
Emerging Therapeutic Targets for Acute Coronary Syndromes: Novel Advancements and Future Directions
 ,
,  , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Antiplatelet Agents

{kind=link}
| Agent Name | Mechanism of Action | Preclinical Efficacy | Clinical Trials Phase |
|---|---|---|---|
| Vorapaxar | PAR-1 antagonist | Reduced platelet aggregation and thrombus formation in animal models | Phase III completed [25]; approved by FDA |
| Cangrelor | P2Y12 receptor antagonist | Rapid onset and offset of action, effective inhibition of platelet aggregation | Phase III completed [28]; approved by FDA |
| Elinogrel | Reversible P2Y12 receptor antagonist | Effective platelet inhibition, reversible effects, and safety in animal models | Phase II completed [30]; further studies needed |
| Selatogrel (ACT-246475) | Selective P2Y12 receptor antagonist | Rapid and potent platelet inhibition, potential for subcutaneous administration | Phase II completed [31]; Phase III ongoing |
| BMS-986141 | Thromboxane receptor antagonist | Reduced thrombus formation, decreased bleeding risk in animal models | Phase I completed [33]; Phase II ongoing |
| RUC-4 | Glycoprotein IIb/IIIa receptor antagonist | Quick inhibition of platelet aggregation, potential for emergency use | Phase I completed [34], Phase II ongoing |
3. Anti-Inflammatory Drugs
4. Agents Targeting Plaque Stabilization
| Agent Name | Mechanism of Action | Preclinical Efficacy | Clinical Trials Phase |
|---|---|---|---|
| ApoA-I Milano | HDL Mimetic, promotes cholesterol efflux | Reduced plaque size and improved plaque stability in animal models | Phase II completed [85] |
| Evolocumab | PCSK9 Inhibitor, reduces LDL levels | Significant reduction in atherosclerotic plaque burden in preclinical studies | Phase III completed [76]; FDA approved |
| Alirocumab | PCSK9 Inhibitor, reduces LDL levels | Decreased plaque size and improved plaque stability in animal models | Phase III completed [79]; FDA approved |
| Inclisiran | PCSK9 Inhibitor, reduces LDL levels | Significantly reduced LDL-C levels by approximately 50% compared to placebo. | Phase III completed [81,82]; FDA approved |
| Torcetrapib | CETP Inhibitor, increases HDL levels | Decreased vascular inflammation and reduced plaque progression in animal models | Discontinued in Phase III due to side effects [90] |
| Doxycycline | MMP Inhibitor, reduces matrix metalloproteinase activity | Reduced plaque instability and inflammation in animal models | Phase II completed [95] |
| MCP-1 Inhibitor | Chemokine Receptor Antagonist, reduces monocyte recruitment | Reduced macrophage infiltration and stabilized plaques in preclinical models | Phase II ongoing [104] |
| D-4F | ApoA-I Mimetic Peptide, promotes cholesterol efflux | Reduced atherosclerosis and stabilized plaques in animal studies | Phase I/II completed [108] |
| AZD5718 | FLAP Inhibitor, reduces leukotriene production | Reduced plaque inflammation and size in preclinical studies | Phase II ongoing [111] |
| Darapladib | Lp-PLA2 Inhibitor, reduces inflammation | Decreased plaque necrosis and inflammation in animal models | Phase III completed [115,116], not approved |
5. Integration of Novel Therapeutic Strategies Into Current Clinical Practice
6. Challenges and Unmet Needs
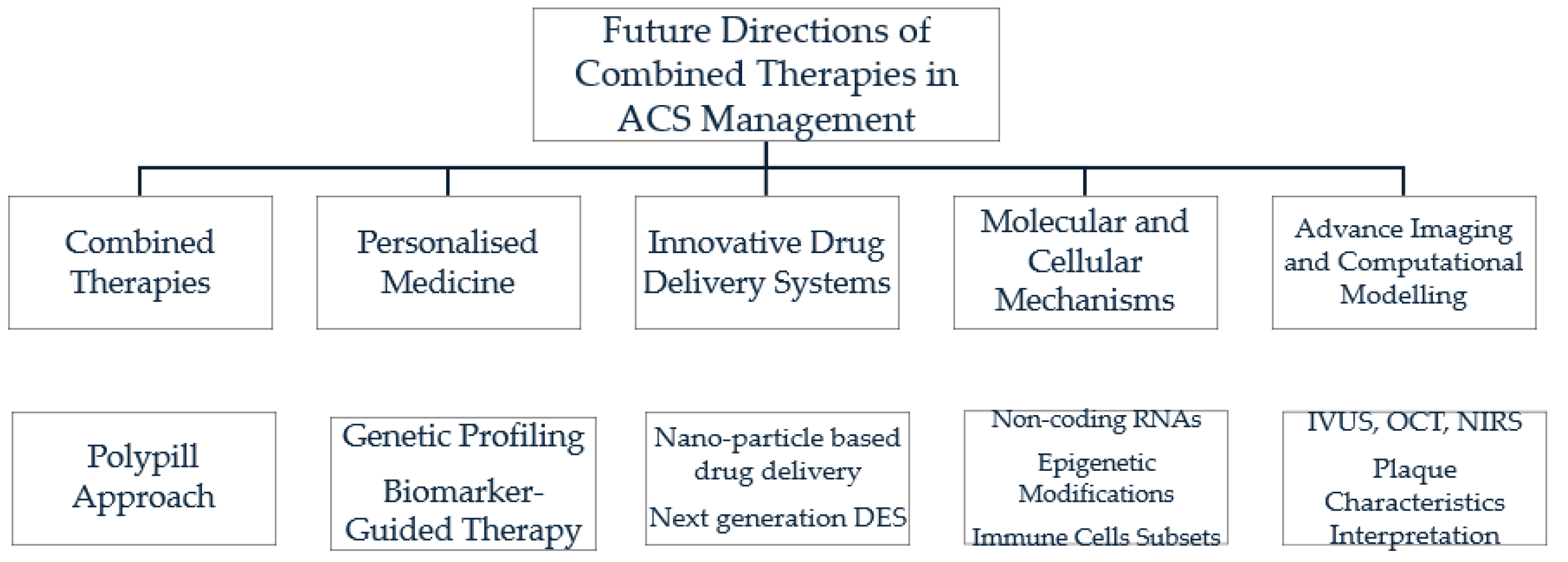
7. Future Directions
8. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Mitsis, A.; Gragnano, F. Myocardial Infarction with and without ST-Segment Elevation: A Contemporary Reappraisal of Similarities and Differences. Curr. Cardiol. Rev. 2021, 17, e230421189013. [Google Scholar] [CrossRef]
- Tsao, C.W.; Aday, A.W.; Almarzooq, Z.I.; Anderson, C.A.M.; Arora, P.; Avery, C.L.; Baker-Smith, C.M.; Beaton, A.Z.; Boehme, A.K.; Buxton, A.E.; et al. Heart Disease and Stroke Statistics-2023 Update: A Report From the American Heart Association. Circulation 2023, 147, e93–e621. [Google Scholar] [CrossRef]
- Licordari, R.; Costa, F.; Garcia-Ruiz, V.; Mamas, M.A.; Marquis-Gravel, G.; de la Torre Hernandez, J.M.; Gomez Doblas, J.J.; Jimenez-Navarro, M.; Rodriguez-Capitan, J.; Urbano-Carrillo, C.; et al. The Evolving Field of Acute Coronary Syndrome Management: A Critical Appraisal of the 2023 European Society of Cardiology Guidelines for the Management of Acute Coronary Syndrome. J. Clin. Med. 2024, 13, 1885. [Google Scholar] [CrossRef]
- Bentzon, J.F.; Otsuka, F.; Virmani, R.; Falk, E. Mechanisms of Plaque Formation and Rupture. Circ. Res. 2014, 114, 1852–1866. [Google Scholar] [CrossRef]
- Meadows, T.A.; Bhatt, D.L. Clinical Aspects of Platelet Inhibitors and Thrombus Formation. Circ. Res. 2007, 100, 1261–1275. [Google Scholar] [CrossRef]
- Davì, G.; Patrono, C. Platelet Activation and Atherothrombosis. N. Engl. J. Med. 2007, 357, 2482–2494. [Google Scholar] [CrossRef]
- Bazzoni, G.; Dejana, E.; Del Maschio, A. Platelet-Neutrophil Interactions. Possible Relevance in the Pathogenesis of Thrombosis and Inflammation. Haematologica 1991, 76, 491–499. [Google Scholar]
- Ghoshal, K.; Bhattacharyya, M. Overview of Platelet Physiology: Its Hemostatic and Nonhemostatic Role in Disease Pathogenesis. Sci. World J. 2014, 2014, 781857. [Google Scholar] [CrossRef] [PubMed]
- O’Donoghue, L.; Smolenski, A. Roles of G Proteins and Their GTPase-Activating Proteins in Platelets. Biosci. Rep. 2024, 44, BSR20231420. [Google Scholar] [CrossRef]
- Bhatt, D.L.; Hulot, J.-S.; Moliterno, D.J.; Harrington, R.A. Antiplatelet and Anticoagulation Therapy for Acute Coronary Syndromes. Circ. Res. 2014, 114, 1929–1943. [Google Scholar] [CrossRef]
