
Treating Cardiovascular Disease in the Inflammatory Setting of Rheumatoid Arthritis: An Ongoing Challenge
 ,
, 
Abstract
:1. Introduction
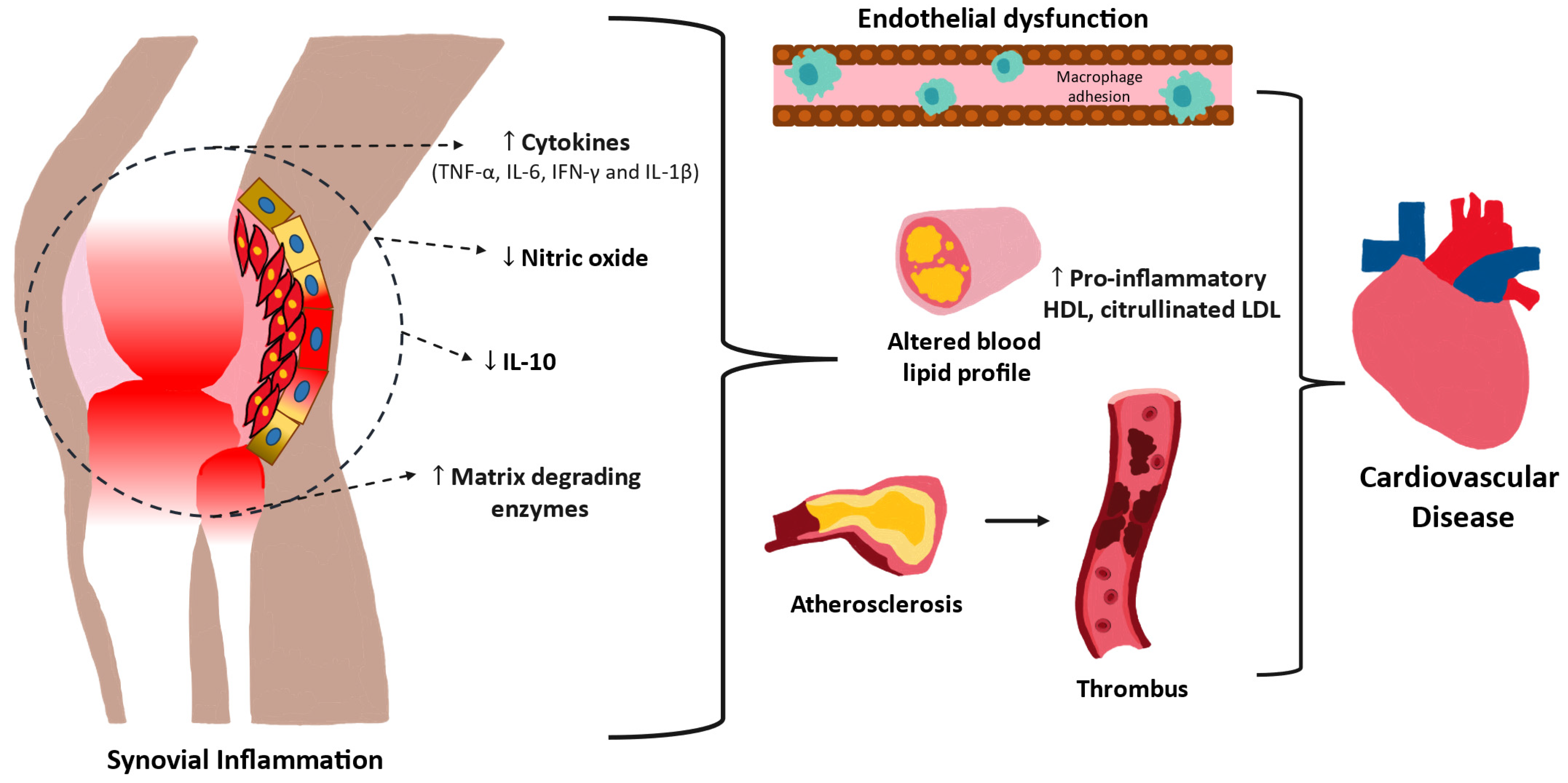
2. Pathophysiology of Cardiovascular Diseases
3. Mechanisms and Role of Inflammation in Cardiovascular Disease (CVD)
4. Current Treatment Options
4.1. Methotrexate
4.2. Non-Steroidal Anti-Inflammatory Drugs (NSAIDs)
4.3. Leflunomide
4.4. Sulfasalazine
4.5. Hydroxychloroquine
4.6. Glucocorticoids
4.7. Janus Kinase Inhibitors
4.8. Abatacept
4.9. TNF-α Inhibitors
4.10. Tocilizumab
4.11. Rituximab
4.12. Statins

{kind=link}
{kind=link}
| Medication Name or Category | Net Effect on Cardiovascular Risk | Putative Mechanism | References |
|---|---|---|---|
| Methotrexate | Beneficial | Increased adenosine via inhibition of adenosine deamination | [74,75,76,77,78,79,80,81] |
| NSAIDs | Harmful | Inhibition of cyclooxygenase enzymes, interferes with reverse cholesterol transport | [83,84,85,86,87,88] |
| Leflunomide | Neutral | Inhibition of dihydroorotate dehydrogenase | [89,90,91,92] |
| Sulfasalazine | Likely Beneficial | Decreased platelet aggregation, inhibition of NF-κB, improved endothelial function | [74,94,95,96,97,98,99,100] |
| Hydroxychloroquine | Beneficial | Overall reduction in inflammation, possibly anti-thrombotic | [101,102,103,104,105,106,107,108,109] |
| Glucocorticoids | Harmful | Increased oxidative stress and decreased nitric oxide. Increased adiposity and insulin resistance. | [85,110,111,112,113,114,115,116] |
| JAK inhibitors | Potential for harm in certain patients | Suppress the JAK-STAT signaling pathway | [117,118,119,120,121,122,123,124,125,126,127,128,129,130] |
| Abatacept | Neutral | CD80/CD86-CD28 costimulatory modulator | [132,133,134,135,136,137,138] |
| TNF-α inhibitors | Likely beneficial | Inhibition of TNFR1, amelioration of endothelial dysfunction | [139,140,141,142,143,144,145,146,147] |
| Tocilizumab | Neutral | Anti-IL-6 receptor antibody, may increase cholesterol levels | [148,149,150,151,152,153] |
| Rituximab | Neutral | Anti-CD20 | [154,155,156,157,158] |
| Statins | Beneficial | Lower LDL cholesterol, improve lipid profile, anti-inflammatory | [159,160,161,162,163,164,165,166] |
5. Clinical Context
6. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cai, Y.; Zhang, J.; Liang, J.; Xiao, M.; Zhang, G.; Jing, Z.; Lv, L.; Nan, K.; Dang, X. The Burden of Rheumatoid Arthritis: Findings from the 2019 Global Burden of Diseases Study and Forecasts for 2030 by Bayesian Age-Period-Cohort Analysis. J. Clin. Med. 2023, 12, 1291. [Google Scholar] [CrossRef]
- Nie, Q.; Luo, Q.; Yan, W.; Zhang, T.; Wang, H.; Wu, J. Rheumatoid arthritis and coronary atherosclerosis: A two-sample Mendelian randomization study. Front. Cardiovasc. Med. 2023, 10, 1033644. [Google Scholar] [CrossRef]
- Turesson, C. Comorbidity in rheumatoid arthritis. Swiss Med. Wkly. 2016, 146, 14290. [Google Scholar] [CrossRef]
- van der Woude, D.; van der Helm-van Mil, A.H.M. Update on the epidemiology, risk factors, and disease outcomes of rheumatoid arthritis. Best Pract. Res. Clin. Rheumatol. 2018, 32, 174–187. [Google Scholar] [CrossRef]
- Lin, Y.J.; Anzaghe, M.; Schülke, S. Update on the Pathomechanism, Diagnosis, and Treatment Options for Rheumatoid Arthritis. Cells 2020, 9, 880. [Google Scholar] [CrossRef]
- Jahid, M.; Khan, K.U.; Rehan-Ul-Haq; Ahmed, R.S. Overview of Rheumatoid Arthritis and Scientific Understanding of the Disease. Mediterr. J. Rheumatol. 2023, 34, 284–291. [Google Scholar] [CrossRef]
- Finckh, A.; Gilbert, B.; Hodkinson, B.; Bae, S.C.; Thomas, R.; Deane, K.D.; Alpizar-Rodriguez, D.; Lauper, K. Global epidemiology of rheumatoid arthritis. Nat. Rev. Rheumatol. 2022, 18, 591–602. [Google Scholar] [CrossRef]
- Yu, F.; Chen, H.; Li, Q.; Tao, M.; Jin, Z.; Geng, L.; Sun, L. Secular trend of mortality and incidence of rheumatoid arthritis in global, 1990-2019: An age period cohort analysis and joinpoint analysis. BMC Pulm. Med. 2023, 23, 356. [Google Scholar] [CrossRef]
- Ali, A.; Ali, A.; Kumar, D.; Kumar, R.; Elahi, K.; Suman, F.; Anjum, Z.; Tharwani, P.; Jahangir, M.; Rizwan, A. Comparison of Incidence of Myocardial Infarction in Patients with Rheumatoid Arthritis and Diabetes Mellitus. Cureus 2021, 13, e15716. [Google Scholar] [CrossRef]
- Taylor, P.C.; Atzeni, F.; Balsa, A.; Gossec, L.; Müller-Ladner, U.; Pope, J. The Key Comorbidities in Patients with Rheumatoid Arthritis: A Narrative Review. J. Clin. Med. 2021, 10, 509. [Google Scholar] [CrossRef]
- Guo, Y.; Chung, W.; Shan, Z.; Zhu, Z.; Costenbader, K.H.; Liang, L. Genome-Wide Assessment of Shared Genetic Architecture Between Rheumatoid Arthritis and Cardiovascular Diseases. J. Am. Heart Assoc. 2023, 12, e030211. [Google Scholar] [CrossRef]
- Widdifield, J.; Paterson, J.M.; Huang, A.; Bernatsky, S. Causes of death in rheumatoid arthritis: How do they compare to the general population? Arthritis Care Res. 2018, 70, 1748–1755. [Google Scholar] [CrossRef]
- Arora, A.; Ingle, V.; Joshi, R.; Malik, R.; Khandelwal, G. Exploring the Subclinical Atherosclerotic Load in Patients With Rheumatoid Arthritis: A Cross-Sectional Study. Cureus 2022, 14, e32644. [Google Scholar] [CrossRef]
- Bernardes, M.; Vieira, T.S.; Martins, M.J.; Lucas, R.; Costa, L.; Pereira, J.G.; Ventura, F.; Martins, E. Myocardial Perfusion in Rheumatoid Arthritis Patients: Associations with Traditional Risk Factors and Novel Biomarkers. BioMed Res Int. 2017, 2017, 6509754. [Google Scholar] [CrossRef]
- Rouached, L.; Tekaya, R.; Ben Ayed, H.; Bouden, S.; Ben Tekaya, A.; Ben Ahmed, H.; Mahmoud, I.; Saidane, O.; Abdelmoula, L. Screening of Silent Myocardial Ischaemia Using a Stress Test in Rheumatoid Arthritis Patients: Association with Traditional Risk Factors and Disease Activity. Musculoskelet. Care 2023, 21, 997–1004. [Google Scholar] [CrossRef]
- Arts, E.E.; Popa, C.; Den Broeder, A.A.; Semb, A.G.; Toms, T.; Kitas, G.D.; van Riel, P.L.; Fransen, J. Performance of four current risk algorithms in predicting cardiovascular events in patients with early rheumatoid arthritis. Ann. Rheum. Dis. 2015, 74, 668–674. [Google Scholar] [CrossRef]
- Chodara, A.M.; Wattiaux, A.; Bartels, C.M. Managing Cardiovascular Disease Risk in Rheumatoid Arthritis: Clinical Updates and Three Strategic Approaches. Curr. Rheumatol. Rep. 2017, 19, 16. [Google Scholar] [CrossRef]
- Nikiphorou, E.; de Lusignan, S.; Mallen, C.D.; Khavandi, K.; Bedarida, G.; Buckley, C.D.; Galloway, J.; Raza, K. Cardiovascular risk factors and outcomes in early rheumatoid arthritis: A population-based study. Heart 2020, 106, 1566–1572. [Google Scholar] [CrossRef]
