- Smythies, J. Redox Aspects of Signaling by Catecholamines and Their Metabolites. Antioxid Redox Signal. 2000, 2, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.J.; Kim, S.W.; Lee, S.Y.; Hwang, O. Dopamine-Dependent Cytotoxicity of Tetrahydrobiopterin: A Possible Mechanism for Selective Neurodegeneration in Parkinson’s Disease. J. Neurochem. 2003, 86, 143–152. [Google Scholar] [CrossRef]
- Patel, M. Targeting Oxidative Stress in Central Nervous System Disorders. Trends Pharmacol. Sci. 2016, 37, 768–778. [Google Scholar] [CrossRef]
- Nicholls, C.; Li, H.; Liu, J.P. GAPDH: A Common Enzyme with Uncommon Functions. Clin. Exp. Pharmacol. Physiol. 2012, 39, 674–679. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Xiong, N.; Chen, C.; Xiong, J.; Jia, M.; Zhang, Z.; Cao, X.; Liang, Z.; Sun, S.; Lin, Z.; et al. Glyceraldehyde-3-Phosphate Dehydrogenase: Activity Inhibition and Protein Overexpression in Rotenone Models for Parkinson’s Disease. Neuroscience 2011, 192, 598–608. [Google Scholar] [CrossRef] [PubMed]
- Ou, X.M.; Lu, D.; Johnson, C.; Chen, K.; Youdim, M.B.H.; Rajkowska, G.; Shih, J.C. Glyceraldehyde-3-Phosphate Dehydrogenase-Monoamine Oxidase B-Mediated Cell Death-Induced by Ethanol Is Prevented by Rasagiline and 1-R-Aminoindan. Neurotox. Res. 2009, 16, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Goyal, A.; Agrawal, A.; Verma, A.; Dubey, N. The PI3K-AKT Pathway: A Plausible Therapeutic Target in Parkinson’s Disease. Exp. Mol. Pathol. 2022, 129, 104846. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.-Y. Neuroprotection Signaling of Nuclear Akt in Neuronal Cells. Exp. Neurobiol. 2014, 23, 200–206. [Google Scholar] [CrossRef]
- Huang, Q.; Lan, F.; Zheng, Z.; Xie, F.; Han, J.; Dong, L.; Xie, Y.; Zheng, F. Akt2 Kinase Suppresses Glyceraldehyde-3-Phosphate Dehydrogenase (GAPDH)-Mediated Apoptosis in Ovarian Cancer Cells via Phosphorylating Gapdh at Threonine 237 and Decreasing Its Nuclear Translocation. J. Biol. Chem. 2011, 286, 42211–42220. [Google Scholar] [CrossRef]
- Boas, S.M.; Joyce, K.L.; Cowell, R.M. The Nrf2-Dependent Transcriptional Regulation of Antioxidant Defense Pathways: Relevance for Cell Type-Specific Vulnerability to Neurodegeneration and Therapeutic Intervention. Antioxidants 2022, 11, 8. [Google Scholar] [CrossRef] [PubMed]
- Villavicencio Tejo, F.; Quintanilla, R.A. Contribution of the Nrf2 Pathway on Oxidative Damage and Mitochondrial Failure in Parkinson and Alzheimer’s Disease. Antioxidants 2021, 10, 1069. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.-R.; Han, J.-H.; Lee, H.-J.; Park, Y.K.; Kang, M. Emerging Functional Cross-Talk between the Keap1 Nrf2 System and Mitochondria. J. Clin. Biochem. Nutr. 2015, 56, 49–56. [Google Scholar] [CrossRef]
- Kumar, H.; Koppula, S.; Kim, I.S.; More, S.V.; Kim, B.W.; Choi, D.K. Nuclear Factor Erythroid 2-Related Factor 2 Signaling in Parkinson Disease: A Promising Multi Therapeutic Target Against Oxidative Stress, Neuroinflammation and Cell Death. CNS Neurol. Disord. Drug Targets. 2012, 11, 1015–1029. [Google Scholar] [CrossRef]