- Russo, I.; Brookles, C.G.; Barale, C.; Melchionda, E.; Mousavi, A.H.; Biolè, C.; Chinaglia, A.; Bianco, M. Current Strategies to Guide the Antiplatelet Therapy in Acute Coronary Syndromes. Int. J. Mol. Sci. 2024, 25, 3981. [Google Scholar] [CrossRef] [PubMed]
- Savi, P.; Labouret, C.; Delesque, N.; Guette, F.; Lupker, J.; Herbert, J.M. P2y(12), a New Platelet ADP Receptor, Target of Clopidogrel. Biochem. Biophys. Res. Commun. 2001, 283, 379–383. [Google Scholar] [CrossRef] [PubMed]
- Mega, J.L.; Close, S.L.; Wiviott, S.D.; Shen, L.; Hockett, R.D.; Brandt, J.T.; Walker, J.R.; Antman, E.M.; Macias, W.; Braunwald, E.; et al. Cytochrome P-450 Polymorphisms and Response to Clopidogrel. N. Engl. J. Med. 2009, 360, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Greenhalgh, J.; Bagust, A.; Boland, A.; Saborido, C.M.; Fleeman, N.; McLeod, C.; Dundar, Y.; Dickson, R.; Proudlove, C.; Kolamunnage-Dona, R.; et al. Prasugrel for the Treatment of Acute Coronary Artery Syndromes with Percutaneous Coronary Intervention. Health Technol. Assess. 2010, 14 (Suppl. S1), 31–38. [Google Scholar] [CrossRef] [PubMed]
- Wiviott, S.D.; Braunwald, E.; McCabe, C.H.; Montalescot, G.; Ruzyllo, W.; Gottlieb, S.; Neumann, F.-J.; Ardissino, D.; De Servi, S.; Murphy, S.A.; et al. Prasugrel versus Clopidogrel in Patients with Acute Coronary Syndromes. N. Engl. J. Med. 2007, 357, 2001–2015. [Google Scholar] [CrossRef]
- Wallentin, L.; Becker, R.C.; Budaj, A.; Cannon, C.P.; Emanuelsson, H.; Held, C.; Horrow, J.; Husted, S.; James, S.; Katus, H.; et al. Ticagrelor versus Clopidogrel in Patients with Acute Coronary Syndromes. N. Engl. J. Med. 2009, 361, 1045–1057. [Google Scholar] [CrossRef] [PubMed]
- Amico, F.; Amico, A.; Mazzoni, J.; Moshiyakhov, M.; Tamparo, W. The Evolution of Dual Antiplatelet Therapy in the Setting of Acute Coronary Syndrome: Ticagrelor versus Clopidogrel. Postgrad. Med. 2016, 128, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Chenna, V.S.H.; Anam, H.; Hassan, M.; Moeez, A.; Reddy, R.; Chaudhari, S.S.; Sapkota, K.; Usama, M. Ticagrelor Versus Clopidogrel in Patients With Acute Coronary Syndrome and on Dialysis: A Meta-Analysis. Cureus 2023, 15, e40211. [Google Scholar] [CrossRef]
- Quinn, M.J.; Plow, E.F.; Topol, E.J. Platelet Glycoprotein IIb/IIIa Inhibitors: Recognition of a Two-Edged Sword? Circulation 2002, 106, 379–385. [Google Scholar] [CrossRef]
- Coller, B.S. Blockade of Platelet GPIIb/IIIa Receptors as an Antithrombotic Strategy. Circulation 1995, 92, 2373–2380. [Google Scholar] [CrossRef]
- EPIC Investigators. Use of a Monoclonal Antibody Directed against the Platelet Glycoprotein IIb/IIIa Receptor in High-Risk Coronary Angioplasty. N. Engl. J. Med. 1994, 330, 956–961. [Google Scholar] [CrossRef]
- Topol, E.J.; Byzova, T.V.; Plow, E.F. Platelet GPIIb-IIIa Blockers. Lancet 1999, 353, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Platelet Receptor Inhibition in Ischemic Syndrome Management in Patients Limited by Unstable Signs and Symptoms (PRISM-PLUS) Study Investigators. Inhibition of the Platelet Glycoprotein IIb/IIIa Receptor with Tirofiban in Unstable Angina and Non-Q-Wave Myocardial Infarction. N. Engl. J. Med. 1998, 338, 1488–1497. [Google Scholar] [CrossRef]
- Gao, L.; Zhao, F.-L.; Li, S.-C. Efficacy and Safety of Thrombin-Receptor Antagonist (Atopaxar and Vorapaxar) in Patients with Acute Coronary Syndrome or Coronary Artery Disease-A Meta-Analysis of Randomized Controlled Trials. Value Health Reg. Issues 2015, 6, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Lam, S.; Tran, T. Vorapaxar: A Protease-Activated Receptor Antagonist for the Prevention of Thrombotic Events. Cardiol. Rev. 2015, 23, 261–267. [Google Scholar] [CrossRef]
- O’Donoghue, M.L.; Bhatt, D.L.; Wiviott, S.D.; Goodman, S.G.; Fitzgerald, D.J.; Angiolillo, D.J.; Goto, S.; Montalescot, G.; Zeymer, U.; Aylward, P.E.; et al. Safety and Tolerability of Atopaxar in the Treatment of Patients with Acute Coronary Syndromes: The Lessons from Antagonizing the Cellular Effects of Thrombin–Acute Coronary Syndromes Trial. Circulation 2011, 123, 1843–1853. [Google Scholar] [CrossRef]
- Wiviott, S.D.; Flather, M.D.; O’Donoghue, M.L.; Goto, S.; Fitzgerald, D.J.; Cura, F.; Aylward, P.; Guetta, V.; Dudek, D.; Contant, C.F.; et al. Randomized Trial of Atopaxar in the Treatment of Patients with Coronary Artery Disease: The Lessons from Antagonizing the Cellular Effect of Thrombin–Coronary Artery Disease Trial. Circulation 2011, 123, 1854–1863. [Google Scholar] [CrossRef]
- Harrington, R.A.; Stone, G.W.; McNulty, S.; White, H.D.; Lincoff, A.M.; Gibson, C.M.; Pollack, C.V.; Montalescot, G.; Mahaffey, K.W.; Kleiman, N.S.; et al. Platelet Inhibition with Cangrelor in Patients Undergoing PCI. N. Engl. J. Med. 2009, 361, 2318–2329. [Google Scholar] [CrossRef] [PubMed]
- Qamar, A.; Bhatt, D.L. Current Status of Data on Cangrelor. Pharmacol. Ther. 2016, 159, 102–109. [Google Scholar] [CrossRef]
- Welsh, R.C.; Rao, S.V.; Zeymer, U.; Thompson, V.P.; Huber, K.; Kochman, J.; McClure, M.W.; Gretler, D.D.; Bhatt, D.L.; Gibson, C.M.; et al. A Randomized, Double-Blind, Active-Controlled Phase 2 Trial to Evaluate a Novel Selective and Reversible Intravenous and Oral P2Y12 Inhibitor Elinogrel versus Clopidogrel in Patients Undergoing Nonurgent Percutaneous Coronary Intervention: The INNOVATE-PCI Trial. Circ. Cardiovasc. Interv. 2012, 5, 336–346. [Google Scholar] [CrossRef]