- Dessein, P.H.; Gonzalez-Gay, M.A. Management of Cardiovascular Disease Risk in Rheumatoid Arthritis. J. Clin. Med. 2022, 11, 3487. [Google Scholar] [CrossRef]
- Crowson, C.S.; Matteson, E.L.; Roger, V.L.; Therneau, T.M.; Gabriel, S.E. Usefulness of risk scores to estimate the risk of cardiovascular disease in patients with rheumatoid arthritis. Am. J. Cardiol. 2012, 110, 420–424. [Google Scholar] [CrossRef]
- Södergren, A.; Karp, K.; Bengtsson, C.; Möller, B.; Rantapää-Dahlqvist, S.; Wållberg-Jonsson, S. Biomarkers associated with cardiovascular disease in patients with early rheumatoid arthritis. PLoS ONE 2019, 14, e0220531. [Google Scholar] [CrossRef]
- Frąk, W.; Wojtasińska, A.; Lisińska, W.; Młynarska, E.; Franczyk, B.; Rysz, J. Pathophysiology of Cardiovascular Diseases: New Insights into Molecular Mechanisms of Atherosclerosis, Arterial Hypertension, and Coronary Artery Disease. Biomedicines 2022, 10, 1938. [Google Scholar] [CrossRef]
- Scott, J. Pathophysiology and biochemistry of cardiovascular disease. Curr. Opin. Genet. Dev. 2004, 14, 271–279. [Google Scholar] [CrossRef]
- Ciumărnean, L.; Milaciu, M.V.; Runcan, O.; Vesa, S.C.; Răchișan, A.L.; Negrean, V.; Perné, M.; Donca, V.I.; Alexescu, T.; Para, I.; et al. The Effects of Flavonoids in Cardiovascular Diseases. Molecules 2020, 25, 4320. [Google Scholar] [CrossRef]
- Baba, M.; Maris, M.; Jianu, D.; Luca, C.T.; Stoian, D.; Mozos, I. The Impact of the Blood Lipids Levels on Arterial Stiffness. J. Cardiovasc. Dev. Dis. 2023, 10, 127. [Google Scholar] [CrossRef]
- Jebari-Benslaiman, S.; Galicia-García, U.; Larrea-Sebal, A.; Olaetxea, J.R.; Alloza, I.; Vandenbroeck, K.; Benito-Vicente, A.; Martin, C. Pathophysiology of Atherosclerosis. Int. J. Mol. Sci. 2022, 3, 3346. [Google Scholar] [CrossRef]
- Mundi, S.; Massaro, M.; Scoditti, E.; Carluccio, M.A.; van Hinsbergh, V.W.M.; Iruela-Arispe, M.L.; De Caterina, R. Endothelial permeability, LDL deposition, and cardiovascular risk factors—A review. Cardiovasc. Res. 2018, 114, 35–52. [Google Scholar] [CrossRef]
- Bergheanu, S.C.; Bodde, M.C.; Jukema, J.W. Pathophysiology and treatment of atherosclerosis: Current view and future perspective on lipoprotein modification treatment. Neth. Heart J. 2017, 25, 231–242. [Google Scholar] [CrossRef]
- Kowara, M.; Cudnoch-Jedrzejewska, A. Pathophysiology of Atherosclerotic Plaque Development–Contemporary Experience and New Directions in Research. Int. J. Mol. Sci. 2021, 22, 3513. [Google Scholar] [CrossRef]
- Mahdinia, E.; Shokri, N.; Taheri, A.T.; Asgharzadeh, S.; Elahimanesh, M.; Najafi, M. Cellular crosstalk in atherosclerotic plaque microenvironment. Cell Commun. Signal. 2023, 21, 125. [Google Scholar] [CrossRef]
- Allahverdian, S.; Chehroudi, A.C.; McManus, B.M.; Abraham, T.; Francis, G.A. Contribution of Intimal Smooth Muscle Cells to Cholesterol Accumulation and Macrophage-Like Cells in Human Atherosclerosis. Circulation 2014, 129, 1551–1559. [Google Scholar] [CrossRef]
- Sakakura, K.; Nakano, M.; Otsuka, F.; Ladich, E.; Kolodgie, F.D.; Virmani, R. Pathophysiology of atherosclerosis plaque progression. Heart Lung Circ. 2013, 22, 399–411. [Google Scholar] [CrossRef]
- Mushenkova, N.V.; Bezsonov, E.E.; Orekhova, V.A.; Popkova, T.V.; Starodubova, A.V.; Orekhov, A.N. Recognition of Oxidized Lipids by Macrophages and Its Role in Atherosclerosis Development. Biomedicines 2021, 9, 915. [Google Scholar] [CrossRef]
- Stary, H.C.; Chandler, A.B.; Glagov, S.; Guyton, J.R.; Insull, W.; Rosenfeld, M.E.; Schaffer, S.A.; Schwartz, C.J.; Wagner, W.D.; Wissler, R.W. A definition of initial, fatty streak, and intermediate lesions of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation 1994, 89, 2462–2478. [Google Scholar] [CrossRef]
- Reiss, A.B.; Cronstein, B.N. Regulation of foam cells by adenosine. Arterioscler. Thromb. Vasc. Biol. 2012, 3, 879–886. [Google Scholar] [CrossRef]
- Cao, G.; Xuan, X.; Hu, J.; Zhang, R.; Jin, H.; Dong, H. How vascular smooth muscle cell phenotype switching contributes to vascular disease. Cell Commun. Signal. 2022, 20, 180. [Google Scholar] [CrossRef]
- Scott, D.L.; Wolfe, F.; Huizinga, T.W. Rheumatoid arthritis. Lancet 2010, 376, 1094–1108. [Google Scholar] [CrossRef]
- Book, C.; Saxne, T.; Jacobsson, L.T. Prediction of mortality in rheumatoid arthritis based on disease activity markers. J. Rheumatol. 2005, 32, 430–434. [Google Scholar]
- Kramer, H.R.; Giles, J.T. Cardiovascular disease risk in rheumatoid arthritis: Progress, debate, and opportunity. Arthritis Care Res. 2011, 63, 484–499. [Google Scholar] [CrossRef]
- Bathon, J.M.; Centola, M.; Liu, X.; Jin, Z.; Ji, W.; Knowlton, N.S.; Ferraz-Amaro, I.; Fu, Q.; Giles, J.T.; Wasko, M.C.; et al. Identification of novel biomarkers for the prediction of subclinical coronary artery atherosclerosis in patients with rheumatoid arthritis: An exploratory analysis. Arthritis Res. Ther. 2023, 25, 213. [Google Scholar] [CrossRef]
- Bamias, G.; Stamatelopoulos, K.; Zampeli, E.; Protogerou, A.; Sigala, F.; Papamichael, C.; Christopoulos, P.; Kitas, G.D.; Sfikakis, P.P. Circulating levels of TNF-like cytokine 1A correlate with the progression of atheromatous lesions in patients with rheumatoid arthritis. Clin. Immunol. 2013, 147, 144–150. [Google Scholar] [CrossRef]
- Arida, A.; Protogerou, A.D.; Kitas, G.D.; Sfikakis, P.P. Systemic Inflammatory Response and Atherosclerosis: The Paradigm of Chronic Inflammatory Rheumatic Diseases. Int. J. Mol. Sci. 2018, 19, 1890. [Google Scholar] [CrossRef]
- Markin, A.M.; Markina, Y.V.; Bogatyreva, A.I.; Tolstik, T.V.; Chakal, D.A.; Breshenkov, D.G.; Charchyan, E.R. The Role of Cytokines in Cholesterol Accumulation in Cells and Atherosclerosis Progression. Int. J. Mol. Sci. 2023, 24, 6426. [Google Scholar] [CrossRef]
- Cavagna, L.; Boffini, N.; Cagnotto, G.; Inverardi, F.; Grosso, V.; Caporali, R. Atherosclerosis and rheumatoid arthritis: More than a simple association. Mediat. Inflamm. 2012, 2012, 147354. [Google Scholar] [CrossRef]
- Popescu, D.; Rezus, E.; Badescu, M.C.; Dima, N.; Seritean Isac, P.N.; Dragoi, I.-T.; Rezus, C. Cardiovascular Risk Assessment in Rheumatoid Arthritis: Accelerated Atherosclerosis, New Biomarkers, and the of Biological Therapy. Life 2023, 13, 319. [Google Scholar] [CrossRef]
- Ku, I.A.; Imboden, J.B.; Hsue, P.Y.; Ganz, P. Rheumatoid arthritis: Model of systemic inflammation driving atherosclerosis. Circ. J. 2009, 73, 977–985. [Google Scholar] [CrossRef]
- Ahmed, S.; Jacob, B.; Carsons, S.E.; De Leon, J.; Reiss, A.B. Treatment of Cardiovascular Disease in Rheumatoid Arthritis: A Complex Challenge with Increased Atherosclerotic Risk. Pharmaceuticals 2021, 15, 11. [Google Scholar] [CrossRef]