- Niu, Y.; Zhang, J.; Dong, M. Nrf2 as a Potential Target for Parkinson’s Disease Therapy. J. Mol. Med. 2021, 99, 917–931. [Google Scholar] [CrossRef]
- Farina, M.; Vieira, L.E.; Buttari, B.; Profumo, E.; Saso, L. The Nrf2 Pathway in Ischemic Stroke: A Review. Molecules 2021, 26, 5001. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.L.; Wang, Z.L.; Zhang, F.Y.; Liu, H.X.; Mao, L.H.; Yuan, L. Biomarkers of Parkinson’s Disease: From Basic Research to Clinical Practice. Aging Dis. 2024, 15, 1–18. [Google Scholar] [CrossRef]
- Maries, E.; Dass, B.; Collier, T.J.; Kordower, J.H.; Steece-Collier, K. The Role of α-Synuclein in Parkinson’s Disease: Insights from Animal Models. Nat. Rev. Neurosci. 2003, 4, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Mehta, S.L.; Kaimal, B.; Lyons, K.; Dempsey, R.J.; Vemuganti, R. Poststroke Induction of α-Synuclein Mediates Ischemic Brain Damage. J. Neurosci. 2016, 36, 7055–7065. [Google Scholar] [CrossRef]
- Sidhu, A.; Wersinger, C.; Moussa, C.E.; Vernier, P. The Role of Alpha-Synuclein in Both Neuroprotection and Neurodegeneration. Ann. NY Acad. Sci. 2004, 1035, 250–270. [Google Scholar] [CrossRef]
- Stefanis, L.; Kholodilov, N.; Rideout, H.J.; Burke, R.E.; Greene, L.A. Synuclein-1 Is Selectively up-Regulated in Response to Nerve Growth Factor Treatment in PC12 Cells. J. Neurochem. 2001, 76, 1165–1176. [Google Scholar] [CrossRef]
- Lecht, S.; Haroutiunian, S.; Hoffman, A.; Lazarovici, P. Rasagiline—A Novel MAO B Inhibitor in Parkinson’s Disease Therapy. Ther. Clin. Risk Manag. 2007, 3, 467–474. [Google Scholar]
- Weinreb, O.; Amit, T.; Riederer, P.; Youdim, M.B.H.; Mandel, S.A. Neuroprotective Profile of the Multitarget Drug Rasagiline in Parkinson’s Disease, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2011; Volume 100, ISBN 9780123864673. [Google Scholar]
- Tabakman, R.; Lecht, S.; Lazarovici, P. Neuroprotection by Monoamine Oxidase B Inhibitors: A Therapeutic Strategy for Parkinson’s Disease? BioEssays 2004, 26, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Chen, Y.; Shohami, E.; Weinstock, M. Neuroprotective Effect of Rasagiline, a Selective Monoamine Oxidase-B Inhibitor, against Closed Head Injury in the Mouse. Eur. J. Pharmacol. 1999, 366, 127–135. [Google Scholar] [CrossRef]
- Carrillo, M.C.; Kanai, S.; Nokubo, M.; Kitani, K. (-) Deprenyl Induces Activities of Both Superoxide Dismutase and Catalase but Not of Glutathione Peroxidase in the Striatum of Young Male Rats. Life Sci. 1991, 48, 517–521. [Google Scholar] [CrossRef] [PubMed]
- Tabakman, R.; Jiang, H.; Shahar, I.; Arien-Zakay, H.; Levine, R.A.; Lazarovici, P. Neuroprotection by NGF in the PC12 In Vitro OGD Model: Involvement of Mitogen-Activated Protein Kinases and Gene Expression. Ann. NY Acad. Sci. 2005, 1053, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Abu-Raya, S.; Tabakman, R.; Blaugrund, E.; Trembovler, V.; Lazarovici, P. Neuroprotective and Neurotoxic Effects of Monoamine Oxidase-B Inhibitors and Derived Metabolites under Ischemia in PC12 Cells. Eur. J. Pharmacol. 2002, 434, 109–116. [Google Scholar] [CrossRef]