- Baldoni, D.; Bruderer, S.; Krause, A.; Gutierrez, M.; Gueret, P.; Astruc, B.; Dingemanse, J. A New Reversible and Potent P2Y12 Receptor Antagonist (ACT-246475): Tolerability, Pharmacokinetics, and Pharmacodynamics in a First-in-Man Trial. Clin. Drug Investig. 2014, 34, 807–818. [Google Scholar] [CrossRef]
- Milluzzo, R.P.; Franchina, G.A.; Capodanno, D.; Angiolillo, D.J. Selatogrel, a Novel P2Y12 Inhibitor: A Review of the Pharmacology and Clinical Development. Expert Opin. Investig. Drugs 2020, 29, 537–546. [Google Scholar] [CrossRef]
- Merali, S.; Wang, Z.; Frost, C.; Meadows-Shropshire, S.; Hawthorne, D.; Yang, J.; Seiffert, D. First-in-Human Study to Assess the Safety, Pharmacokinetics, and Pharmacodynamics of BMS-986141, a Novel, Reversible, Small-Molecule, PAR4 Agonist in Non-Japanese and Japanese Healthy Participants. Platelets 2023, 34, 2222846. [Google Scholar] [CrossRef]
- Bor, W.L.; Zheng, K.L.; Tavenier, A.H.; Gibson, C.M.; Granger, C.B.; Bentur, O.; Lobatto, R.; Postma, S.; Coller, B.S.; van ’t Hof, A.W.J.; et al. Pharmacokinetics, Pharmacodynamics, and Tolerability of Subcutaneous Administration of a Novel Glycoprotein IIb/IIIa Inhibitor, RUC-4, in Patients with ST-Segment Elevation Myocardial Infarction. EuroIntervention 2021, 17, e401–e410. [Google Scholar] [CrossRef]
- Muhlestein, J.B. Effect of Antiplatelet Therapy on Inflammatory Markers in Atherothrombotic Patients. Thromb. Haemost. 2010, 103, 71–82. [Google Scholar] [CrossRef]
- Steinhubl, S.R.; Berger, P.B.; Mann, J.T.; Fry, E.T.A.; DeLago, A.; Wilmer, C.; Topol, E.J.; CREDO Investigators. Early and Sustained Dual Oral Antiplatelet Therapy Following Percutaneous Coronary Intervention: A Randomized Controlled Trial. JAMA 2002, 288, 2411–2420. [Google Scholar] [CrossRef]
- Bhatt, D.L.; Fox, K.A.A.; Hacke, W.; Berger, P.B.; Black, H.R.; Boden, W.E.; Cacoub, P.; Cohen, E.A.; Creager, M.A.; Easton, J.D.; et al. Clopidogrel and Aspirin versus Aspirin Alone for the Prevention of Atherothrombotic Events. N. Engl. J. Med. 2006, 354, 1706–1717. [Google Scholar] [CrossRef]
- Mehta, S.R.; Yusuf, S.; Peters, R.J.; Bertrand, M.E.; Lewis, B.S.; Natarajan, M.K.; Malmberg, K.; Rupprecht, H.; Zhao, F.; Chrolavicius, S.; et al. Effects of Pretreatment with Clopidogrel and Aspirin Followed by Long-Term Therapy in Patients Undergoing Percutaneous Coronary Intervention: The PCI-CURE Study. Lancet 2001, 358, 527–533. [Google Scholar] [CrossRef]
- van der Hoeven, N.W.; Janssens, G.N.; Everaars, H.; Nap, A.; Lemkes, J.S.; de Waard, G.A.; van de Ven, P.M.; van Rossum, A.C.; Escaned, J.; Mejia-Renteria, H.; et al. Platelet Inhibition, Endothelial Function, and Clinical Outcome in Patients Presenting With ST-Segment-Elevation Myocardial Infarction Randomized to Ticagrelor Versus Prasugrel Maintenance Therapy: Long-Term Follow-Up of the REDUCE-MVI Trial. J. Am. Heart Assoc. 2020, 9, e014411. [Google Scholar] [CrossRef] [PubMed]
- Zeng, S.; Zhou, X.; Ge, L.; Ji, W.-J.; Shi, R.; Lu, R.-Y.; Sun, H.-Y.; Guo, Z.-Z.; Zhao, J.-H.; Jiang, T.-M.; et al. Monocyte Subsets and Monocyte-Platelet Aggregates in Patients with Unstable Angina. J. Thromb. Thrombolysis 2014, 38, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Shantsila, E.; Tapp, L.D.; Wrigley, B.J.; Montoro-García, S.; Lip, G.Y.H. CXCR4 Positive and Angiogenic Monocytes in Myocardial Infarction. Thromb. Haemost. 2013, 109, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Tapp, L.D.; Shantsila, E.; Wrigley, B.J.; Pamukcu, B.; Lip, G.Y.H. The CD14++CD16+ Monocyte Subset and Monocyte-Platelet Interactions in Patients with ST-Elevation Myocardial Infarction. J. Thromb. Haemost. 2012, 10, 1231–1241. [Google Scholar] [CrossRef]
- Layne, K.; Di Giosia, P.; Ferro, A.; Passacquale, G. Anti-Platelet Drugs Attenuate the Expansion of Circulating CD14highCD16+ Monocytes under pro-Inflammatory Conditions. Cardiovasc. Res. 2016, 111, 26–33. [Google Scholar] [CrossRef]
- Myrianthefs, M.; Kafkas, N.; Demponeras, C.; Venetsanou, K.; Kyroudi, A.; Kouloukousa, M.; Myrianthefs, P.; Baltopoulos, G. Diagnostic Coronary Angiography Is Related to Decreased TNF-Alpha Production after Ex-Vivo Whole Blood Stimulation with LPS. Cytokine 2010, 51, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Mitsis, A.; Kadoglou, N.P.E.; Lambadiari, V.; Alexiou, S.; Theodoropoulos, K.C.; Avraamides, P.; Kassimis, G. Prognostic Role of Inflammatory Cytokines and Novel Adipokines in Acute Myocardial Infarction: An Updated and Comprehensive Review. Cytokine 2022, 153, 155848. [Google Scholar] [CrossRef] [PubMed]
- Hansson, G.K. Inflammation, Atherosclerosis, and Coronary Artery Disease. N. Engl. J. Med. 2005, 352, 1685–1695. [Google Scholar] [CrossRef]
- Libby, P.; Ridker, P.M.; Hansson, G.K. Progress and Challenges in Translating the Biology of Atherosclerosis. Nature 2011, 473, 317–325. [Google Scholar] [CrossRef]
- Gutiérrez, E.; Flammer, A.J.; Lerman, L.O.; Elízaga, J.; Lerman, A.; Fernández-Avilés, F. Endothelial Dysfunction over the Course of Coronary Artery Disease. Eur. Heart. J. 2013, 34, 3175–3181. [Google Scholar] [CrossRef]
- Medina-Leyte, D.J.; Zepeda-García, O.; Domínguez-Pérez, M.; González-Garrido, A.; Villarreal-Molina, T.; Jacobo-Albavera, L. Endothelial Dysfunction, Inflammation and Coronary Artery Disease: Potential Biomarkers and Promising Therapeutical Approaches. Int. J. Mol. Sci. 2021, 22, 3850. [Google Scholar] [CrossRef]
- Henein, M.Y.; Vancheri, S.; Longo, G.; Vancheri, F. The Role of Inflammation in Cardiovascular Disease. Int. J. Mol. Sci. 2022, 23, 12906. [Google Scholar] [CrossRef]