- Gimbrone, M.A., Jr.; García-Cardeña, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620. [Google Scholar] [CrossRef]
- Searles, C.D. Transcriptional and posttranscriptional regulation of endothelial nitric oxide synthase expression. Am. J. Physiol. Cell Physiol. 2006, 291, C803–C816. [Google Scholar] [CrossRef]
- Forstermann, U.; Xia, N.; Li, H. Roles of vascular oxidative stress and nitric oxide in the pathogenesis of atherosclerosis. Circ. Res. 2017, 120, 713–735. [Google Scholar] [CrossRef]
- Page, M.J.; Bester, J.; Pretorius, E. The inflammatory effects of TNF and complement component 3 on coagulation. Sci. Rep. 2018, 8, 1812. [Google Scholar] [CrossRef] [PubMed]
- Schunk, S.J.; Triem, S.; Schmit, D.; Zewinger, S.; Sarakpi, T.; Becker, E.; Hütter, G.; Wrublewsky, S.; Küting, F.; Hohl, M.; et al. Interleukin-1α Is a Central Regulator of Leukocyte-Endothelial Adhesion in Myocardial Infarction and in Chronic Kidney Disease. Circulation 2021, 144, 893–908. [Google Scholar] [CrossRef] [PubMed]
- Poznyak, A.V.; Nikiforov, N.G.; Starodubova, A.V.; Popkova, T.V.; Orekhov, A.N. Macrophages and Foam Cells: Brief Overview of Their Role, Linkage, and Targeting Potential in Atherosclerosis. Biomedicines 2021, 9, 1221. [Google Scholar] [CrossRef]
- Voloshyna, I.; Littlefield, M.J.; Reiss, A.B. Atherosclerosis and interferon-γ: New insights and therapeutic targets. Trends Cardiovasc. Med. 2014, 24, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Elyasi, A.; Voloshyna, I.; Ahmed, S.; Kasselman, L.J.; Behbodikhah, J.; De Leon, J.; Reiss, A.B. The role of interferon-γ in cardiovascular disease: An update. Inflamm. Res. 2020, 69, 975–988. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Sahu, A.; Hwang, Y.; Kim, G.B.; Nam, G.H.; Kim, I.S.; Chan Kwon, I.; Tae, G. Targeted delivery of anti-inflammatory cytokine by nanocarrier reduces atherosclerosis in Apo E-/- mice. Biomaterials 2020, 226, 119550. [Google Scholar] [CrossRef] [PubMed]
- Tabares-Guevara, J.H.; Villa-Pulgarin, J.A.; Hernandez, J.C. Atherosclerosis: Immunopathogenesis and strategies for immunotherapy. Immunotherapy 2021, 13, 1231–1244. [Google Scholar] [CrossRef]
- Filatova, A.Y.; Pylaeva, E.A.; Potekhina, A.V.; Ruleva, N.Y.; Klesareva, E.A.; Radyukhina, N.V.; Masenko, V.P.; Shchinova, A.M.; Noeva, E.A.; Provatorov, S.I.; et al. Low Blood content of IL-10-producing C4+ T cells as a risk factor for progression of coronary atherosclerosis. Bull. Exp. Biol. Med. 2019, 166, 330–333. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, P.; Rajasingh, J.; Lambers, E.; Qin, G.; Losordo, D.W.; Kishore, R. IL-10 inhibits inflammation and attenuates left ventricular remodeling after myocardial infarction via activation of STAT3 and suppression of HuR. Circ. Res. 2009, 104, e9–e18. [Google Scholar] [CrossRef]
- Orecchioni, M.; Wolf, D.; Suryawanshi, V.; Winkels, H.; Kobiyama, K.; Makings, J.; Kiosses, W.B.; Ley, K. Deleting interleukin-10 from myeloid cells exacerbates atherosclerosis in Apoe-/- mice. Cell. Mol. Life Sci. 2022, 80, 10. [Google Scholar] [CrossRef]
- Coomes, E.; Chan, E.S.; Reiss, A.B. Methotrexate in atherogenesis and cholesterol metabolism. Cholesterol 2011, 2011, 503028. [Google Scholar] [CrossRef] [PubMed]
- Daidone, M.; Del Cuore, A.; Casuccio, A.; Di Chiara, T.; Guggino, G.; Di Raimondo, D.; Puleo, M.G.; Ferrante, A.; Scaglione, R.; Pinto, A.; et al. Vascular health in subjects with rheumatoid arthritis: Assessment of endothelial function indices and serum biomarkers of vascular damage. Intern. Emerg. Med. 2023, 18, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Voloshyna, I.; Modayil, S.; Littlefield, M.J.; Belilos, E.; Belostocki, K.; Bonetti, L.; Rosenblum, G.; Carsons, S.E.; Reiss, A.B. Plasma from rheumatoid arthritis patients promotes pro-atherogenic cholesterol transport gene expression in THP-1 human macrophages. Exp. Biol. Med. 2013, 238, 1192–1197. [Google Scholar] [CrossRef]
- Alisik, T.; Alisik, M.; Nacir, B.; Ayhan, F.F.; Genc, H.; Erel, O. Evaluation of dysfunctional high-density lipoprotein levels with myeloperoxidase/paraoxonase-1 ratio in rheumatoid arthritis. Int. J. Clin. Pract. 2021, 75, e14172. [Google Scholar] [CrossRef] [PubMed]
- Vyletelová, V.; Nováková, M.; Pašková, Ľ. Alterations of HDL’s to piHDL’s Proteome in Patients with Chronic Inflammatory Diseases, and HDL-Targeted Therapies. Pharmaceuticals 2022, 15, 1278. [Google Scholar] [CrossRef] [PubMed]
- García-Gómez, M.C.; Padró, T.; Muñoz-García, N.; Bianchi, M.; Álvarez, L.; Badimon, L.; Corbella, E.; Pintó, X. Dysfunctional antioxidant capacity of high-density lipoprotein in rheumatoid arthritis. Eur. J. Clin. Investig. 2023, 53, e13999. [Google Scholar] [CrossRef] [PubMed]
- Hahn, B.H.; Grossman, J.; Ansell, B.J.; Skaggs, B.J.; McMahon, M. Altered lipoprotein metabolism in chronic inflammatory states: Proinflammatory high-density lipoprotein and accelerated atherosclerosis in systemic lupus erythematosus and rheumatoid arthritis. Arthritis Res. Ther. 2008, 10, 213. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.K.; Cheng, W.C.; Ma, W.L.; Chen, P.K.; Chen, C.H.; Shen, P.C.; Chen, C.C.; Chang, S.H.; Lai, Y.H.; Chen, D.Y. The Potential Role of Electronegative High-Density Lipoprotein H5 Subfraction in RA-Related Atherosclerosis. Int. J. Mol. Sci. 2021, 22, 11419. [Google Scholar] [CrossRef] [PubMed]
- Charles-Schoeman, C.; Lee, Y.Y.; Grijalva, V.; Amjadi, S.; FitzGerald, J.; Ranganath, V.K.; Taylor, M.; McMahon, M.; Paulus, H.E.; Reddy, S.T. Cholesterol efflux by high density lipoproteins is impaired in patients with active rheumatoid arthritis. Ann. Rheum. Dis. 2012, 71, 1157–1162. [Google Scholar] [CrossRef]
- Parada-Turska, J.; Wójcicka, G.; Beltowski, J. Paraoxonase 1 Phenotype and Protein N-Homocysteinylation in Patients with Rheumatoid Arthritis: Implications for Cardiovascular Disease. Antioxidants 2020, 9, 899. [Google Scholar] [CrossRef]
- de Groot, L.; Hinkema, H.; Westra, J.; Smit, A.J.; Kallenberg, C.G.; Bijl, M.; Posthumus, M.D. Advanced glycation endproducts are increased in rheumatoid arthritis patients with controlled disease. Arthritis Res. Ther. 2011, 13, R205. [Google Scholar] [CrossRef]
- Rajamohan, A.; Heit, B.; Cairns, E.; Barra, L. Citrullinated and homocitrullinated low-density lipoprotein in rheumatoid arthritis. Scand. J. Rheumatol. 2021, 50, 343–350. [Google Scholar] [CrossRef]
- Giles, J.T. Cardiovascular disease in rheumatoid arthritis: Current perspectives on assessing and mitigating risk in clinical practice. Best Pract. Res. Clin. Rheumatol. 2015, 29, 597–613. [Google Scholar] [CrossRef]