- Lahiani, A.; Hidmi, A.; Katzhendler, J.; Yavin, E.; Lazarovici, P. Novel Synthetic PEGylated Conjugate of α-Lipoic Acid and Tempol Reduces Cell Death in a Neuronal PC12 Clonal Line Subjected to Ischemia. ACS Chem. Neurosci. 2016, 7, 1452–1462. [Google Scholar] [CrossRef] [PubMed]
- Maatuf, Y.; Priel, A.; Lazarovici, P. Measurements of Cell Death Induced by Snake and Spider’s Venoms and Derived Toxins. Methods Mol Biol. 2020, 2068, 239–268. [Google Scholar] [CrossRef] [PubMed]
- Gincberg, G.; Shohami, E.; Trembovler, V.; Alexandrovich, A.G.; Lazarovici, P.; Elchalal, U. Nerve Growth Factor Plays a Role in the Neurotherapeutic Effect of a CD45+ Pan-Hematopoietic Subpopulation Derived from Human Umbilical Cord Blood in a Traumatic Brain Injury Model. Cytotherapy 2018, 20, 245–261. [Google Scholar] [CrossRef] [PubMed]
- Ettinger, K.; Lecht, S.; Arien-Zakay, H.; Cohen, G.; Aga-Mizrachi, S.; Yanay, N.; Saragovi, H.U.; Nedev, H.; Marcinkiewicz, C.; Nevo, Y.; et al. Nerve Growth Factor Stimulation of ERK1/2 Phosphorylation Requires Both P75NTR and A9β1 Integrin and Confers Myoprotection towards Ischemia in C2C12 Skeletal Muscle Cell Model. Cell. Signal. 2012, 24, 2378–2388. [Google Scholar] [CrossRef] [PubMed]
- Bruni, F.; Polosa, P.L.; Gadaleta, M.N.; Cantatore, P.; Roberti, M. Nuclear Respiratory Factor 2 Induces the Expression of Many but Not All Human Proteins Acting in Mitochondrial DNA Transcription and Replication. J. Biol. Chem. 2010, 285, 3939–3948. [Google Scholar] [CrossRef] [PubMed]
- Shilo, D.; Cohen, G.; Blumenfeld, A.; Goren, K.; Hanhan, S.; Sharon, S.; Haze, A.; Deutsch, D.; Lazarovici, P. Tuftelin Is Required for NGF-Induced Differentiation of PC12 Cells. J. Mol. Neurosci. 2019, 68, 135–143. [Google Scholar] [CrossRef]
- Qi, H.; Han, Y.; Rong, J. Potential Roles of PI3K/Akt and Nrf2-Keap1 Pathways in Regulating Hormesis of Z-Ligustilide in PC12 Cells against Oxygen and Glucose Deprivation. Neuropharmacology 2012, 62, 1659–1670. [Google Scholar] [CrossRef]
- Sawa, A.; Khan, A.A.; Hester, L.D.; Snyder, S.H. Glyceraldehyde-3-Phosphate Dehydrogenase: Nuclear Translocation Participates in Neuronal and Nonneuronal Cell Death. Proc. Natl. Acad. Sci. USA 1997, 94, 11669–11674. [Google Scholar] [CrossRef] [PubMed]
- Zamanian, M.Y.; Parra, R.M.R.; Soltani, A.; Kujawska, M.; Mustafa, Y.F.; Raheem, G.; Al-Awsi, L.; Lafta, H.A.; Taheri, N.; Heidari, M.; et al. Targeting Nrf2 Signaling Pathway and Oxidative Stress by Resveratrol for Parkinson’s Disease: An Overview and Update on New Developments. Mol. Biol. Rep. 2023, 50, 5455–5464. [Google Scholar] [CrossRef] [PubMed]
- Tahavvori, A.; Gargari, M.K.; Yazdani, Y.; Mamalo, A.S.; Beilankouhi, E.A.V.; Valilo, M. Involvement of Antioxidant Enzymes in Parkinson’s Disease. Pathol. Res. Pract. 2023, 249, 154757. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, X.; Xiong, X.; Zhu, H.; Chen, R.; Zhang, S.; Chen, G.; Jian, Z. Nrf2 Regulates Oxidative Stress and Its Role in Cerebral Ischemic Stroke. Antioxidants 2022, 11, 2377. [Google Scholar] [CrossRef] [PubMed]