- Mitsis, A.; Kyriakou, M.; Sokratous, S.; Karmioti, G.; Drakomathioulakis, M.; Myrianthefs, M.; Ziakas, A.; Tzikas, S.; Kassimis, G. Exploring the Landscape of Anti-Inflammatory Trials: A Comprehensive Review of Strategies for Targeting Inflammation in Acute Myocardial Infraction. Biomedicines 2024, 12, 701. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; MacFadyen, J.G.; Everett, B.M.; Libby, P.; Thuren, T.; Glynn, R.J.; Ridker, P.M.; MacFadyen, J.G.; Everett, B.M.; Libby, P.; et al. Relationship of C-Reactive Protein Reduction to Cardiovascular Event Reduction Following Treatment with Canakinumab: A Secondary Analysis from the CANTOS Randomised Controlled Trial. Lancet 2018, 391, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.R.; Abbasi, Z.; Fatima, M.; Ochani, R.K.; Shahnawaz, W.; Asim Khan, M.; Shah, S.A. Canakinumab and Cardiovascular Outcomes: Results of the CANTOS Trial. J. Community Hosp. Intern. Med. Perspect. 2018, 8, 21–22. [Google Scholar] [CrossRef] [PubMed]
- Hemkens, L.G.; Ewald, H.; Gloy, V.L.; Arpagaus, A.; Olu, K.K.; Nidorf, M.; Glinz, D.; Nordmann, A.J.; Briel, M. Colchicine for Prevention of Cardiovascular Events. Cochrane Database Syst. Rev. 2016, 2016, CD011047. [Google Scholar] [CrossRef] [PubMed]
- Nelson, K.; Fuster, V.; Ridker, P.M. Low-Dose Colchicine for Secondary Prevention of Coronary Artery Disease: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2023, 82, 648–660. [Google Scholar] [CrossRef] [PubMed]
- Nidorf, S.M.; Fiolet, A.T.L.; Mosterd, A.; Eikelboom, J.W.; Schut, A.; Opstal, T.S.J.; The, S.H.K.; Xu, X.-F.; Ireland, M.A.; Lenderink, T.; et al. Colchicine in Patients with Chronic Coronary Disease. N. Engl. J. Med. 2020, 383, 1838–1847. [Google Scholar] [CrossRef] [PubMed]
- Tardif, J.-C.; Kouz, S.; Waters, D.D.; Bertrand, O.F.; Diaz, R.; Maggioni, A.P.; Pinto, F.J.; Ibrahim, R.; Gamra, H.; Kiwan, G.S.; et al. Efficacy and Safety of Low-Dose Colchicine after Myocardial Infarction. N. Engl. J. Med. 2019, 381, 2497–2505. [Google Scholar] [CrossRef] [PubMed]
- Banach, M.; Penson, P.E. Colchicine and Cardiovascular Outcomes: A Critical Appraisal of Recent Studies. Curr. Atheroscler. Rep. 2021, 23, 32. [Google Scholar] [CrossRef]
- Liao, J.K.; Laufs, U. Pleiotropic Effects of Statins. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 89–118. [Google Scholar] [CrossRef]
- Singh, N.; Tamariz, J.; Chamorro, G.; Medina-Franco, J.L. Inhibitors of HMG-CoA Reductase: Current and Future Prospects. Mini Rev. Med. Chem. 2009, 9, 1272–1283. [Google Scholar] [CrossRef] [PubMed]
- Nakagomi, A.; Shibui, T.; Kohashi, K.; Kosugi, M.; Kusama, Y.; Atarashi, H.; Shimizu, W. Differential Effects of Atorvastatin and Pitavastatin on Inflammation, Insulin Resistance, and the Carotid Intima-Media Thickness in Patients with Dyslipidemia. J. Atheroscler. Thromb. 2015, 22, 1158–1171. [Google Scholar] [CrossRef] [PubMed]
- Diamantis, E.; Kyriakos, G.; Quiles-Sanchez, L.V.; Farmaki, P.; Troupis, T. The Anti-Inflammatory Effects of Statins on Coronary Artery Disease: An Updated Review of the Literature. Curr. Cardiol. Rev. 2017, 13, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; JUPITER Study Group. Rosuvastatin in the Primary Prevention of Cardiovascular Disease among Patients with Low Levels of Low-Density Lipoprotein Cholesterol and Elevated High-Sensitivity C-Reactive Protein: Rationale and Design of the JUPITER Trial. Circulation 2003, 108, 2292–2297. [Google Scholar] [CrossRef] [PubMed]
- Cannon, C.P.; Braunwald, E.; McCabe, C.H.; Rader, D.J.; Rouleau, J.L.; Belder, R.; Joyal, S.V.; Hill, K.A.; Pfeffer, M.A.; Skene, A.M. Intensive versus Moderate Lipid Lowering with Statins after Acute Coronary Syndromes. N. Engl. J. Med. 2004, 350, 1495–1504. [Google Scholar] [CrossRef]
- Nissen, S.E.; Tuzcu, E.M.; Schoenhagen, P.; Brown, B.G.; Ganz, P.; Vogel, R.A.; Crowe, T.; Howard, G.; Cooper, C.J.; Brodie, B.; et al. Effect of Intensive Compared with Moderate Lipid-Lowering Therapy on Progression of Coronary Atherosclerosis: A Randomized Controlled Trial. JAMA 2004, 291, 1071–1080. [Google Scholar] [CrossRef] [PubMed]
- Albert, M.A.; Staggers, J.; Chew, P.; Ridker, P.M.; PRINCE Investigators. The Pravastatin Inflammation CRP Evaluation (PRINCE): Rationale and Design. Am. Heart J. 2001, 141, 893–898. [Google Scholar] [CrossRef] [PubMed]
- Albert, M.A.; Danielson, E.; Rifai, N.; Ridker, P.M.; for the PRINCE Investigators. Effect of Statin Therapy on C-Reactive Protein LevelsThe Pravastatin Inflammation/CRP Evaluation (PRINCE): A Randomized Trial and Cohort Study. JAMA 2001, 286, 64. [Google Scholar] [CrossRef] [PubMed]
- Libby, P. Mechanisms of Acute Coronary Syndromes and Their Implications for Therapy. N. Engl. J. Med. 2013, 368, 2004–2013. [Google Scholar] [CrossRef]
- Waksman, R.; Di Mario, C.; Torguson, R.; Ali, Z.A.; Singh, V.; Skinner, W.H.; Artis, A.K.; Cate, T.T.; Powers, E.; Kim, C.; et al. Identification of Patients and Plaques Vulnerable to Future Coronary Events with Near-Infrared Spectroscopy Intravascular Ultrasound Imaging: A Prospective, Cohort Study. Lancet 2019, 394, 1629–1637. [Google Scholar] [CrossRef]
- Goldstein, J.A. Multifocal Coronary Plaque Instability. Prog. Cardiovasc. Dis. 2002, 44, 449–454. [Google Scholar] [CrossRef] [PubMed]
- Dave, T.; Ezhilan, J.; Vasnawala, H.; Somani, V. Plaque Regression and Plaque Stabilisation in Cardiovascular Diseases. Indian J. Endocrinol. Metab. 2013, 17, 983–989. [Google Scholar] [CrossRef] [PubMed]
- Mazhar, F.; Haider, N. Proprotein Convertase Subtilisin/Kexin Type 9 Enzyme Inhibitors: An Emerging New Therapeutic Option for the Treatment of Dyslipidemia. J. Pharmacol. Pharmacother. 2016, 7, 190–193. [Google Scholar] [CrossRef] [PubMed]