- Fraenkel, L.; Bathon, J.M.; England, B.R.; St Clair, E.W.; Arayssi, T.; Carandang, K.; Deane, K.D.; Genovese, M.; Huston, K.K.; Kerr, G.; et al. 2021 American College of Rheumatology Guideline for the Treatment of Rheumatoid Arthritis. Arthritis Care Res. 2021, 73, 924–939. [Google Scholar] [CrossRef]
- Sramek, M.; Neradil, J.; Veselska, R. Much more than you expected: The non-DHFR-mediated effects of methotrexate. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 499–503. [Google Scholar] [CrossRef]
- Friedman, B.; Cronstein, B. Methotrexate mechanism in treatment of rheumatoid arthritis. Jt. Bone Spine 2019, 86, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Reiss, A.B.; Grossfeld, D.; Kasselman, L.J.; Renna, H.A.; Vernice, N.A.; Drewes, W.; Konig, J.; Carsons, S.E.; DeLeon, J. Adenosine and the Cardiovascular System. Am. J. Cardiovasc. Drugs 2019, 19, 449–464. [Google Scholar] [CrossRef] [PubMed]
- Reiss, A.B.; Carsons, S.E.; Anwar, K.; Rao, S.; Edelman, S.D.; Zhang, H.; Fernandez, P.; Cronstein, B.N.; Chan, E.S. Atheroprotective effects of methotrexate on reverse cholesterol transport proteins and foam cell transformation in human THP-1 monocyte/macrophages. Arthritis Rheum. 2008, 58, 3675–3683. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z. The use of THP-1 cells as a model for mimicking the function and regulation of monocytes and macrophages in the vasculature. Atherosclerosis 2012, 221, 2–11. [Google Scholar] [CrossRef]
- Yang, D.; Haemmig, S.; Zhou, H.; Perez-Cremades, D.; Sun, X.; Chen, L.; Li, J.; Haneo-Mejia, J.; Yang, T.; Hollan, I.; et al. Methotrexate attenuates vascular inflammation through an adenosine-microRNA-dependent pathway. eLife 2021, 10, e58064. [Google Scholar] [CrossRef]
- Choi, H.K.; Hernán, M.A.; Seeger, J.D.; Robins, J.M.; Wolfe, F. Methotrexate and mortality in patients with rheumatoid arthritis: A prospective study. Lancet 2002, 359, 1173–1177. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.M.; Sayles, H.R.; Baker, J.F.; George, M.D.; Roul, P.; Zheng, C.; Sauer, B.; Liao, K.P.; Anderson, D.R.; Mikuls, T.R.; et al. Investigating changes in disease activity as a mediator of cardiovascular risk reduction with methotrexate use in rheumatoid arthritis. Ann. Rheum. Dis. 2021, 80, 1385–1392. [Google Scholar] [CrossRef]
- Nissen, S.E.; Yeomans, N.D.; Solomon, D.H.; Lüscher, T.F.; Libby, P.; Husni, M.E.; Graham, D.Y.; Borer, J.S.; Wisniewski, L.M.; Wolski, K.E.; et al. Cardiovascular Safety of Celecoxib, Naproxen, or Ibuprofen for Arthritis. N. Engl. J. Med. 2016, 375, 2519–2529. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, R.; Alajbegovic, A.; Gomes, A.V. NSAIDs and cardiovascular diseases: Role of reactive oxygen species. Oxid. Med. Cell. Longev. 2015, 2015, 536962. [Google Scholar] [CrossRef] [PubMed]
- Roubille, C.; Richer, V.; Starnino, T.; McCourt, C.; McFarlane, A.; Fleming, P.; Siu, S.; Kraft, J.; Lynde, C.; Pope, J.; et al. The effects of tumour necrosis factor inhibitors, methotrexate, non-steroidal anti-inflammatory drugs and corticosteroids on cardiovascular events in rheumatoid arthritis, psoriasis and psoriatic arthritis: A systematic review and meta-analysis. Ann. Rheum. Dis. 2015, 74, 480–489. [Google Scholar] [CrossRef]
- Chan, E.S.; Zhang, H.; Fernandez, P.; Edelman, S.D.; Pillinger, M.H.; Ragolia, L.; Palaia, T.; Carsons, S.; Reiss, A.B. Effect of COX inhibition on cholesterol efflux proteins and atheromatous foam cell transformation in THP-1 human macrophages: A possible mechanism for increased cardiovascular risk. Arthritis Res. Ther. 2007, 9, R4. [Google Scholar] [CrossRef]
- Voloshyna, I.; Kasselman, L.J.; Carsons, S.E.; Littlefield, M.J.; Gomolin, I.H.; De Leon, J.; Reiss, A.B. COX-2-dependent and independent effects of COX-2 inhibitors and NSAIDs on proatherogenic changes in human monocytes/macrophages. J. Investig. Med. 2017, 65, 694–704. [Google Scholar] [CrossRef]
- MacDonald, T.M.; Hawkey, C.J.; Ford, I.; McMurray, J.J.V.; Scheiman, J.M.; Hallas, J.; Findlay, E.; Grobbee, D.E.; Hobbs, F.D.R.; Ralston, S.H.; et al. Randomized trial of switching from prescribed non-selective non-steroidal anti-inflammatory drugs to prescribed celecoxib: The Standard care vs. Celecoxib Outcome Trial (SCOT). Eur. Heart J. 2016, 38, 1843–1850. [Google Scholar] [CrossRef]
- Alamri, R.D.; Elmeligy, M.A.; Albalawi, G.A.; Alquayr, S.M.; Alsubhi, S.S.; El-Ghaiesh, S.H. Leflunomide an immunomodulator with antineoplastic and antiviral potentials but drug-induced liver injury: A comprehensive review. Int. Immunopharmacol. 2021, 93, 107398. [Google Scholar] [CrossRef]
- Rho, Y.H.; Oeser, A.; Chung, C.P.; Milne, G.L.; Stein, C.M. Drugs Used in the Treatment of Rheumatoid Arthritis: Relationship between Current Use and Cardiovascular Risk Factors. Arch. Drug Inf. 2009, 2, 34–40. [Google Scholar] [CrossRef]
- Ji, X.; Chen, J.; You, C.; Sun, J.; Xu, X. Leflunomide alleviates obesity via activation of the TAK1-AMPK pathway and induction of lipophagy. FASEB J. 2023, 37, e23227. [Google Scholar] [CrossRef] [PubMed]
- Nurmohamed, M.T.; van Halm, V.P.; Dijkmans, B.A.C. Cardiovascular Risk Profile of Antirheumatic Agents in Patients with Osteoarthritis and Rheumatoid Arthritis. Drugs 2002, 62, 1599–1609. [Google Scholar] [CrossRef]
- Jenks, K.A.; Stamp, L.K.; O’Donnell, J.L.; Savage, R.L.; Chapman, P.T. Leflunomide-associated infections in rheumatoid arthritis. J. Rheumatol. 2007, 34, 2201–2203. [Google Scholar]
- MacMullan, P.A.; Madigan, A.M.; Paul, N.; Peace, A.J.; Alagha, A.; Nolan, K.B.; McCarthy, G.M.; Kenny, D. Sulfasalazine and its metabolites inhibit platelet function in patients with inflammatory arthritis. Clin. Rheumatol. 2016, 35, 447–455. [Google Scholar] [CrossRef]
- Park, Y.B.; Choi, H.K.; Kim, M.Y.; Lee, W.K.; Song, J.; Kim, D.K.; Lee, W.K.; Song, J.; Kim, D.K.; Lee, S.K. Effects of antirheumatic therapy on serum lipid levels in patients with rheumatoid arthritis: A prospective study. Am. J. Med. 2002, 113, 188–193. [Google Scholar] [CrossRef]
- Tabit, C.E.; Holbrook, M.; Shenouda, S.M.; Dohadwala, M.M.; Widlansky, M.E.; Frame, A.A.; Kim, B.H.; Duess, M.A.; Kluge, M.A.; Levit, A.; et al. Effect of sulfasalazine on inflammation and endothelial function in patients with established coronary artery disease. Vasc. Med. 2012, 17, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Naranjo, A.; Sokka, T.; Descalzo, M.A.; Calvo-Alén, J.; Hørslev-Petersen, K.; Luukkainen, R.K.; Combe, B.; Burmester, G.R.; Devlin, J.; Ferraccioli, G.; et al. Cardiovascular disease in patients with rheumatoid arthritis: Results from the QUEST-RA study. Arthritis Res. Ther. 2008, 10, R30. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Cho, H.J.; Sir, J.J.; Kim, B.K.; Hur, J.; Youn, S.W.; Yang, H.M.; Jun, S.I.; Park, K.W.; Hwang, S.J.; et al. Sulfasalazine induces haem oxygenase-1 via ROS-dependent Nrf2 signalling, leading to control of neointimal hyperplasia. Cardiovasc. Res. 2009, 82, 550–560. [Google Scholar] [CrossRef]