- Saberzadeh, J.; Arabsolghar, R.; Takhshid, M.A. Alpha Synuclein Protein Is Involved in Aluminum-Induced Cell Death and Oxidative Stress in PC12 Cells. Brain Res. 2016, 1635, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Regensburger, M.; Ip, C.W.; Kohl, Z.; Schrader, C.; Urban, P.P.; Kassubek, J.; Jost, W.H. Clinical Benefit of MAO-B and COMT Inhibition in Parkinson’s Disease: Practical Considerations. J. Neural Transm. 2023, 130, 847–861. [Google Scholar] [CrossRef] [PubMed]
- Moustafa, B.; Testai, F.D. Navigating Antiplatelet Treatment Options for Stroke: Evidence-Based and Pragmatic Strategies. Curr. Neurol. Neurosci. Rep. 2022, 22, 789–802. [Google Scholar] [CrossRef]
- McFarthing, K.; Buff, S.; Rafaloff, G.; Fiske, B.; Mursaleen, L.; Fuest, R.; Wyse, R.K.; Stott, S.R.W. Parkinson’s Disease Drug Therapies in the Clinical Trial Pipeline: 2023 Update. J. Parkinsons. Dis. 2023, 13, 427–439. [Google Scholar] [CrossRef]
- Safouris, A.; Magoufis, G.; Tsivgoulis, G. Emerging Agents for the Treatment and Prevention of Stroke: Progress in Clinical Trials. Expert Opin. Investig. Drugs. 2021, 30, 1025–1035. [Google Scholar] [CrossRef]
- Alborghetti, M.; Bianchini, E.; De Carolis, L.; Galli, S.; Pontieri, F.E.; Rinaldi, D. Type-B Monoamine Oxidase Inhibitors in Neurological Diseases. Neural Regen. Res. 2024, 19, 16–21. [Google Scholar] [CrossRef]
- Beghi, E.; Binder, H.; Birle, C.; Bornstein, N.; Diserens, K.; Groppa, S.; Homberg, V.; Lisnic, V.; Pugliatti, M.; Randall, G.; et al. European Academy of Neurology and European Federation of Neurorehabilitation Societies Guideline on Pharmacological Support in Early Motor Rehabilitation after Acute Ischaemic Stroke. Eur. J. Neurol. 2021, 28, 2831–2845. [Google Scholar] [CrossRef] [PubMed]
- Lahiani, A.; Brand-Yavin, A.; Yavin, E.; Lazarovici, P. Neuroprotective Effects of Bioactive Compounds and Mapk Pathway Modulation in “Ischemia”—Stressed PC12 Pheochromocytoma Cells. Brain Sci. 2018, 8, 32. [Google Scholar] [CrossRef] [PubMed]
- Peng, T.; Jiang, Y.; Farhan, M.; Lazarovici, P.; Chen, L.; Zheng, W. Anti-Inflammatory Effects of Traditional Chinese Medicines on Preclinical in Vivo Models of Brain Ischemia-Reperfusion-Injury: Prospects for Neuroprotective Drug Discovery and Therapy. Front. Pharmacol. 2019, 10, 204. [Google Scholar] [CrossRef] [PubMed]
- Aluf, Y.; Vaya, J.; Khatib, S.; Loboda, Y.; Finberg, J.P.M. Selective Inhibition of Monoamine Oxidase A or B Reduces Striatal Oxidative Stress in Rats with Partial Depletion of the Nigro-Striatal Dopaminergic Pathway. Neuropharmacology 2013, 65, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Seif-El-Nasr, M.; Atia, A.S.; Abdelsalam, R.M. Effect of MAO-B Inhibition Against Ischemia-Induced Oxidative Stress in the Rat Nrain. Comparison with a Rational Antioxidant. Arzneimittelforschung 2008, 58, 160–167. [Google Scholar] [CrossRef]