- Sabatine, M.S.; Giugliano, R.P.; Keech, A.; Honarpour, N.; Wang, H.; Liu, T.; Wasserman, S.M.; Scott, R.; Sever, P.S.; Pedersen, T.R. Rationale and Design of the Further Cardiovascular OUtcomes Research with PCSK9 Inhibition in Subjects with Elevated Risk Trial. Am. Heart J. 2016, 173, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Sabatine, M.S.; Giugliano, R.P.; Wiviott, S.D.; Raal, F.J.; Blom, D.J.; Robinson, J.; Ballantyne, C.M.; Somaratne, R.; Legg, J.; Wasserman, S.M.; et al. Efficacy and Safety of Evolocumab in Reducing Lipids and Cardiovascular Events. N. Engl. J. Med. 2015, 372, 1500–1509. [Google Scholar] [CrossRef] [PubMed]
- Sabatine, M.S.; Giugliano, R.P.; Keech, A.C.; Honarpour, N.; Wiviott, S.D.; Murphy, S.A.; Kuder, J.F.; Wang, H.; Liu, T.; Wasserman, S.M.; et al. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. N. Engl. J. Med. 2017, 376, 1713–1722. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, G.G.; Bessac, L.; Berdan, L.G.; Bhatt, D.L.; Bittner, V.; Diaz, R.; Goodman, S.G.; Hanotin, C.; Harrington, R.A.; Jukema, J.W.; et al. Effect of Alirocumab, a Monoclonal Antibody to PCSK9, on Long-Term Cardiovascular Outcomes Following Acute Coronary Syndromes: Rationale and Design of the ODYSSEY Outcomes Trial. Am. Heart J. 2014, 168, 682–689. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.G.; Farnier, M.; Krempf, M.; Bergeron, J.; Luc, G.; Averna, M.; Stroes, E.S.; Langslet, G.; Raal, F.J.; El Shahawy, M.; et al. Efficacy and Safety of Alirocumab in Reducing Lipids and Cardiovascular Events. N. Engl. J. Med. 2015, 372, 1489–1499. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, G.G.; Steg, P.G.; Szarek, M.; Bhatt, D.L.; Bittner, V.A.; Diaz, R.; Edelberg, J.M.; Goodman, S.G.; Hanotin, C.; Harrington, R.A.; et al. Alirocumab and Cardiovascular Outcomes after Acute Coronary Syndrome. N. Engl. J. Med. 2018, 379, 2097–2107. [Google Scholar] [CrossRef]
- Ray, K.K.; Landmesser, U.; Leiter, L.A.; Kallend, D.; Dufour, R.; Karakas, M.; Hall, T.; Troquay, R.P.T.; Turner, T.; Visseren, F.L.J.; et al. Inclisiran in Patients at High Cardiovascular Risk with Elevated LDL Cholesterol. N. Engl. J. Med. 2017, 376, 1430–1440. [Google Scholar] [CrossRef]
- Raal, F.J.; Kallend, D.; Ray, K.K.; Turner, T.; Koenig, W.; Wright, R.S.; Wijngaard, P.L.J.; Curcio, D.; Jaros, M.J.; Leiter, L.A.; et al. Inclisiran for the Treatment of Heterozygous Familial Hypercholesterolemia. N. Engl. J. Med. 2020, 382, 1520–1530. [Google Scholar] [CrossRef] [PubMed]
- Ray, K.K.; Wright, R.S.; Kallend, D.; Koenig, W.; Leiter, L.A.; Raal, F.J.; Bisch, J.A.; Richardson, T.; Jaros, M.; Wijngaard, P.L.J.; et al. Two Phase 3 Trials of Inclisiran in Patients with Elevated LDL Cholesterol. N. Engl. J. Med. 2020, 382, 1507–1519. [Google Scholar] [CrossRef] [PubMed]
- Frampton, J.E. Inclisiran: A Review in Hypercholesterolemia. Am. J. Cardiovasc. Drugs 2023, 23, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Sirtori, C.R.; Calabresi, L.; Franceschini, G.; Baldassarre, D.; Amato, M.; Johansson, J.; Salvetti, M.; Monteduro, C.; Zulli, R.; Muiesan, M.L.; et al. Cardiovascular Status of Carriers of the Apolipoprotein A-I(Milano) Mutant: The Limone Sul Garda Study. Circulation 2001, 103, 1949–1954. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, M.; Petrlova, J.; Dalla-Riva, J.; Wasserstrom, S.; Rippe, C.; Domingo-Espin, J.; Kotowska, D.; Krupinska, E.; Berggreen, C.; Jones, H.A.; et al. ApoA-I Milano Stimulates Lipolysis in Adipose Cells Independently of cAMP/PKA Activation. J. Lipid Res. 2015, 56, 2248–2259. [Google Scholar] [CrossRef] [PubMed]
- Abudukeremu, A.; Huang, C.; Li, H.; Sun, R.; Liu, X.; Wu, X.; Xie, X.; Huang, J.; Zhang, J.; Bao, J.; et al. Efficacy and Safety of High-Density Lipoprotein/Apolipoprotein A1 Replacement Therapy in Humans and Mice With Atherosclerosis: A Systematic Review and Meta-Analysis. Front. Cardiovasc. Med. 2021, 8, 700233. [Google Scholar] [CrossRef] [PubMed]
- Kalayci, A.; Gibson, C.M.; Ridker, P.M.; Wright, S.D.; Kingwell, B.A.; Korjian, S.; Chi, G.; Lee, J.J.; Tricoci, P.; Kazmi, S.H.; et al. ApoA-I Infusion Therapies Following Acute Coronary Syndrome: Past, Present, and Future. Curr. Atheroscler. Rep. 2022, 24, 585–597. [Google Scholar] [CrossRef] [PubMed]
- Santos-Gallego, C.G.; Ibanez, B.; Badimon, J.J. HDL-Cholesterol: Is It Really Good? Differences between apoA-I and HDL. Biochem. Pharmacol. 2008, 76, 443–452. [Google Scholar] [CrossRef]
- de Haan, W.; de Vries-van der Weij, J.; van der Hoorn, J.W.A.; Gautier, T.; van der Hoogt, C.C.; Westerterp, M.; Romijn, J.A.; Jukema, J.W.; Havekes, L.M.; Princen, H.M.G.; et al. Torcetrapib Does Not Reduce Atherosclerosis beyond Atorvastatin and Induces More Proinflammatory Lesions than Atorvastatin. Circulation 2008, 117, 2515–2522. [Google Scholar] [CrossRef]
- Barter, P.J.; Caulfield, M.; Eriksson, M.; Grundy, S.M.; Kastelein, J.J.P.; Komajda, M.; Lopez-Sendon, J.; Mosca, L.; Tardif, J.-C.; Waters, D.D.; et al. Effects of Torcetrapib in Patients at High Risk for Coronary Events. N. Engl. J. Med. 2007, 357, 2109–2122. [Google Scholar] [CrossRef]
- Rosenson, R.S. Off-Target Toxicity: Risks Associated with Adrenal Corticoid Activation in ILLUMINATE. Curr. Atheroscler. Rep. 2008, 10, 227–229. [Google Scholar] [CrossRef]