- Brownfoot, F.C.; Hannan, N.J.; Cannon, P.; Nguyen, V.; Hastie, R.; Parry, L.J.; Senadheera, S.; Tuohey, L.; Tong, S.; Kaitu’u-Lino, T. Sulfasalazine reduces placental secretion of antiangiogenic factors, up-regulates the secretion of placental growth factor and rescues endothelial dysfunction. EBioMedicine 2019, 41, 636–648. [Google Scholar] [CrossRef]
- Sonmez, M.I.; Shahzadi, A.; Kose, C.; Sonmez, H.; Ozyazgan, S.; Akkan, A.G. Effect of sulfasalazine on endothelium-dependent vascular response by the activation of Nrf2 signalling pathway. Front. Pharmacol. 2022, 13, 979300. [Google Scholar] [CrossRef]
- Ponticelli, C.; Moroni, G. Hydroxychloroquine in systemic lupus erythematosus (SLE). Expert Opin. Drug Saf. 2017, 16, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Kuznik, A.; Bencina, M.; Svajger, U.; Jeras, M.; Rozman, B.; Jerala, R. Mechanism of endosomal TLR inhibition by antimalarial drugs and imidazoquinolines. J. Immunol. 2011, 186, 4794–4804. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.J.; Gudsoorkar, V.S.; Weisman, M.H.; Venuturupalli, S.R. New insights into mechanisms of therapeutic effects of antimalarial agents in SLE. Nat. Rev. Rheumatol. 2012, 8, 522–533. [Google Scholar] [CrossRef] [PubMed]
- Nirk, E.L.; Reggiori, F.; Mauthe, M. Hydroxychloroquine in rheumatic autoimmune disorders and beyond. EMBO Mol. Med. 2020, 12, e12476. [Google Scholar] [CrossRef] [PubMed]
- Sharma, T.S.; Wasko, M.C.; Tang, X.; Vedamurthy, D.; Yan, X.; Cote, J.; Bili, A. Hydroxychloroquine Use Is Associated with Decreased Incident Cardiovascular Events in Rheumatoid Arthritis Patients. J. Am. Heart Assoc. 2016, 5, e002867. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.C.; Mariz, H.A.; Rocha, L.F.; Oliveira, P.S.; Dantas, A.T.; Duarte, A.L.; da Rocha Pitta, I.; Galdino, S.L.; Pitta, M.G. Hydroxychloroquine decreases Th17-related cytokines in systemic lupus erythematosus and rheumatoid arthritis patients. Clinics 2013, 68, 766–771. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Irastorza, G.; Egurbide, M.V.; Pijoan, J.I.; e Garmendia, M.; Villar, I.; Martinez-Berriotxoa, A.; Erdozain, J.G.; Aguirre, C. Effect of antimalarials on thrombosis and survival in patients with systemic lupus erythematosus. Lupus 2006, 15, 77–583. [Google Scholar] [CrossRef] [PubMed]
- Shi, N.; Zhang, S.; Silverman, G.; Mengtao, L.; Cai, J.; Niu, H. Protective effect of hydroxychloroquine on rheumatoid arthritis-associated atherosclerosis. Anim. Model. Exp. Med. 2019, 2, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Hage, M.P.; Al-Badri, M.R.; Azar, S.T. A favorable effect of hydroxychloroquine on glucose and lipid metabolism beyond its anti-inflammatory role. Ther. Adv. Endocrinol. Metab. 2014, 5, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Verhoeven, F.; Prati, C.; Maguin-Gaté, K.; Wendling, D.; Demougeot, C. Glucocorticoids and endothelial function in inflammatory diseases: Focus on rheumatoid arthritis. Arthritis Res. Ther. 2016, 18, 258. [Google Scholar] [CrossRef]
- Ramamoorthy, S.; Cidlowski, J.A. Corticosteroids: Mechanisms of Action in Health and Disease. Rheum. Dis. Clin N. Am. 2016, 42, 15–31. [Google Scholar] [CrossRef] [PubMed]
- Ocon, A.J.; Reed, G.; Pappas, D.A.; Curtis, J.R.; Kremer, J.M. Short-term dose and duration-dependent glucocorticoid risk for cardiovascular events in glucocorticoid-naive patients with rheumatoid arthritis. Ann. Rheum. Dis. 2021, 80, 1522–1529. [Google Scholar] [CrossRef] [PubMed]
- del Rincón, I.; Battafarano, D.F.; Restrepo, J.F.; Erikson, J.M.; Escalante, A. Glucocorticoid dose thresholds assoiated with all-cause and cardiovascular mortality in rheumatoid arthritis. Arthritis Rheumatol. 2014, 66, 264–272. [Google Scholar] [CrossRef] [PubMed]
- Atzeni, F.; Rodríguez-Carrio, J.; Popa, C.D.; Nurmohamed, M.T.; Szűcs, G.; Szekanecz, Z. Cardiovascular effects of approved drugs for rheumatoid arthritis. Nat. Rev. Rheumatol. 2021, 17, 270–290. [Google Scholar] [CrossRef] [PubMed]
- Petersons, C.J.; Mangelsdorf, B.L.; Poljak, A.; Smith, M.D.; Greenfield, J.R.; Thompson, C.H.; Burt, M.G. Low dose prednisolone and insulin sensitivity differentially affect arterial stiffness and endothelial function: An open interventional and cross-sectional study. Atherosclerosis 2017, 258, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Pofi, R.; Caratti, G.; Ray, D.W.; Tomlinson, J.W. Treating the Side Effects of Exogenous Glucocorticoids; Can We Separate the Good From the Bad? Endocr. Rev. 2023, 44, 975–1011. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Shen, P.; Yu, B.; Xu, X.; Ge, R.; Cheng, X.; Chen, Q.; Bian, J.; Li, Z.; Wang, J. Janus kinases (JAKs): The efficient therapeutic targets for autoimmune diseases and myeloproliferative disorders. Eur. J. Med. Chem. 2020, 192, 112155. [Google Scholar] [CrossRef] [PubMed]
- Ytterberg, S.R.; Bhatt, D.L.; Mikuls, T.R.; Koch, G.G.; Fleischmann, R.; Rivas, J.L.; Germino, R.; Menon, S.; Sun, Y.; Wang, C.; et al. Cardiovascular and Cancer Risk with Tofacitinib in Rheumatoid Arthritis. N. Engl. J. Med. 2022, 386, 316–326. [Google Scholar] [CrossRef] [PubMed]
- Sakai, R.; Tanaka, E.; Inoue, E.; Harigai, M. Increased risk of cardiovascular events under the treatments with Janus kinase inhibitors versus biological disease-modifying antirheumatic drugs in patients with rheumatoid arthritis: A retrospective longitudinal population-based study using the Japanese health insurance database. RMD Open 2024, 10, e003885. [Google Scholar] [CrossRef] [PubMed]
- Hoisnard, L.; Pina Vegas, L.; Dray-Spira, R.; Weill, A.; Zureik, M.; Sbidian, E. Risk of major adverse cardiovascular and venous thromboembolism events in patients with rheumatoid arthritis exposed to JAK inhibitors versus adalimumab: A nationwide cohort study. Ann. Rheum. Dis. 2023, 82, 182–188. [Google Scholar] [CrossRef]
- Corrao, S. Crucial safety issues on Janus kinase inhibitors in rheumatoid arthritis might be associated with the lack of LDL-cholesterol management: A reasoned literature analysis. Intern. Emerg. Med. 2023, 18, 2157–2161. [Google Scholar] [CrossRef] [PubMed]
- Alzahrani, Z.; Alhazmi, A.; Almalki, H.; Aljehani, N.; Dumyati, M.; Alabdali, H. Efficacy and Safety of Tofacitinib in Rheumatoid Arthritis (RA): A Retrospective Study from Two Centers in Jeddah, Saudi Arabia. Cureus 2022, 14, e32240. [Google Scholar] [CrossRef]