- Naoi, M.; Maruyama, W.; Shamoto-Nagai, M. Neuroprotective Function of Rasagiline and Selegiline, Inhibitors of Type B Monoamine Oxidase, and Role of Monoamine Oxidases in Synucleinopathies. Int. J. Mol. Sci. 2022, 23, 1059. [Google Scholar] [CrossRef]
- Zhai, D.; Chin, K.; Wang, M.; Liu, F. Disruption of the Nuclear P53-GAPDH Complex Protects against Ischemia-Induced Neuronal Damage. Mol. Brain 2014, 7, 20. [Google Scholar] [CrossRef]
- Maruyama, W.; Akao, Y.; Youdim, M.B.; Davis, B.A.; Naoi, M. Transfection-enforced Bcl-2 Overexpression and an Anti-Parkinson Drug, Rasagiline, Prevent Nuclear Accumulation of Glyceraldehyde-3-phosphate Dehydrogenase Induced by an Endogenous Dopaminergic Neurotoxin, N-methyl(R)salsolinol. J Neurochem. 2001, 78, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Hara, M.R.; Thomas, B.; Cascio, M.B.; Bae, B.I.; Hester, L.D.; Dawson, V.L.; Dawson, T.M.; Sawa, A.; Snyder, S.H. Neuroprotection by Pharmacologic Blockade of the GAPDH Death Cascade. Proc. Natl. Acad. Sci. USA 2006, 103, 3887–3889. [Google Scholar] [CrossRef]
- Tatton, W.; Chalmers-Redman, R.; Tatton, N. Neuroprotection by Deprenyl and Other Propargylamines: Glyceraldehyde-3-Phosphate Dehydrogenase Rather than Monoamine Oxidase B. J. Neural Transm. 2003, 110, 509–515. [Google Scholar] [CrossRef]
- Sharma, S.K.; Carlson, E.C.; Ebadi, M. Neuroprotective Actions of Selegiline in Inhibiting 1-Methyl, 4-Phenyl, Pyridinium Ion (MPP+)-Induced Apoptosis in SK-N-SH Neurons. J. Neurocytol. 2003, 32, 329–343. [Google Scholar] [CrossRef] [PubMed]
- Czerniczyniec, A.; Bustamante, J.; Lores-Arnaiz, S. Modulation of Brain Mitochondrial Function by Deprenyl. Neurochem. Int. 2006, 48, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Irer, S.V.; Alper, G.E.; Sezer, E.D.; Duman, E.; Saatcioglu, F.; Yilmaz, C. The Effect of L-Deprenyl on Tissue mRNA Expressions of NOS Isoforms and NO Levels in an Experimental Diabetes Mellitus Model. J. Neural Transm. 2007, 114, 811–815. [Google Scholar] [CrossRef]
- Nakaso, K.; Nakamura, C.; Sato, H.; Imamura, K.; Takeshima, T.; Nakashima, K. Novel Cytoprotective Mechanism of Anti-Parkinsonian Drug Deprenyl: PI3K and Nrf2-Derived Induction of Antioxidative Proteins. Biochem. Biophys. Res. Commun. 2006, 339, 915–922. [Google Scholar] [CrossRef]
- Andoh, T.; Boon Chock, P.; Murphy, D.L.; Chiueh, C.C. Role of the Redox Protein Thioredoxin in Cytoprotective Mechanism Evoked by (-)-Deprenyl. Mol. Pharmacol. 2005, 68, 1408–1414. [Google Scholar] [CrossRef]
- Cui, Y.; Liu, K.W.K.; Liang, Y.; Ip, M.S.M.; Mak, J.C.W. Inhibition of Monoamine Oxidase-B by Selegiline Reduces Cigarette Smoke-Induced Oxidative Stress and Inflammation in Airway Epithelial Cells. Toxicol. Lett. 2017, 268, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Liu, N.; Liu, X.X.; Yang, Y.Y.; Zhou, M.W. α-Synuclein in Traumatic and Vascular Diseases of the Central Nervous System. Aging 2020, 12, 22313–22334. [Google Scholar] [CrossRef]