- Lindsey, M.L.; Zamilpa, R. Temporal and Spatial Expression of Matrix Metalloproteinases and Tissue Inhibitors of Metalloproteinases Following Myocardial Infarction. Cardiovasc. Ther. 2012, 30, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Galis, Z.S.; Sukhova, G.K.; Lark, M.W.; Libby, P. Increased Expression of Matrix Metalloproteinases and Matrix Degrading Activity in Vulnerable Regions of Human Atherosclerotic Plaques. J. Clin. Investig. 1994, 94, 2493–2503. [Google Scholar] [CrossRef] [PubMed]
- Halade, G.V.; Jin, Y.-F.; Lindsey, M.L. Matrix Metalloproteinase (MMP)-9: A Proximal Biomarker for Cardiac Remodeling and a Distal Biomarker for Inflammation. Pharmacol. Ther. 2013, 139, 32–40. [Google Scholar] [CrossRef]
- Brown, D.L.; Desai, K.K.; Vakili, B.A.; Nouneh, C.; Lee, H.-M.; Golub, L.M. Clinical and Biochemical Results of the Metalloproteinase Inhibition with Subantimicrobial Doses of Doxycycline to Prevent Acute Coronary Syndromes (MIDAS) Pilot Trial. Arter. Thromb. Vasc. Biol. 2004, 24, 733–738. [Google Scholar] [CrossRef]
- Axisa, B.; Loftus, I.M.; Naylor, A.R.; Goodall, S.; Jones, L.; Bell, P.R.F.; Thompson, M.M. Prospective, Randomized, Double-Blind Trial Investigating the Effect of Doxycycline on Matrix Metalloproteinase Expression within Atherosclerotic Carotid Plaques. Stroke 2002, 33, 2858–2864. [Google Scholar] [CrossRef] [PubMed]
- Molloy, K.J.; Thompson, M.M.; Jones, J.L.; Schwalbe, E.C.; Bell, P.R.F.; Naylor, A.R.; Loftus, I.M. Unstable Carotid Plaques Exhibit Raised Matrix Metalloproteinase-8 Activity. Circulation 2004, 110, 337–343. [Google Scholar] [CrossRef]
- Brew, K.; Dinakarpandian, D.; Nagase, H. Tissue Inhibitors of Metalloproteinases: Evolution, Structure and Function1. Biochim. Biophys. Acta Protein Struct. Mol. Enzymol. 2000, 1477, 267–283. [Google Scholar] [CrossRef]
- Cavusoglu, E.; Ruwende, C.; Chopra, V.; Yanamadala, S.; Eng, C.; Clark, L.T.; Pinsky, D.J.; Marmur, J.D. Tissue Inhibitor of Metalloproteinase-1 (TIMP-1) Is an Independent Predictor of All-Cause Mortality, Cardiac Mortality, and Myocardial Infarction. Am. Heart J. 2006, 151, e1–e1101. [Google Scholar] [CrossRef]
- Lahdentausta, L.; Leskelä, J.; Winkelmann, A.; Tervahartiala, T.; Sorsa, T.; Pesonen, E.; Pussinen, P.J. Serum MMP-9 Diagnostics, Prognostics, and Activation in Acute Coronary Syndrome and Its Recurrence. J. Cardiovasc. Transl. Res. 2018, 11, 210–220. [Google Scholar] [CrossRef]
- Johnson, J.L.; George, S.J.; Newby, A.C.; Jackson, C.L. Divergent Effects of Matrix Metalloproteinases 3, 7, 9, and 12 on Atherosclerotic Plaque Stability in Mouse Brachiocephalic Arteries. Proc. Natl. Acad. Sci. USA 2005, 102, 15575–15580. [Google Scholar] [CrossRef]
- Deshmane, S.L.; Kremlev, S.; Amini, S.; Sawaya, B.E. Monocyte Chemoattractant Protein-1 (MCP-1): An Overview. J. Interferon Cytokine Res. 2009, 29, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Georgakis, M.K.; van der Laan, S.W.; Asare, Y.; Mekke, J.M.; Haitjema, S.; Schoneveld, A.H.; de Jager, S.C.A.; Nurmohamed, N.S.; Kroon, J.; Stroes, E.S.G.; et al. Monocyte-Chemoattractant Protein-1 Levels in Human Atherosclerotic Lesions Associate With Plaque Vulnerability. Arter. Thromb. Vasc. Biol. 2021, 41, 2038–2048. [Google Scholar] [CrossRef]
- Gilbert, J.; Lekstrom-Himes, J.; Donaldson, D.; Lee, Y.; Hu, M.; Xu, J.; Wyant, T.; Davidson, M.; MLN1202 Study Group. Effect of CC Chemokine Receptor 2 CCR2 Blockade on Serum C-Reactive Protein in Individuals at Atherosclerotic Risk and with a Single Nucleotide Polymorphism of the Monocyte Chemoattractant Protein-1 Promoter Region. Am. J. Cardiol. 2011, 107, 906–911. [Google Scholar] [CrossRef]
- Van Lenten, B.J.; Wagner, A.C.; Jung, C.-L.; Ruchala, P.; Waring, A.J.; Lehrer, R.I.; Watson, A.D.; Hama, S.; Navab, M.; Anantharamaiah, G.M.; et al. Anti-Inflammatory apoA-I-Mimetic Peptides Bind Oxidized Lipids with Much Higher Affinity than Human apoA-I. J. Lipid Res. 2008, 49, 2302–2311. [Google Scholar] [CrossRef]
- Weihrauch, D.; Xu, H.; Shi, Y.; Wang, J.; Brien, J.; Jones, D.W.; Kaul, S.; Komorowski, R.A.; Csuka, M.E.; Oldham, K.T.; et al. Effects of D-4F on Vasodilation, Oxidative Stress, Angiostatin, Myocardial Inflammation, and Angiogenic Potential in Tight-Skin Mice. Am. Physiol. Soc. 2007, 293, H1432–H1441. [Google Scholar] [CrossRef]
- Xie, Q.; Zhao, S.; Li, F. D-4F, an Apolipoprotein A-I Mimetic Peptide, Promotes Cholesterol Efflux from Macrophages via ATP-Binding Cassette Transporter A1. Tohoku J. Exp. Med. 2010, 220, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Yao, S.; Tian, H.; Miao, C.; Zhang, D.-W.; Zhao, L.; Li, Y.; Yang, N.; Jiao, P.; Sang, H.; Guo, S.; et al. D4F Alleviates Macrophage-Derived Foam Cell Apoptosis by Inhibiting CD36 Expression and ER Stress-CHOP Pathway. J. Lipid Res. 2015, 56, 836–847. [Google Scholar] [CrossRef] [PubMed]
- Lemurell, M.; Ulander, J.; Emtenäs, H.; Winiwarter, S.; Broddefalk, J.; Swanson, M.; Hayes, M.A.; Prieto Garcia, L.; Westin Eriksson, A.; Meuller, J.; et al. Novel Chemical Series of 5-Lipoxygenase-Activating Protein Inhibitors for Treatment of Coronary Artery Disease. J. Med. Chem. 2019, 62, 4325–4349. [Google Scholar] [CrossRef] [PubMed]
- Ericsson, H.; Nelander, K.; Lagerstrom-Fermer, M.; Balendran, C.; Bhat, M.; Chialda, L.; Gan, L.-M.; Heijer, M.; Kjaer, M.; Lambert, J.; et al. Initial Clinical Experience with AZD5718, a Novel Once Daily Oral 5-Lipoxygenase Activating Protein Inhibitor. Clin. Transl. Sci. 2018, 11, 330–338. [Google Scholar] [CrossRef]