- Czókolyová, M.; Hamar, A.; Pusztai, A.; Tajti, G.; Végh, E.; Pethő, Z.; Bodnár, N.; Horváth, Á.; Soós, B.; Szamosi, S.; et al. Effects of One-Year Tofacitinib Therapy on Lipids and Adipokines in Association with Vascular Pathophysiology in Rheumatoid Arthritis. Biomolecules 2022, 12, 1483. [Google Scholar] [CrossRef]
- McInnes, I.B.; Kim, H.Y.; Lee, S.H.; Mandel, D.; Song, Y.W.; Connell, C.A.; Luo, Z.; Brosnan, M.J.; Zuckerman, A.; Zwillich, S.H.; et al. Open-label and double-blind atorvastatin in rheumatoid arthritis patients: A randomised study. Ann. Rheum. Dis. 2014, 73, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Charles-Schoeman, C.; DeMasi, R.; Valdez, H.; Soma, K.; Hwang, L.J.; Boy, M.G.; Biswas, P.; McInnes, I.B. Risk factors for major adverse cardiovascular events in phase III and long-term extension studies of tofacitinib in patients with rheumatoid arthritis. Arthritis Rheumatol. 2019, 71, 1450–1459. [Google Scholar] [CrossRef]
- Chang, C.K.; Chiang, E.I.; Chang, K.H.; Tang, K.T.; Chen, P.K.; Yip, H.T.; Chen, C.H.; Chen, D.Y. The Sizes and Composition of HDL-Cholesterol Are Significantly Associated with Inflammation in Rheumatoid Arthritis Patients. Int. J. Mol. Sci. 2023, 24, 10645. [Google Scholar] [CrossRef]
- Kume, K.; Amano, K.; Yamada, S.; Kanazawa, T.; Ohta, H.; Hatta, K.; Amano, K.; Kuwaba, N. Tofacitinib improves atherosclerosis despite up-regulating serum cholesterol in patients with active rheumatoid arthritis: A cohort study. Rheumatol. Int. 2017, 37, 2079–2085. [Google Scholar] [CrossRef] [PubMed]
- Adorni, M.P.; Papotti, B.; Borghi, M.O.; Raschi, E.; Zimetti, F.; Bernini, F.; Meroni, P.L.; Ronda, N. Effect of the JAK/STAT Inhibitor Tofacitinib on Macrophage Cholesterol Metabolism. Int. J. Mol. Sci. 2023, 24, 12571. [Google Scholar] [CrossRef]
- Hongo, S.; Watanabe, T.; Arita, S.; Kanome, T.; Kageyama, H.; Shioda, S.; Miyazaki, A. Leptin modulates ACAT1 expression and cholesterol efflux from human macrophages. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E474–E482. [Google Scholar] [CrossRef]
- Anyfanti, P.; Angeloudi, E.; Dara, A.; Pagkopoulou, E.; Moysidou, G.S.; Deuteraiou, K.; Boutel, M.; Bekiari, E.; Doumas, M.; Kitas, G.D.; et al. Non-Invasive Assessment of Micro- and Macrovascular Function after Initiation of JAK Inhibitors in Patients with Rheumatoid Arthritis. Diagnostics 2024, 14, 834. [Google Scholar] [CrossRef]
- Conigliaro, P.; Minerba, C.; Vendola, A.; Fiannacca, L.; Triggianese, P.; Kroegler, B.; Greco, E.; Bergamini, A.; Chimenti, M.S. The steroid-sparing effect of JAK inhibitors across multiple patient populations. Front. Immunol. 2024, 15, 1376476. [Google Scholar] [CrossRef] [PubMed]
- Bonelli, M.; Scheinecker, C. How does abatacept really work in rheumatoid arthritis? Curr. Opin. Rheumatol. 2018, 30, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Ursini, F.; Russo, E.; Letizia Hribal, M.; Mauro, D.; Savarino, F.; Bruno, C.; Tripolino, C.; Rubino, M.; Naty, S.; Grembiale, R.D. Abatacept improves whole-body insulin sensitivity in rheumatoid arthritis: An observational study. Medicine 2015, 94, e888. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Kang, E.H.; Brill, G.; Desai, R.J.; Kim, S.C. Cardiovascular (CV) risk after initiation of abatacept versus TNF inhibitors in rheumatoid arthritis patients with and without baseline CV disease. J. Rheumatol. 2018, 45, 1240–1248. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Z.; Muraoka, S.; Kawazoe, M.; Hirose, W.; Kono, H.; Yasuda, S.; Sugihara, T.; Nanki, T. Long-term effects of abatacept on atherosclerosis and arthritis in older vs. younger patients with rheumatoid arthritis: 3-year results of a prospective, multicenter, observational study. Arthritis Res. Ther. 2024, 26, 87. [Google Scholar] [CrossRef] [PubMed]
- Delcoigne, B.; Ljung, L.; Provan, S.A.; Glintborg, B.; Hetland, M.L.; Grøn, K.L.; Peltomaa, R.; Relas, H.; Turesson, C.; Gudbjornsson, B.; et al. Short-term, intermediate-term and long-term risks of acute coronary syndrome in cohorts of patients with RA starting biologic DMARDs: Results from four Nordic countries. Ann. Rheum. Dis. 2022, 81, 789–797. [Google Scholar] [CrossRef] [PubMed]
- Charles-Schoeman, C.; Wang, J.; Shahbazian, A.; Wilhalme, H.; Brook, J.; Kaeley, G.S.; Oganesian, B.; Ben-Artzi, A.; Elashoff, D.A.; Ranganath, V.K. Power doppler ultrasound signal predicts abnormal HDL function in patients with rheumatoid arthritis. Rheumatol. Int. 2023, 43, 1041–1053. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Han, X.; Lobo, F.; Nanji, S.; Patel, D. A Cost per Responder Model for Abatacept versus Adalimumab Among Rheumatoid Arthritis Patients with Seropositivity. Clinicoecon. Outcomes Res. 2020, 12, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Jang, D.I.; Lee, A.H.; Shin, H.Y.; Song, H.R.; Park, J.H.; Kang, T.B.; Lee, S.R.; Yang, S.H. The role of tumor necrosis factor alpha (TNF-α) in autoimmune disease and current TNF-α inhibitors in therapeutics. Int. J. Mol. Sci. 2021, 22, 2719. [Google Scholar] [CrossRef]
- Delbaere, Q.; Chapet, N.; Huet, F.; Delmas, C.; Mewton, N.; Prunier, F.; Angoulvant, D.; Roubille, F. Anti-Inflammatory Drug Candidates for Prevention and Treatment of Cardiovascular Diseases. Pharmaceuticals 2023, 16, 78. [Google Scholar] [CrossRef]
- Ljung, L.; Rantapää-Dahlqvist, S.; Jacobsson, L.T.; Askling, J. Response to biological treatment and subsequent risk of coronary events in rheumatoid arthritis. Ann. Rheum. Dis. 2016, 75, 2087–2094. [Google Scholar] [CrossRef] [PubMed]
- Hussain, A.; Tarahomi, T.; Singh, L.; Bollampally, M.; Heydari-Kamjani, M.; Kesselman, M.M. Cardiovascular Risk Associated With TNF Alpha Inhibitor Use in Patients With Rheumatoid Arthritis. Cureus 2021, 13, e17938. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.; Singh Kahlon, S.; Sikandar, R.; Peddemul, A.; Tejovath, S.; Hassan, D.; Patel, K.K.; Mostafa, J.A. Tumor Necrosis Factor-Alpha Inhibitors and Cardiovascular Risk in Rheumatoid Arthritis: A Systematic Review. Cureus 2022, 14, e26430. [Google Scholar] [CrossRef] [PubMed]
- Avouac, J.; Allanore, Y. Cardiovascular risk in rheumatoid arthritis: Effects of anti-TNF drugs. Expert Opin. Pharmacother. 2008, 9, 1121–1128. [Google Scholar] [CrossRef] [PubMed]
- Calvo Alén, J.; Lavin-Gomez, B.A.; Aurrecoechea, E.; Guerra Ruiz, A.R.; Martínez Taboada, V.; Gómez Gerique, J. TNF Inhibitors Exert a “Hidden” Beneficial Effect in the Cardiovascular Lipoprotein Profile of RA Patients. Biologics 2022, 16, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Szeremeta, A.; Jura-Półtorak, A.; Zoń-Giebel, A.; Olczyk, K.; Komosińska-Vassev, K. TNF-α Inhibitors in Combination with MTX Reduce Circulating Levels of Heparan Sulfate/Heparin and Endothelial Dysfunction Biomarkers (sVCAM-1, MCP-1, MMP-9 and ADMA) in Women with Rheumatoid Arthritis. J. Clin. Med. 2022, 11, 4213. [Google Scholar] [CrossRef] [PubMed]