- Speiser, Z.; Mayk, A.; Litinetsky, L.; Fine, T.; Nyska, A.; Blaugrund, E.; Cohen, S. Rasagiline is neuroprotective in an experimental model of brain ischemia in the rat. J. Neural Transm. 2007, 114, 595–605. [Google Scholar] [CrossRef]
- Chang, H.Y.; Li, Y.Y.; Hong, C.T.; Kuan, Y.C. Efficacy of rasagiline monotherapy for early Parkinson disease: A systematic review and meta-analysis of randomized controlled trials. J Psychopharmacol. 2022, 36, 704–714. [Google Scholar] [CrossRef]
- Hauser, R.A.; Li, R.; Perez, A.; Ren, X.; Weintraub, D.; Elm, J.; Goudreau, J.L.; Morgan, J.C.; Fang, J.Y.; Aminoff, M.J.; et al. Longer duration of MAO-B inhibitor exposure is associated with less clinical decline in Parkinson’s disease: An Analysis of NET-PD LS1. J Parkinsons Dis. 2017, 7, 117–127. [Google Scholar] [CrossRef]
- Sivenius, J.; Sarasoja, T.; Aaltonen, H.; Heinonen, E.; Kilkku, O.; Reinikainen, K. Selegiline treatment facilitates recovery after stroke. Neurorehabil Neural Repair. 2001, 15, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Gouda, N.A.; Cho, J. Omarigliptin mitigates 6-hydroxydopamine- or rotenone-induced oxidative toxicity in PC12 cells by antioxidant, anti-inflammatory, and anti-apoptotic actions. Antioxidants 2022, 11, 1940. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | LDH Release (% of Total) | Neuroprotection (% of Rasagiline) | |
|---|---|---|---|
| OGD | OGD + Rasagiline | ||
| Control 1 | 38 ± 9 | 10 ± 3 | 74 ± 7 |
| LY294002 1 | 42 ± 7 | 22 ± 6 1,* | 48 ± 9 |
| Scramble RNA 2 | 47 ± 5 | 15 ± 5 2 | 68 ± 6 |
| Nrf2 siRNA 2 | 36 ± 10 | 28 ± 6 2,* | 20 ± 4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lecht, S.; Lahiani, A.; Klazas, M.; Naamneh, M.S.; Rubin, L.; Dong, J.; Zheng, W.; Lazarovici, P. Rasagiline Exerts Neuroprotection towards Oxygen–Glucose-Deprivation/Reoxygenation-Induced GAPDH-Mediated Cell Death by Activating Akt/Nrf2 Signaling. Biomedicines 2024, 12, 1592. https://doi.org/10.3390/biomedicines12071592
Lecht S, Lahiani A, Klazas M, Naamneh MS, Rubin L, Dong J, Zheng W, Lazarovici P. Rasagiline Exerts Neuroprotection towards Oxygen–Glucose-Deprivation/Reoxygenation-Induced GAPDH-Mediated Cell Death by Activating Akt/Nrf2 Signaling. Biomedicines. 2024; 12(7):1592. https://doi.org/10.3390/biomedicines12071592
Chicago/Turabian StyleLecht, Shimon, Adi Lahiani, Michal Klazas, Majdi Saleem Naamneh, Limor Rubin, Jiayi Dong, Wenhua Zheng, and Philip Lazarovici. 2024. "Rasagiline Exerts Neuroprotection towards Oxygen–Glucose-Deprivation/Reoxygenation-Induced GAPDH-Mediated Cell Death by Activating Akt/Nrf2 Signaling" Biomedicines 12, no. 7: 1592. https://doi.org/10.3390/biomedicines12071592
APA StyleLecht, S., Lahiani, A., Klazas, M., Naamneh, M. S., Rubin, L., Dong, J., Zheng, W., & Lazarovici, P. (2024). Rasagiline Exerts Neuroprotection towards Oxygen–Glucose-Deprivation/Reoxygenation-Induced GAPDH-Mediated Cell Death by Activating Akt/Nrf2 Signaling. Biomedicines, 12(7), 1592. https://doi.org/10.3390/biomedicines12071592