- Prescott, E.; Angerås, O.; Erlinge, D.; Grove, E.L.; Hedman, M.; Jensen, L.O.; Pernow, J.; Saraste, A.; Åkerblom, A.; Svedlund, S.; et al. Safety and Efficacy of the 5-Lipoxygenase-Activating Protein Inhibitor AZD5718 in Patients with Recent Myocardial Infarction: The Phase 2a FLAVOUR Study. Int. J. Cardiol. 2022, 365, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Bui, Q.T.; Wilensky, R.L. Darapladib. Expert Opin. Investig. Drugs 2010, 19, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Serruys, P.W.; García-García, H.M.; Buszman, P.; Erne, P.; Verheye, S.; Aschermann, M.; Duckers, H.; Bleie, O.; Dudek, D.; Bøtker, H.E.; et al. Effects of the Direct Lipoprotein-Associated Phospholipase A(2) Inhibitor Darapladib on Human Coronary Atherosclerotic Plaque. Circulation 2008, 118, 1172–1182. [Google Scholar] [CrossRef] [PubMed]
- Lp-PLA(2) Studies Collaboration; Thompson, A.; Gao, P.; Orfei, L.; Watson, S.; Di Angelantonio, E.; Kaptoge, S.; Ballantyne, C.; Cannon, C.P.; Criqui, M.; et al. Lipoprotein-Associated Phospholipase A(2) and Risk of Coronary Disease, Stroke, and Mortality: Collaborative Analysis of 32 Prospective Studies. Lancet 2010, 375, 1536–1544. [Google Scholar] [CrossRef]
- STABILITY Investigators; White, H.D.; Held, C.; Stewart, R.; Tarka, E.; Brown, R.; Davies, R.Y.; Budaj, A.; Harrington, R.A.; Steg, P.G.; et al. Darapladib for Preventing Ischemic Events in Stable Coronary Heart Disease. N. Engl. J. Med. 2014, 370, 1702–1711. [Google Scholar] [CrossRef] [PubMed]
- O’Donoghue, M.L.; Braunwald, E.; White, H.D.; Steen, D.P.; Lukas, M.A.; Tarka, E.; Steg, P.G.; Hochman, J.S.; Bode, C.; Maggioni, A.P.; et al. Effect of Darapladib on Major Coronary Events After an Acute Coronary Syndrome: The SOLID-TIMI 52 Randomized Clinical Trial. JAMA 2014, 312, 1006. [Google Scholar] [CrossRef] [PubMed]
- Jankowski, P.; Safar, M.E.; Benetos, A. Pleiotropic Effects of Drugs Inhibiting the Renin-Angiotensin-Aldosterone System. Curr. Pharm. Des. 2009, 15, 571–584. [Google Scholar] [CrossRef] [PubMed]
- Heart Outcomes Prevention Evaluation Study Investigators; Yusuf, S.; Sleight, P.; Pogue, J.; Bosch, J.; Davies, R.; Dagenais, G. Effects of an Angiotensin-Converting-Enzyme Inhibitor, Ramipril, on Cardiovascular Events in High-Risk Patients. N. Engl. J. Med. 2000, 342, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Dahlöf, B.; Devereux, R.B.; Kjeldsen, S.E.; Julius, S.; Beevers, G.; de Faire, U.; Fyhrquist, F.; Ibsen, H.; Kristiansson, K.; Lederballe-Pedersen, O.; et al. Cardiovascular Morbidity and Mortality in the Losartan Intervention For Endpoint Reduction in Hypertension Study (LIFE): A Randomised Trial against Atenolol. Lancet 2002, 359, 995–1003. [Google Scholar] [CrossRef]
- Byrne, R.A.; Rossello, X.; Coughlan, J.J.; Barbato, E.; Berry, C.; Chieffo, A.; Claeys, M.J.; Dan, G.-A.; Dweck, M.R.; Galbraith, M.; et al. 2023 ESC Guidelines for the Management of Acute Coronary Syndromes. Eur. Heart. J. Acute Cardiovasc. Care 2024, 13, 55–161. [Google Scholar] [CrossRef]
- Lawton, J.S.; Tamis-Holland, J.E.; Bangalore, S.; Bates, E.R.; Beckie, T.M.; Bischoff, J.M.; Bittl, J.A.; Cohen, M.G.; DiMaio, J.M.; Don, C.W.; et al. 2021 ACC/AHA/SCAI Guideline for Coronary Artery Revascularization: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation 2022, 145, e18–e114. [Google Scholar] [CrossRef]
- Whitebread, S.; Dumotier, B.; Armstrong, D.; Fekete, A.; Chen, S.; Hartmann, A.; Muller, P.Y.; Urban, L. Secondary Pharmacology: Screening and Interpretation of off-Target Activities—Focus on Translation. Drug Discov. Today 2016, 21, 1232–1242. [Google Scholar] [CrossRef] [PubMed]
- Bender, A.; Scheiber, J.; Glick, M.; Davies, J.W.; Azzaoui, K.; Hamon, J.; Urban, L.; Whitebread, S.; Jenkins, J.L. Analysis of Pharmacology Data and the Prediction of Adverse Drug Reactions and Off-Target Effects from Chemical Structure. ChemMedChem 2007, 2, 861–873. [Google Scholar] [CrossRef] [PubMed]
- Moore, R.A.; Derry, S.; McQuay, H.J.; Straube, S.; Aldington, D.; Wiffen, P.; Bell, R.F.; Kalso, E.; Rowbotham, M.C.; ACTINPAIN writing group of the IASP Special Interest Group (SIG) on Systematic Reviews in Pain Relief. Clinical Effectiveness: An Approach to Clinical Trial Design More Relevant to Clinical Practice, Acknowledging the Importance of Individual Differences. Pain 2010, 149, 173–176. [Google Scholar] [CrossRef]
- Senn, S.; Rolfe, K.; Julious, S.A. Investigating Variability in Patient Response to Treatment—A Case Study from a Replicate Cross-over Study. Stat. Methods Med. Res. 2011, 20, 657–666. [Google Scholar] [CrossRef]
- Gewandter, J.S.; McDermott, M.P.; He, H.; Gao, S.; Cai, X.; Farrar, J.T.; Katz, N.P.; Markman, J.D.; Senn, S.; Turk, D.C.; et al. Demonstrating Heterogeneity of Treatment Effects among Patients: An Overlooked but Important Step toward Precision Medicine. Clin. Pharmacol. Ther. 2019, 106, 204–210. [Google Scholar] [CrossRef]
- Berchialla, P.; Lanera, C.; Sciannameo, V.; Gregori, D.; Baldi, I. Prediction of Treatment Outcome in Clinical Trials under a Personalized Medicine Perspective. Sci. Rep. 2022, 12, 4115. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Sechidis, K.; Chen, Y.; Lu, J.; Ma, C.; Mirshani, A.; Ohlssen, D.; Vandemeulebroecke, M.; Bornkamp, B. Comparing Algorithms for Characterizing Treatment Effect Heterogeneity in Randomized Trials. Biom. J. 2024, 66, e2100337. [Google Scholar] [CrossRef]
- Ling, Y.; Upadhyaya, P.; Chen, L.; Jiang, X.; Kim, Y. Emulate Randomized Clinical Trials Using Heterogeneous Treatment Effect Estimation for Personalized Treatments: Methodology Review and Benchmark. J. Biomed. Inf. 2023, 137, 104256. [Google Scholar] [CrossRef]
- Lipkovich, I.; Svensson, D.; Ratitch, B.; Dmitrienko, A. Overview of Modern Approaches for Identifying and Evaluating Heterogeneous Treatment Effects from Clinical Data. Clin. Trials 2023, 20, 380–393. [Google Scholar] [CrossRef]