- Thota, L.N.R.; Chignalia, A.Z. The role of the glypican and syndecan families of heparan sulfate proteoglycans in cardiovascular function and disease. Am. J. Physiol. Cell Physiol. 2022, 323, C1052–C1060. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Chen, L.; He, J.; Xia, W.; Ye, Z.; She, J. Structural insights into IL-6 signaling inhibition by therapeutic antibodies. Cell Rep. 2024, 43, 113819. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M. From C-Reactive Protein to Interleukin-6 to Interleukin-1: Moving Upstream to Identify Novel Targets for Atheroprotection. Circ. Res. 2016, 118, 145–156. [Google Scholar] [CrossRef]
- Singh, S.; Fumery, M.; Singh, A.G.; Singh, N.; Prokop, L.J.; Dulai, P.S.; Sandborn, W.J.; Curtis, J.R. Comparative Risk of Cardiovascular Events with Biologic and Synthetic Disease-Modifying Antirheumatic Drugs in Patients With Rheumatoid Arthritis: A Systematic Review and Meta-Analysis. Arthritis Care Res. 2020, 72, 561–576. [Google Scholar] [CrossRef]
- Giles, J.T.; Sattar, N.; Gabriel, S.; Ridker, P.M.; Gay, S.; Warne, C.; Musselman, D.; Brockwell, L.; Shittu, E.; Klearman, M.; et al. Cardiovascular Safety of Tocilizumab Versus Etanercept in Rheumatoid Arthritis: A Randomized Controlled Trial. Arthritis Rheumatol. 2020, 72, 31–40. [Google Scholar] [CrossRef]
- Castagné, B.; Viprey, M.; Martin, J.; Schott, A.M.; Cucherat, M.; Soubrier, M. Cardiovascular safety of tocilizumab: A systematic review and network meta-analysis. PLoS ONE 2019, 14, e02s20178. [Google Scholar] [CrossRef] [PubMed]
- Alsulaim, T.; Alhassan, N.; Khalil, H.; Almutlaq, A. Tocilizumab Effect on Lipid Profile in Correlation to Cardiovascular Events: A Retrospective Cohort Study. Int. J. Rheumatol. 2021, 2021, 5535486. [Google Scholar] [CrossRef] [PubMed]
- Tavakolpour, S.; Alesaeidi, S.; Darvishi, M.; GhasemiAdl, M.; Darabi-Monadi, S.; Akhlaghdoust, M.; likaei Behjati, S.; Jafarieh, A. A comprehensive review of rituximab therapy in rheumatoid arthritis patients. Clin. Rheumatol. 2019, 38, 2977–2994. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.D.; Keystone, E. Rituximab for Rheumatoid Arthritis. Rheumatol. Ther. 2015, 2, 99–111. [Google Scholar] [CrossRef]
- Provan, S.A.; Berg, I.J.; Hammer, H.B.; Mathiessen, A.; Kvien, T.K.; Semb, A.G. The Impact of Newer Biological Disease Modifying Anti-Rheumatic Drugs on Cardiovascular Risk Factors: A 12-Month Longitudinal Study in Rheumatoid Arthritis Patients Treated with Rituximab, Abatacept and Tociliziumab. PLoS ONE 2015, 10, e0130709. [Google Scholar] [CrossRef] [PubMed]
- Hsue, P.Y.; Scherzer, R.; Grunfeld, C.; Imboden, J.; Wu, Y.; Del Puerto, G.; Nitta, E.; Shigenaga, J.; Schnell Heringer, A.; Ganz, P.; et al. Depletion of B-cells with rituximab improves endothelial function and reduces inflammation among individuals with rheumatoid arthritis. J. Am. Heart Assoc. 2014, 3, e001267. [Google Scholar] [CrossRef] [PubMed]
- Novikova, D.S.; Popkova, T.V.; Lukina, G.V.; Luchikhina, E.L.; Karateev, D.E.; Volkov, A.V.; Novikov, A.A.; Aleksandrova, E.N.; Nasonov, E.L. The Effects of Rituximab on Lipids, Arterial Stiffness and Carotid Intima-Media Thickness in Rheumatoid Arthritis. J. Korean Med. Sci. 2016, 31, 202–207. [Google Scholar] [CrossRef] [PubMed]
- Ward, N.C.; Watts, G.F.; Eckel, R.H. Statin Toxicity. Circ. Res. 2019, 124, 328–350. [Google Scholar] [CrossRef]
- Lüftjohann, D.; Sigit, J.I.; Locatelli, S.; von Bergmann, K.; Schmidt, H.H. High-dose simvastin (80 mg/day) decreases plasma concentrations of total homocysteine in patients with hypercholesteromia. Atherosclerosis 2001, 155, 265–266. [Google Scholar] [CrossRef]
- Jougasaki, M.; Ichiki, T.; Takenoshita, Y.; Setoguchi, M. Statins suppress interleukin-6-induced monocyte chemo-attractant protein-1 by inhibiting Janus kinase/signal transducers and activators of transcription pathways in human vascular endothelial cells. Br. J. Pharmacol. 2010, 159, 1294–1303. [Google Scholar] [CrossRef]
- Tremoulet, A.H. The role of statins in inflammatory vasculitides. Autoimmunity 2015, 48, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Koushki, K.; Shahbaz, S.K.; Mashayekhi, K.; Sadeghi, M.; Zayeri, Z.D.; Taba, M.Y.; Banach, M.; Al-Rasadi, K.; Johnston, T.P.; Sahebkar, A. Anti-inflammatory Action of Statins in Cardiovascular Disease: The Role of Inflammasome and Toll-Like Receptor Pathways. Clinic. Rev. Allerg. Immunol. 2021, 60, 175–199. [Google Scholar] [CrossRef] [PubMed]
- Venetsanopoulou, A.I.; Pelechas, E.; Voulgari, P.V.; Drosos, A.A. The lipid paradox in rheumatoid arthritis: The dark horse of the augmented cardiovascular risk. Rheumatol. Int. 2020, 40, 1181–1191. [Google Scholar] [CrossRef] [PubMed]
- Kitas, G.D.; Nightingale, P.; Armitage, J.; Sattar, N.; Belch, J.J.F.; Symmons, D.P.M.; TRACE RA Consortium. A Multicenter, Randomized, Placebo-Controlled Trial of Atorvastatin for the Primary Prevention of Cardiovascular Events in Patients With Rheumatoid Arthritis. Arthritis Rheumatol. 2019, 71, 1437–1449. [Google Scholar] [CrossRef]
- De Vera, M.A.; Choi, H.; Abrahamowicz, M.; Kopec, J.; Lacaille, D. Impact of statin discontinuation on mortality in patients with rheumatoid arthritis: A population-based study. Arthritis Care Res. 2012, 64, 809–816. [Google Scholar] [CrossRef] [PubMed]
- Karpouzas, G.A.; Ormseth, S.R.; Hernandez, E.; Budoff, M.J. The impact of statins on coronary atherosclerosis progression and long-term cardiovascular disease risk in rheumatoid arthritis. Rheumatology 2022, 61, 1857–1866. [Google Scholar] [CrossRef] [PubMed]
- Grundy, S.M.; Stone, N.J.; Bailey, A.L.; Beam, C.; Birtcher, K.K.; Blumenthal, R.S.; Braun, L.T.; de Ferranti, S.; Faiella-Tommasino, J.; Forman, D.E.; et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA guideline on the management of blood cholesterol: A report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 2019, 139, e1082–e1143. [Google Scholar] [CrossRef]
- Tinggaard, A.B.; de Thurah, A.; Andersen, I.T.; Riis, A.H.; Therkildsen, J.; Winther, S.; Hauge, E.M.; Bøttcher, M. Rheumatoid Arthritis as a Risk Factor for Coronary Artery Calcification and Obstructive Coronary Artery Disease in Patients with Chest Pain: A Registry Based Cross-Sectional Study. Clin. Epidemiol. 2020, 12, 679–689. [Google Scholar] [CrossRef]
- Lindhardsen, J.; Ahlehoff, O.; Gislason, G.H.; Madsen, O.R.; Olesen, J.B.; Torp-Pedersen, C.; Hansen, P.R. The risk of myocardial infarction in rheumatoid arthritis and diabetes mellitus: A Danish nationwide cohort study. Ann. Rheum. Dis. 2011, 70, 929. [Google Scholar] [CrossRef]