- Duan, X.-P.; Qin, B.-D.; Jiao, X.-D.; Liu, K.; Wang, Z.; Zang, Y.-S. New Clinical Trial Design in Precision Medicine: Discovery, Development and Direction. Signal Transduct. Target. Ther. 2024, 9, 57. [Google Scholar] [CrossRef]
- Webster, R.; Murphy, A.; Bygrave, H.; Ansbro, É.; Grobbee, D.E.; Perel, P. Implementing Fixed Dose Combination Medications for the Prevention and Control of Cardiovascular Diseases. Glob. Heart 2020, 15, 57. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, D.; Uzoije, P.; Reynolds, C.; Miller, R.; Walkley, D.; Pappalardo, S.; Tousey, P.; Munro, H.; Gonzales, H.; Song, W.; et al. Polypill for Cardiovascular Disease Prevention in an Underserved Population. N. Engl. J. Med. 2019, 381, 1114–1123. [Google Scholar] [CrossRef] [PubMed]
- Webster, R.; Castellano, J.M.; Onuma, O.K. Putting Polypills into Practice: Challenges and Lessons Learned. Lancet 2017, 389, 1066–1074. [Google Scholar] [CrossRef] [PubMed]
- Shchekochikhin, D.; Kopylov, P. Personalized Medicine in Coronary Artery Disease: Where Are We in 2022? J. Pers Med. 2022, 12, 1446. [Google Scholar] [CrossRef] [PubMed]
- Strisciuglio, T.; Franco, D.; Di Gioia, G.; De Biase, C.; Morisco, C.; Trimarco, B.; Barbato, E. Impact of Genetic Polymorphisms on Platelet Function and Response to Anti Platelet Drugs. Cardiovasc. Diagn. Ther. 2018, 8, 610–620. [Google Scholar] [CrossRef] [PubMed]
- Vavuranakis, M.; Kariori, M.G.; Kalogeras, K.I.; Vrachatis, D.A.; Moldovan, C.; Tousoulis, D.; Stefanadis, C. Biomarkers as a Guide of Medical Treatment in Cardiovascular Diseases. Curr. Med. Chem. 2012, 19, 2485–2496. [Google Scholar] [CrossRef] [PubMed]
- Pruett, A.E.; Lee, A.K.; Patterson, H.; Schwartz, T.A.; Glotzer, J.M.; Adams, K.F., Jr. Evolution of Biomarker Guided Therapy for Heart Failure: Current Concepts and Trial Evidence. Curr. Cardiol. Rev. 2015, 11, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Xue, J.; Wang, G.; Diao, Q. Nanoparticle-Based Drug Delivery Systems for the Treatment of Cardiovascular Diseases. Front. Pharmacol. 2022, 13, 999404. [Google Scholar] [CrossRef]
- Jeewandara, T.M.; Wise, S.G.; Ng, M.K.C. Biocompatibility of Coronary Stents. Materials 2014, 7, 769–786. [Google Scholar] [CrossRef]
- Waterhouse, A.; Yin, Y.; Wise, S.G.; Bax, D.V.; McKenzie, D.R.; Bilek, M.M.M.; Weiss, A.S.; Ng, M.K.C. The Immobilization of Recombinant Human Tropoelastin on Metals Using a Plasma-Activated Coating to Improve the Biocompatibility of Coronary Stents. Biomaterials 2010, 31, 8332–8340. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Jin, Y. Noncoding RNAs as Biomarkers for Acute Coronary Syndrome. Biomed. Res. Int. 2020, 2020, 3298696. [Google Scholar] [CrossRef] [PubMed]
- Rizzacasa, B.; Amati, F.; Romeo, F.; Novelli, G.; Mehta, J.L. Epigenetic Modification in Coronary Atherosclerosis: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2019, 74, 1352–1365. [Google Scholar] [CrossRef] [PubMed]
- Cimmino, G.; Loffredo, F.S.; Morello, A.; D’Elia, S.; De Palma, R.; Cirillo, P.; Golino, P. Immune-Inflammatory Activation in Acute Coronary Syndromes: A Look into the Heart of Unstable Coronary Plaque. Curr. Cardiol. Rev. 2017, 13, 110–117. [Google Scholar] [CrossRef] [PubMed]
| Name of the Study | Year | Studied Statin | Number of Participants | Disease of the Participants | Results |
|---|---|---|---|---|---|
| JUPITER (Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin) [64] | 2008 | Rosuvastatin | 17,802 | Elevated CRP, normal LDL | HR for major cardiovascular events: 0.56 (95% CI: 0.46–0.69); significant reduction in events |
| PROVE-IT TIMI 22 (Pravastatin or Atorvastatin Evaluation and Infection Therapy—Thrombolysis in Myocardial Infarction 22) [65] | 2004 | Atorvastatin | 4162 | Acute coronary syndromes | HR for death or major cardiovascular events: 0.84 (95% CI: 0.70–1.00); greater reduction in events with intensive therapy |
| REVERSAL (Reversal of Atherosclerosis with Aggressive Lipid Lowering) [66] | 2004 | Atorvastatin | 502 | Coronary artery disease | 0.4% reduction in atherosclerosis progression vs. 2.7% progression with pravastatin; p < 0.001 |
| PRINCE (Pravastatin Inflammation/CRP Evaluation) [67] | 2001 | Pravastatin | 1702 | Coronary artery disease | Significant reduction in CRP levels; improved clinical outcomes |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mitsis, A.; Myrianthefs, M.; Sokratous, S.; Karmioti, G.; Kyriakou, M.; Drakomathioulakis, M.; Tzikas, S.; Kadoglou, N.P.E.; Karagiannidis, E.; Nasoufidou, A.; et al. Emerging Therapeutic Targets for Acute Coronary Syndromes: Novel Advancements and Future Directions. Biomedicines 2024, 12, 1670. https://doi.org/10.3390/biomedicines12081670
Mitsis A, Myrianthefs M, Sokratous S, Karmioti G, Kyriakou M, Drakomathioulakis M, Tzikas S, Kadoglou NPE, Karagiannidis E, Nasoufidou A, et al. Emerging Therapeutic Targets for Acute Coronary Syndromes: Novel Advancements and Future Directions. Biomedicines. 2024; 12(8):1670. https://doi.org/10.3390/biomedicines12081670
Chicago/Turabian StyleMitsis, Andreas, Michael Myrianthefs, Stefanos Sokratous, Georgia Karmioti, Michaela Kyriakou, Michail Drakomathioulakis, Stergios Tzikas, Nikolaos P. E. Kadoglou, Efstratios Karagiannidis, Athina Nasoufidou, and et al. 2024. "Emerging Therapeutic Targets for Acute Coronary Syndromes: Novel Advancements and Future Directions" Biomedicines 12, no. 8: 1670. https://doi.org/10.3390/biomedicines12081670