- Myasoedova, E.; Crowson, C.S.; Kremers, H.M.; Roger, V.L.; Fitz-Gibbon, P.D.; Therneau, T.M.; Gabriel, S.E. Lipid paradox in rheumatoid arthritis: The impact of serum lipid measures and systemic inflammation on the risk of cardiovascular disease. Ann. Rheum. Dis. 2011, 70, 482–487. [Google Scholar] [CrossRef] [PubMed]
- Ronda, N.; Favari, E.; Borghi, M.O.; Ingegnoli, F.; Gerosa, M.; Chighizola, C.; Zimetti, F.; Adorni, M.P.; Bernini, F.; Meroni, P.L. Impaired serum cholesterol efflux capacity in rheumatoid arthritis and systemic lupus erythematosus. Ann. Rheum. Dis. 2014, 73, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Semb, A.G.; Kvien, T.K.; Aastveit, A.H.; Jungner, I.; Pedersen, T.R.; Walldius, G.; Holme, I. Lipids, myocardial infarction and ischaemic stroke in patients with rheumatoid arthritis in the apolipoprotein-related mortality RISk (AMORIS) study. Ann. Rheum. Dis. 2010, 69, 1996–2001. [Google Scholar] [CrossRef] [PubMed]
- Bedeković, D.; Bošnjak, I.; Šarić, S.; Kirner, D.; Novak, S. Role of Inflammatory Cytokines in Rheumatoid Arthritis and Development of Atherosclerosis: A Review. Medicina 2023, 59, 1550. [Google Scholar] [CrossRef]
- Hall, F.C.; Dalbeth, N. Disease modification and cardiovascular risk reduction: Two sides of the same coin? Rheumatology 2005, 44, 1473–1482. [Google Scholar] [CrossRef] [PubMed]
- Barber, C.E.; Smith, A.; Esdaile, J.M.; Barnabe, C.; Martin, L.O.; Faris, P.; Hazlewood, G.; Noormohamed, R.; Alvarez, N.; Mancini, G.B.; et al. Best Practices for Cardiovascular Disease Prevention in Rheumatoid Arthritis: A Systematic Review of Guideline Recommendations and Quality Indicators. Arthritis Care Res. 2015, 67, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Farhat, H.; Irfan, H.; Muthiah, K.; Pallipamu, N.; Taheri, S.; Thiagaraj, S.S.; Shukla, T.S.; Gutlapalli, S.D.; Giva, S.; Penumetcha, S.S. Increased Risk of Cardiovascular Diseases in Rheumatoid Arthritis: A Systematic Review. Cureus 2022, 14, e32308. [Google Scholar] [CrossRef]
- Ikdahl, E.; Semb, A.G.; Kerola, A.M. No sign of elevated cardiovascular risk in patients with seropositive rheumatoid arthritis who remain on stable antirheumatic therapy: Results from the nationwide Norwegian Cardio-Rheuma Register. Ann. Rheum. Dis. 2024. advance online publication. [Google Scholar] [CrossRef] [PubMed]
- Giles, J.T.; Solomon, D.H.; Liao, K.P.; Rist, P.M.; Fayad, Z.A.; Tawakol, A.; Bathon, J.M. Association of the multi-biomarker disease activity score with arterial 18-fluorodeoxyglucose uptake in rheumatoid arthritis. Rheumatology 2024. advance online publication. [Google Scholar] [CrossRef] [PubMed]
- Mandel, A.; Schwarting, A.; Cavagna, L.; Triantafyllias, K. Novel Surrogate Markers of Cardiovascular Risk in the Setting of Autoimmune Rheumatic Diseases: Current Data and Implications for the Future. Front. Med. 2022, 9, 820263. [Google Scholar] [CrossRef]
- Jamthikar, A.D.; Gupta, D.; Puvvula, A.; Johri, A.M.; Khanna, N.N.; Saba, L.; Mavrogeni, S.; Laird, J.R.; Pareek, G.; Miner, M.; et al. Cardiovascular risk assessment in patients with rheumatoid arthritis using carotid ultrasound B-mode imaging. Rheumatol. Int. 2020, 40, 1921–1939. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.F.; Hunt, N.B.; Gordillo-Marañón, M.; Charoen, P.; Drenos, F.; Kivimaki, M.; Lawlor, D.A.; Giambartolomei, C.; Papacosta, O.; Chaturvedi, N.; et al. Cholesteryl ester transfer protein (CETP) as a drug target for cardiovascular disease. Nat. Commun. 2021, 12, 5640. [Google Scholar] [CrossRef] [PubMed]
- Biolo, G.; Vinci, P.; Mangogna, A.; Landolfo, M.; Schincariol, P.; Fiotti, N.; Mearelli, F.; Di Girolamo, F.G. Mechanism of action and therapeutic use of bempedoic acid in atherosclerosis and metabolic syndrome. Front. Cardiovasc. Med. 2022, 9, 1028355. [Google Scholar] [CrossRef]
- Yuan, W.; Ernst, K.; Kuai, R.; Morin, E.E.; Yu, M.; Sviridov, D.O.; Tang, J.; Mei, L.; Li, D.; Ackermann, R.; et al. Systematic evaluation of the effect of different apolipoprotein A-I mimetic peptides on the performance of synthetic high-density lipoproteins in vitro and in vivo. Nanomedicine 2023, 48, 102646. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Lei, L.; Ray, K.K.; Ballantyne, C.M.; Bradwin, G.; Rifai, N. Effects of bempedoic acid on CRP, IL-6, fibrinogen and lipoprotein(a) in patients with residual inflammatory risk: A secondary analysis of the CLEAR harmony trial. J. Clin. Lipidol. 2023, 17, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Bu, T.; Li, Z.; Hou, Y.; Sun, W.; Zhang, R.; Zhao, L.; Wei, M.; Yang, G.; Yuan, L. Exosome-mediated delivery of inflammation-responsive Il-10 mRNA for controlled atherosclerosis treatment. Theranostics 2021, 11, 9988–10000. [Google Scholar] [CrossRef] [PubMed]
- Barozet, M.; Le Tilly, O.; Bejan-Angoulvant, T.; Fesler, P.; Roubille, C. Hypertension and Cardiovascular Outcomes in Inflammatory and Autoimmune Diseases: A Systematic Review and Meta-analysis. Curr. Hypertens. Rep. 2024. advance online publication. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.P.; Giles, J.T.; Petri, M.; Szklo, M.; Post, W.; Blumenthal, R.S.; Gelber, A.C.; Ouyang, P.; Jenny, N.S.; Bathon, J.M. Prevalence of traditional modifiable cardiovascular risk factors in patients with rheumatoid arthritis: Comparison with control subjects from the multi-ethnic study of atherosclerosis. Semin. Arthritis Rheum. 2012, 41, 535–544. [Google Scholar] [CrossRef]
- Løgstrup, B.B.; Olesen, K.K.W.; Masic, D.; Gyldenkerne, C.; Thrane, P.G.; Ellingsen, T.; Bøtker, H.E.; Maeng, M. Impact of rheumatoid arthritis on major cardiovascular events in patients with and without coronary artery disease. Ann. Rheum. Dis. 2020, 79, 1182–1188. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Godbole, S.; Solomon, J.L.; Johnson, M.; Srivastava, A.; Carsons, S.E.; Belilos, E.; De Leon, J.; Reiss, A.B. Treating Cardiovascular Disease in the Inflammatory Setting of Rheumatoid Arthritis: An Ongoing Challenge. Biomedicines 2024, 12, 1608. https://doi.org/10.3390/biomedicines12071608
Godbole S, Solomon JL, Johnson M, Srivastava A, Carsons SE, Belilos E, De Leon J, Reiss AB. Treating Cardiovascular Disease in the Inflammatory Setting of Rheumatoid Arthritis: An Ongoing Challenge. Biomedicines. 2024; 12(7):1608. https://doi.org/10.3390/biomedicines12071608
Chicago/Turabian StyleGodbole, Saloni, Jenny Lue Solomon, Maryann Johnson, Ankita Srivastava, Steven E. Carsons, Elise Belilos, Joshua De Leon, and Allison B. Reiss. 2024. "Treating Cardiovascular Disease in the Inflammatory Setting of Rheumatoid Arthritis: An Ongoing Challenge" Biomedicines 12, no. 7: 1608. https://doi.org/10.3390/biomedicines12071608